Design, Engineering and Discovery of Novel α-Helical and β-Boomerang Antimicrobial Peptides against Drug Resistant Bacteria



Abstract
:1. Introduction
2. Engineering Temporins for Superior Activity
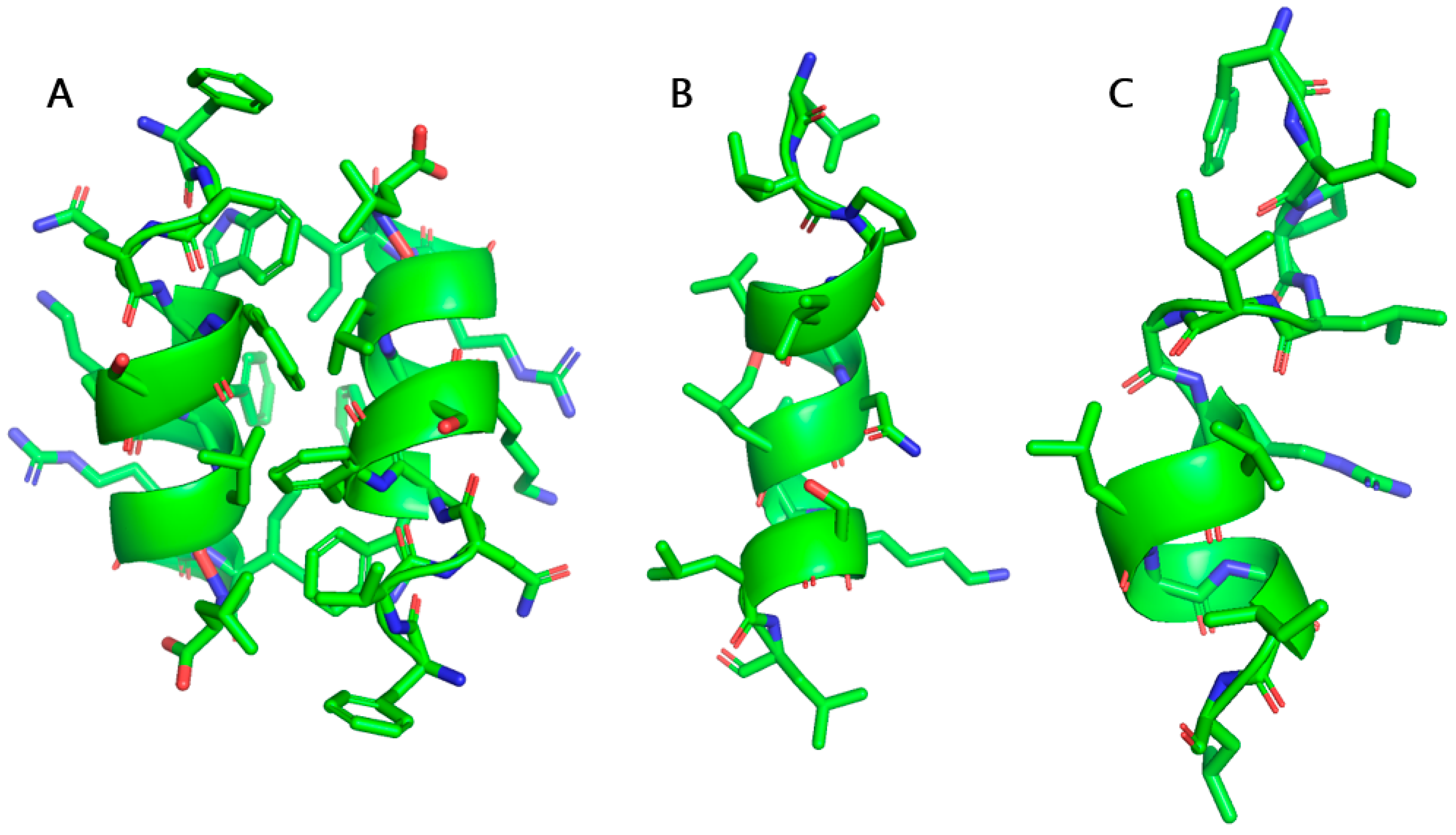
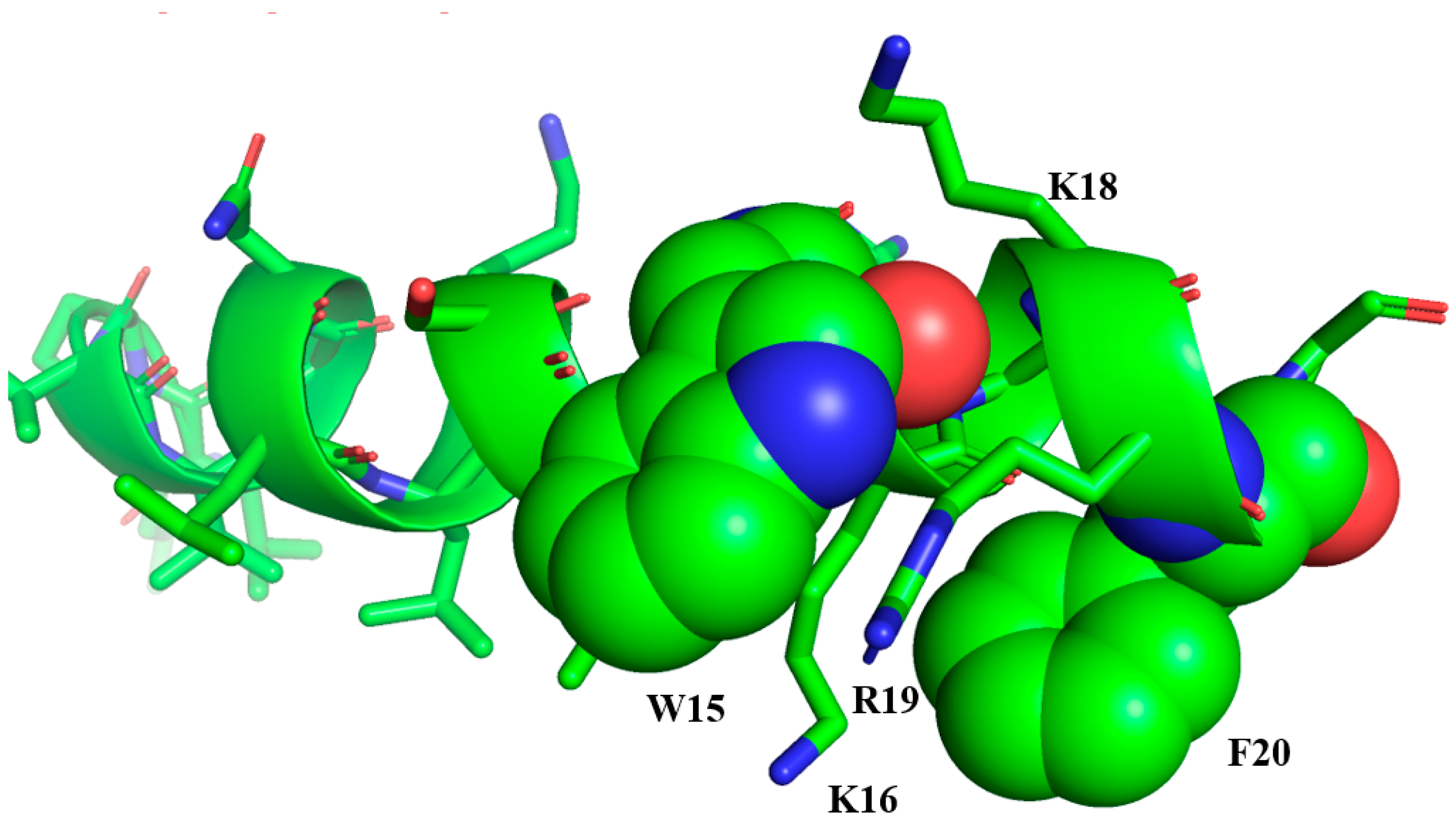
3. Engineering Aurein 2.2 for Superior Activity and Bioavailability
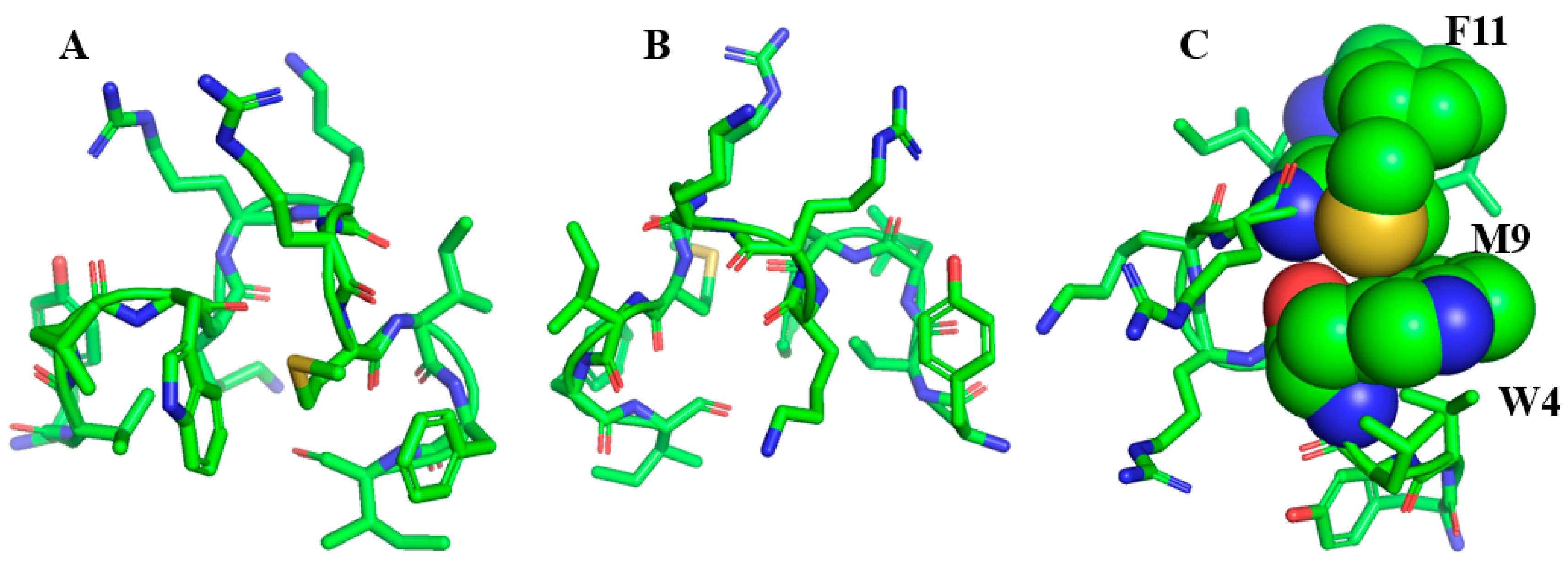
4. De Novo Designed β-Boomerang LPS Binding Antimicrobial and Antiendotoxic Peptides
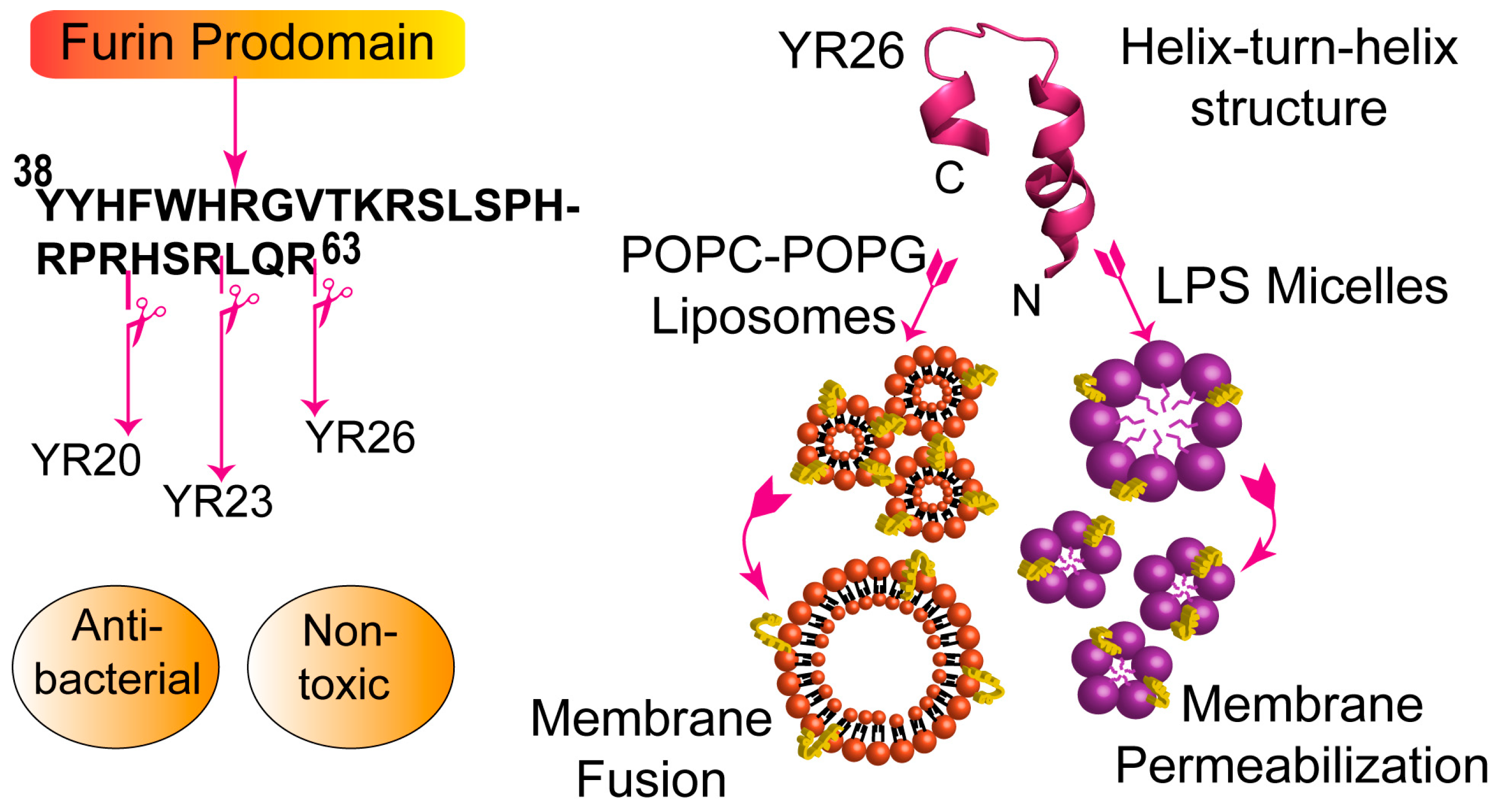
5. Mining the Human Proteome for Novel AMPs
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hancock, R.E.W.; Sahl, H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Haney, E.F.; Hancock, R.E.W. Peptide design for antimicrobial and immunomodulatory applications. Biopolymers 2013, 100, 572–583. [Google Scholar] [CrossRef]
- Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Reassessing the Host Defense Peptide Landscape. Front. Chem. 2019, 7, 43. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial peptides: Diversity, mechanism of action and strategies to improve the activity and biocompatibility in vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Dale, B.A.; Tao, R.; Kimball, J.R.; Jurevic, R.J. Oral antimicrobial peptides and biological control of caries. BMC Oral Health 2006, 6 (Suppl. S1), S13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidopoulou, S.; Diza, E.; Menexes, G.; Kalfas, S. Salivary concentration of the antimicrobial peptide LL-37 in children. Arch. Oral Biol. 2012, 57, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Pütsep, K.; Carlsson, G.; Boman, H.G.; Andersson, M. Deficiency of antibacterial peptides in patients with morbus Kostmann: An observation study. Lancet 2002, 360, 1144–1149. [Google Scholar] [CrossRef]
- Bowdish, D.M.E.; Davidson, D.J.; Hancock, R.E.W. A re-evaluation of the role of host defence peptides in mammalian immunity. Curr. Protein Pept. Sci. 2005, 6, 35–51. [Google Scholar] [CrossRef] [Green Version]
- Nijnik, A.; Hancock, R. Host defence peptides: Antimicrobial and immunomodulatory activity and potential applications for tackling antibiotic-resistant infections. Emerg. Health Threats J. 2009, 2, e1. [Google Scholar] [CrossRef] [PubMed]
- Felício, M.R.; Silva, O.N.; Gonçalves, S.; Santos, N.C.; Franco, O.L. Peptides with Dual Antimicrobial and Anticancer Activities. Front. Chem. 2017, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Hoskin, D.W.; Ramamoorthy, A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta 2008, 1778, 357–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernysh, S.; Kim, S.I.; Bekker, G.; Pleskach, V.A.; Filatova, N.A.; Anikin, V.B.; Platonov, V.G.; Bulet, P. Antiviral and antitumor peptides from insects. Proc. Natl. Acad. Sci. USA 2002, 99, 12628–12632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pletzer, D.; Hancock, R.E.W. Antibiofilm peptides: Potential as broadspectrum agents. J. Bacteriol. 2016, 198, 2572–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasir, M.; Willcox, M.D.P.; Dutta, D. Action of antimicrobial peptides against bacterial biofilms. Materials 2018, 11, 2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haney, E.F.; Brito-Sánchez, Y.; Trimble, M.J.; Mansour, S.C.; Cherkasov, A.; Hancock, R.E.W. Computer-aided Discovery of Peptides that Specifically Attack Bacterial Biofilms. Sci. Rep. 2018, 8, 1871. [Google Scholar] [CrossRef] [Green Version]
- Pletzer, D.; Mansour, S.C.; Hancock, R.E.W. Synergy between conventional antibiotics and anti-biofilm peptides in a murine, sub-cutaneous abscess model caused by recalcitrant ESKAPE pathogens. PLoS Pathog. 2018, 14, e1007084. [Google Scholar] [CrossRef]
- Uhlig, T.; Kyprianou, T.; Martinelli, F.G.; Oppici, C.A.; Heiligers, D.; Hills, D.; Calvo, X.R.; Verhaert, P. The emergence of peptides in the pharmaceutical business: From exploration to exploitation. EuPA Open Proteom. 2014. [Google Scholar] [CrossRef] [Green Version]
- Kosikowska, P.; Lesner, A. Antimicrobial peptides (AMPs) as drug candidates: A patent review (2003–2015). Expert Opin. Ther. Pat. 2016, 26, 689–702. [Google Scholar] [CrossRef]
- WHO Antimicrobial resistance. Bull. World Health Organ. 2014, 61, 383–394. [CrossRef]
- CDC. Antibiotic Resistance Threats in the United States, 2019; U.S. Department of Health and Human Services, CDC: Atlanta, GA, USA, 2019.
- Abdelraouf, K.; Braggs, K.H.; Yin, T.; Truong, L.D.; Hu, M.; Tam, V.H. Characterization of polymyxin B-induced nephrotoxicity: Implications for dosing regimen design. Antimicrob. Agents Chemother. 2012, 56, 4625–4629. [Google Scholar] [CrossRef] [Green Version]
- Dubashynskaya, N.V.; Skorik, Y.A. Polymyxin delivery systems: Recent advances and challenges. Pharmaceuticals 2020, 13, 83. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, M.M.; Rovig, J.; Weber, S.; Hilton, B.; Forouzan, M.M.; Savage, B. Susceptibility of Colistin-Resistant, Gram-Negative Bacteria to Antimicrobial Peptides and Ceragenins. Antimicrob. Agents Chemother. 2017, 61, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Zhou, D.; Steitz, T.A.; Polikanov, Y.S.; Gagnon, M.G. Ribosome-Targeting Antibiotics: Modes of Action, Mechanisms of Resistance, and Implications for Drug Design. Annu. Rev. Biochem. 2018, 87, 451–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, M.S.; Paterson, D.L. Antibiotics in the clinical pipeline in October 2019. J. Antibiot. (Tokyo) 2020, 73, 329. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Zasloff, M. Mysteries that still remain. Biochim. Biophys. Acta Biomembr. 2009, 1788, 1693–1694. [Google Scholar] [CrossRef] [Green Version]
- Lázár, V.; Martins, A.; Spohn, R.; Daruka, L.; Grézal, G. Antibiotic-resistant bacteria show widespread collateral sensitivity to antimicrobial peptides. Nat. Microbiol. 2018, 3, 718–731. [Google Scholar] [CrossRef] [Green Version]
- Raheem, N.; Straus, S.K. Mechanisms of Action for Antimicrobial Peptides with Antibacterial and Antibiofilm Functions. Front. Microbiol. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Park, C.B.; Kim, H.S.; Kim, S.C. Mechanism of action of the antimicrobial peptide buforin II: Buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 1998, 244, 253–257. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Chikushi, A.; Tougu, S.; Imura, Y.; Nishida, M.; Yano, Y.; Matsuzaki, K. Membrane Translocation Mechanism of the Antimicrobial Peptide Buforin 2. Biochemistry 2004, 43, 15610–15616. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papo, N.; Shai, Y. A molecular mechanism for lipopolysaccharide protection of Gram-negative bacteria from antimicrobial peptides. J. Biol. Chem. 2005, 280, 10378–10387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, S.; Kim, D.; McIntosh, T.J. Lipopolysaccharide bilayer structure: Effect of chemotype, core mutations, divalent cations, and temperature. Biochemistry 1999, 38, 10758–10767. [Google Scholar] [CrossRef] [PubMed]
- Snyder, D.S.; McIntosh, T.J. The lipopolysaccharide barrier: Correlation of antibiotic susceptibility with antibiotic permeability and fluorescent probe binding kinetics. Biochemistry 2000, 39, 11777–11787. [Google Scholar] [CrossRef]
- Nikaido, H.; Vaara, M. Molecular basis of bacterial outer membrane permeability. Microbiol. Rev. 1985, 49, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Nikaido, H. Prevention of drug access to bacterial targets: Permeability barriers and active efflux. Science 1994, 264, 382–388. [Google Scholar] [CrossRef] [Green Version]
- Nikaido, H. Molecular Basis of Bacterial Outer Membrane Permeability Revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjya, S. De novo Designed Lipopolysaccharide Binding Peptides: Structure Based Development of Antiendotoxic and Antimicrobial Drugs. Curr. Med. Chem. 2010, 17, 3080–3093. [Google Scholar] [CrossRef]
- Bhattacharjya, S.; Ramamoorthy, A. Multifunctional host defense peptides: Functional and mechanistic insights from NMR structures of potent antimicrobial peptides. FEBS J. 2009, 276, 6465–6473. [Google Scholar] [CrossRef] [Green Version]
- Allende, D.; McIntosh, T.J. Lipopolysaccharides in bacterial membranes act like cholesterol in eukaryotic plasma membranes in providing protection against melittin-induced bilayer lysis. Biochemistry 2003, 42, 1101–1108. [Google Scholar] [CrossRef]
- Cama, J.; Henney, A.M.; Winterhalter, M. Breaching the barrier: Quantifying antibiotic permeability across gram-negative bacterial membranes. J. Mol. Biol. 2019, 431, 3531–3546. [Google Scholar] [CrossRef] [PubMed]
- Bhunia, A.; Ramamoorthy, A.; Bhattacharjya, S. Helical hairpin structure of a potent antimicrobial peptide MSI-594 in lipopolysaccharide micelles by NMR spectroscopy. Chem. A Eur. J. 2009, 15, 2036–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhunia, A.; Domadia, P.N.; Torres, J.; Hallock, K.J.; Ramamoorthy, A.; Bhattacharjya, S. NMR structure of pardaxin, a pore-forming antimicrobial peptide, in lipopolysaccharide micelles: Mechanism of outer membrane permeabilization. J. Biol. Chem. 2010, 285, 3883–3895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domadia, P.N.; Bhunia, A.; Ramamoorthy, A.; Bhattacharjya, S. Structure, interactions, and antibacterial activities of MSI-594 derived mutant peptide MSI-594F5A in lipopolysaccharide micelles: Role of the helical hairpin conformation in outer-membrane permeabilization. J. Am. Chem. Soc. 2010, 132, 18417–18428. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjya, S. NMR Structures and Interactions of Antimicrobial Peptides with Lipopolysaccharide: Connecting Structures to Functions. Curr. Top. Med. Chem. 2015, 16, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Ilyas, H.; Kim, J.W.; Lee, D.K.; Malmsten, M.; Bhunia, A. Structural insights into the combinatorial effects of antimicrobial peptides reveal a role of aromatic-aromatic interactions in antibacterial synergism. J. Biol. Chem. 2019, 294, 14615–14633. [Google Scholar] [CrossRef]
- Bello, G.; Bodin, A.; Lawrence, M.J.; Barlow, D.; Mason, A.J.; Barker, R.D.; Harvey, R.D. The influence of rough lipopolysaccharide structure on molecular interactions with mammalian antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2016, 1858, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Zheng, L.; Mu, Y.; Ng, W.J.; Bhattacharjya, S. Structure and interactions of a host defense antimicrobial peptide thanatin in lipopolysaccharide micelles reveal mechanism of bacterial cell agglutination. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Swarbrick, J.D.; Karas, J.A.; Li, J.; Velkov, T. Structure of micelle bound cationic peptides by NMR spectroscopy using a lanthanide shift reagent. Chem. Commun. 2020, 56, 2897–2900. [Google Scholar] [CrossRef]
- Saravanan, R.; Holdbrook, D.A.; Petrlova, J.; Singh, S.; Berglund, N.A.; Choong, Y.K.; Kjellström, S.; Bond, P.J.; Malmsten, M.; Schmidtchen, A. Structural basis for endotoxin neutralisation and anti-inflammatory activity of thrombin-derived C-terminal peptides. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Mahajan, M.; Awasthi, B.; Tandon, A.; Harioudh, M.K.; Shree, S.; Singh, P.; Shukla, P.K.; Ramachandran, R.; Mitra, K.; et al. Piscidin-1-analogs with double L- and D-lysine residues exhibited different conformations in lipopolysaccharide but comparable anti-endotoxin activities. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, A.; Ghosh, A.; Airoldi, C.; Sperandeo, P.; Mroue, K.H.; Jimenez-Barbero, J.; Kundu, P.; Ramamoorthy, A.; Bhunia, A. Antimicrobial peptides: Insights into membrane permeabilization, lipopolysaccharide fragmentation and application in plant disease control. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzel, M.; Chiriac, A.I.; Otto, A.; Zweytick, D.; May, C.; Schumacher, C.; Gust, R.; Albada, H.B.; Penkova, M.; Krämer, U.; et al. Small cationic antimicrobial peptides delocalize peripheral membrane proteins. Proc. Natl. Acad. Sci. USA 2014, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, H.; Nguyen, L.T.; Gopal, R.; Aizawa, T.; Vogel, H.J. Overexpression of Antimicrobial, Anticancer, and Transmembrane Peptides in Escherichia coli through a Calmodulin-Peptide Fusion System. J. Am. Chem. Soc. 2016, 138, 11318–11326. [Google Scholar] [CrossRef] [PubMed]
- Abdel Monaim, S.A.H.; Somboro, A.M.; El-Faham, A.; de la Torre, B.G.; Albericio, F. Bacteria Hunt Bacteria through an Intriguing Cyclic Peptide. ChemMedChem 2019, 14, 24–51. [Google Scholar] [CrossRef]
- Wang, G. Post-Translational Modifications of Natural Antimicrobial Peptides and Strategies for Peptide Engineering. Curr. Biotechnol. 2012, 1, 72–79. [Google Scholar] [CrossRef]
- Ting, D.S.J.; Beuerman, R.W.; Dua, H.S.; Lakshminarayanan, R.; Mohammed, I. Strategies in Translating the Therapeutic Potentials of Host Defense Peptides. Front. Immunol. 2020, 11, 983. [Google Scholar] [CrossRef]
- Mourtada, R.; Herce, H.D.; Yin, D.J.; Moroco, J.A.; Wales, T.E.; Engen, J.R.; Walensky, L.D. Design of stapled antimicrobial peptides that are stable, nontoxic and kill antibiotic-resistant bacteria in mice. Nat. Biotechnol. 2019, 37, 1186–1197. [Google Scholar] [CrossRef]
- Kumar, P.; Pletzer, D.; Haney, E.F.; Rahanjam, N.; Cheng, J.T.J.; Yue, M.; Aljehani, W.; Hancock, R.E.W.; Kizhakkedathu, J.N.; Straus, S.K. Aurein-Derived Antimicrobial Peptides Formulated with Pegylated Phospholipid Micelles to Target Methicillin-Resistant Staphylococcus aureus Skin Infections. ACS Infect. Dis. 2019, 5, 443–453. [Google Scholar] [CrossRef]
- Wani, N.A.; Singh, G.; Shankar, S.; Sharma, A.; Katoch, M.; Rai, R. Short hybrid peptides incorporating β- and γ-amino acids as antimicrobial agents. Peptides 2017, 97, 46–53. [Google Scholar] [CrossRef]
- Hicks, R.P.; Abercrombie, J.J.; Wong, R.K.; Leung, K.P. Antimicrobial peptides containing unnatural amino acid exhibit potent bactericidal activity against ESKAPE pathogens. Bioorg. Med. Chem. 2013, 21, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Qvit, N.; Rubin, S.J.S.; Urban, T.J.; Mochly-Rosen, D.; Gross, E.R. Peptidomimetic therapeutics: Scientific approaches and opportunities. Drug Discov. Today 2017, 22, 454–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drayton, M.; Kizhakkedathu, J.N.; Straus, S.K. Towards Robust Delivery of Antimicrobial Peptides to Combat Bacterial Resistance. Molecules 2020, 25, 3048. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.L.; Papo, N.; Saugar, J.M.; Barra, D.; Shai, Y.; Simmaco, M.; Rivas, L. Effect of natural L- to D-amino acid conversion on the organization, membrane binding, and biological function of the antimicrobial peptides bombinins H. Biochemistry 2006, 45, 4266–4276. [Google Scholar] [CrossRef]
- Saravanan, R.; Bhunia, A.; Bhattacharjya, S. Micelle-bound structures and dynamics of the hinge deleted analog of melittin and its diastereomer: Implications in cell selective lysis by d-amino acid containing antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2010, 1798, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Oren, Z.; Shai, Y. Selective lysis of bacteria but not mammalian cells by diastereomers of melittin: Structure-function study. Biochemistry 1997, 36, 1826–1835. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Kumari, T.; Harioudh, M.K.; Yadav, P.K.; Kathuria, M.; Shukla, P.K.; Mitra, K.; Ghosh, J.K. Identification of GXXXXG motif in Chrysophsin-1 and its implication in the design of analogs with cell-selective antimicrobial and anti-endotoxin activities. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar Tripathi, A.; Kathuria, M.; Shree, S.; Kumar Tripathi, J.; Purshottam, R.K.; Ramachandran, R.; Mitra, K.; Kanti Ghosh, J. Single amino acid substitutions at specific positions of the heptad repeat sequence of piscidin-1 yielded novel analogs that show low cytotoxicity and in vitro and in vivo antiendotoxin activity. Antimicrob. Agents Chemother. 2016, 60, 3687–3699. [Google Scholar] [CrossRef] [Green Version]
- Saravanan, R.; Li, X.; Lim, K.; Mohanram, H.; Peng, L.; Mishra, B.; Basu, A.; Lee, J.M.; Bhattacharjya, S.; Leong, S.S.J. Design of short membrane selective antimicrobial peptides containing tryptophan and arginine residues for improved activity, salt-resistance, and biocompatibility. Biotechnol. Bioeng. 2014, 111, 37–49. [Google Scholar] [CrossRef]
- Kumar, P.; Takayesu, A.; Abbasi, U.; Kalathottukaren, M.T.; Abbina, S.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptide-Polymer Conjugates with High Activity: Influence of Polymer Molecular Weight and Peptide Sequence on Antimicrobial Activity, Proteolysis, and Biocompatibility. ACS Appl. Mater. Interfaces 2017, 9. [Google Scholar] [CrossRef]
- Kumar, P.; Shenoi, R.A.; Lai, B.F.L.; Nguyen, M.; Kizhakkedathu, J.N.; Straus, S.K. Conjugation of Aurein 2.2 to HPG Yields an Antimicrobial with Better Properties. Biomacromolecules 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Ladram, A.; Nicolas, P. Antimicrobial peptides from frog skin: Biodiversity and therapeutic promises. Front. Biosci. Landmark 2016, 21, 1341–1371. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Lai, R. The chemistry and biological activities of peptides from amphibian skin secretions. Chem. Rev. 2015, 115, 1760–1846. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, A.C. Antimicrobial peptides from amphibian skin: An expanding scenario: Commentary. Curr. Opin. Chem. Biol. 2002, 6, 799–804. [Google Scholar] [CrossRef]
- Pukala, T.L.; Bowie, J.H.; Maselli, V.M.; Musgrave, I.F.; Tyler, M.J. Host-defence peptides from the glandular secretions of amphibians: Structure and activity. Nat. Prod. Rep. 2006, 23, 368–393. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangoni, M.L.; Di Grazia, A.; Cappiello, F.; Casciaro, B.; Luca, V. Naturally Occurring Peptides from Rana temporaria: Antimicrobial Properties and More. Curr. Top. Med. Chem. 2015, 16, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Simmaco, M.; Mignogna, G.; Canofeni, S.; Miele, R.; Mangoni, M.L.; Barra, D. Temporins, antimicrobial peptides from the European red frog Rana temporaria. Eur. J. Biochem. 1996, 242, 788–792. [Google Scholar] [CrossRef]
- Mangoni, M.L.; Shai, Y. Temporins and their synergism against Gram-negative bacteria and in lipopolysaccharide detoxification. Biochim. Biophys. Acta Biomembr. 2009, 1788, 1610–1619. [Google Scholar] [CrossRef] [Green Version]
- Mangoni, M.L. Temporins, anti-infective peptides with expanding properties. Cell. Mol. Life Sci. 2006, 63, 1060–1069. [Google Scholar] [CrossRef]
- Mahalka, A.K.; Kinnunen, P.K.J. Binding of amphipathic α-helical antimicrobial peptides to lipid membranes: Lessons from temporins B and L. Biochim. Biophys. Acta Biomembr. 2009, 1788, 1600–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Rinaldi, A.C.; Di Giulio, A.; Simmaco, M.; Kinnunen, P.K.J. Interactions of the antimicrobial peptides temporins with model biomembranes. Comparison of temporins B and L. Biochemistry 2002, 41, 4425–4436. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, A.C.; Mangoni, M.L.; Rufo, A.; Luzi, C.; Barra, D.; Zhao, H.; Kinnunen, P.K.J.; Bozzi, A.; Di Giulio, A.; Simmaco, M. Temporin L: Antimicrobial, haemolytic and cytotoxic activities, and effects on membrane permeabilization in lipid vesicles. Biochem. J. 2002, 368, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangoni, M.L.; Papo, N.; Barra, D.; Simmaco, M.; Bozzi, A.; Di Giulio, A.; Rinaldi, A.C. Effects of the antimicrobial peptide temporin L on cell morphology, membrane permeability and viability of Escherichia coli. Biochem. J. 2004, 380, 859–865. [Google Scholar] [CrossRef] [Green Version]
- Giacometti, A.; Cirioni, O.; Kamysz, W.; D’Amato, G.; Silvestri, C.; Del Prete, M.S.; Licci, A.; Łukasiak, J.; Scalise, G. In vitro activity and killing effect of temporin A on nosocomial isolates of Enterococcus faecalis and interactions with clinically used antibiotics. J. Antimicrob. Chemother. 2005, 55, 272–274. [Google Scholar] [CrossRef]
- Giacometti, A.; Cirioni, O.; Ghiselli, R.; Mocchegiani, F.; Orlando, F.; Silvestri, C.; Bozzi, A.; Di Giulio, A.; Luzi, C.; Mangoni, M.L.; et al. Interaction of antimicrobial peptide temporin L with lipopolysaccharide in vitro and in experimental rat models of septic shock caused by gram-negative bacterial. Antimicrob. Agents Chemother. 2006, 50, 2478–2486. [Google Scholar] [CrossRef] [Green Version]
- Carotenuto, A.; Malfi, S.; Saviello, M.R.; Campiglia, P.; Gomez-Monterrey, I.; Mangoni, M.L.; Gaddi, L.M.H.; Novellino, E.; Grieco, P. A different molecular mechanism underlying antimicrobial and hemolytic actions of temporins A and L. J. Med. Chem. 2008, 51, 2354–2362. [Google Scholar] [CrossRef]
- Mangoni, M.L.; Carotenuto, A.; Auriemma, L.; Saviello, M.R.; Campiglia, P.; Gomez-Monterrey, I.; Malfi, S.; Marcellini, L.; Barra, D.; Novellino, E.; et al. Structure-activity relationship, conformational and biological studies of temporin l analogues. J. Med. Chem. 2011, 54, 1298–1307. [Google Scholar] [CrossRef]
- Saviello, M.R.; Malfi, S.; Campiglia, P.; Cavalli, A.; Grieco, P.; Novellino, E.; Carotenuto, A. New insight into the mechanism of action of the temporin antimicrobial peptides. Biochemistry 2010, 49, 1477–1485. [Google Scholar] [CrossRef]
- Srivastava, S.; Ghosh, J.K. Introduction of a lysine residue promotes aggregation of temporin L in lipopolysaccharides and augmentation of its antiendotoxin property. Antimicrob. Agents Chemother. 2013, 57, 2457–2466. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Zhu, X.; Zhou, X.; Yang, Y.; Yan, X.; Sun, L.; Shang, D. Potential role of a series of lysine-/leucine-rich antimicrobial peptide in inhibiting lipopolysaccharide-induced inflammation. Biochem. J. 2018, 475, 3687–3706. [Google Scholar] [CrossRef] [PubMed]
- Crépin, A.; Jégou, J.F.; André, S.; Ecale, F.; Croitoru, A.; Cantereau, A.; Berjeaud, J.M.; Ladram, A.; Verdon, J. In vitro and intracellular activities of frog skin temporins against Legionella pneumophila and its eukaryotic hosts. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadbourne, F.L.; Raleigh, C.; Ali, H.Z.; Denny, P.W.; Cobb, S.L. Studies on the antileishmanial properties of the antimicrobial peptides temporin A, B and 1Sa. J. Pept. Sci. 2011, 17, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.L.; Saugar, J.M.; Dellisanti, M.; Barra, D.; Simmaco, M.; Rivas, L. Temporins, small antimicrobial peptides with leishmanicidal activity. J. Biol. Chem. 2005, 280, 984–990. [Google Scholar] [CrossRef] [Green Version]
- Marcocci, M.E.; Amatore, D.; Villa, S.; Casciaro, B.; Aimola, P.; Franci, G.; Grieco, P.; Galdiero, M.; Palamara, A.T.; Mangoni, M.L.; et al. The amphibian antimicrobial peptide temporin b inhibits in vitro herpes simplex virus 1 infection. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Simonetti, O.; Cirioni, O.; Goteri, G.; Ghiselli, R.; Kamysz, W.; Kamysz, E.; Silvestri, C.; Orlando, F.; Barucca, C.; Scalise, A.; et al. Temporin A is effective in MRSA-infected wounds through bactericidal activity and acceleration of wound repair in a murine model. Peptides 2008, 29, 520–528. [Google Scholar] [CrossRef]
- Grazia, A.D.; Luca, V.; Segev-Zarko, L.A.T.; Shai, Y.; Mangoni, M.L. Temporins a and b stimulate migration of hacat keratinocytes and kill intracellular staphylococcus aureus. Antimicrob. Agents Chemother. 2014, 58, 2520–2527. [Google Scholar] [CrossRef] [Green Version]
- Grieco, P.; Carotenuto, A.; Auriemma, L.; Saviello, M.R.; Campiglia, P.; Gomez-Monterrey, I.M.; Marcellini, L.; Luca, V.; Barra, D.; Novellino, E.; et al. The effect of d-amino acid substitution on the selectivity of temporin L towards target cells: Identification of a potent anti-Candida peptide. Biochim. Biophys. Acta Biomembr. 2013, 1828, 652–660. [Google Scholar] [CrossRef]
- Diao, Y.; Han, W.; Zhao, H.; Zhu, S.; Liu, X.; Feng, X.; Gu, J.; Yao, C.; Liu, S.; Sun, C.; et al. Designed synthetic analogs of the α-helical peptide temporin-La with improved antitumor efficacies via charge modification and incorporation of the integrin αvβ3 homing domain. J. Pept. Sci. 2012, 18, 476–486. [Google Scholar] [CrossRef]
- Malgieri, G.; Avitabile, C.; Palmieri, M.; D’Andrea, L.D.; Isernia, C.; Romanelli, A.; Fattorusso, R. Structural basis of a temporin 1b analogue antimicrobial activity against gram negative bacteria determined by CD and NMR techniques in cellular environment. ACS Chem. Biol. 2015, 10, 965–969. [Google Scholar] [CrossRef]
- Mangoni, M.L.; Shai, Y. Short native antimicrobial peptides and engineered ultrashort lipopeptides: Similarities and differences in cell specificities and modes of action. Cell. Mol. Life Sci. 2011, 68, 2267–2280. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, Y.; Barra, D.; Simmaco, M.; Shai, Y.; Mangoni, M.L. A synergism between temporins toward Gram-negative bacteria overcomes resistance imposed by the lipopolysaccharide protective layer. J. Biol. Chem. 2006, 281, 28565–28574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangoni, M.L.; Epand, R.F.; Rosenfeld, Y.; Peleg, A.; Barra, D.; Epand, R.M.; Shai, Y. Lipopolysaccharide, a key molecule involved in the synergism between temporins in inhibiting bacterial growth and in endotoxin neutralization. J. Biol. Chem. 2008, 283, 22907–22917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avitabile, C.; Netti, F.; Orefice, G.; Palmieri, M.; Nocerino, N.; Malgieri, G.; D’Andrea, L.D.; Capparelli, R.; Fattorusso, R.; Romanelli, A. Design, structural and functional characterization of a Temporin-1b analog active against Gram-negative bacteria. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3767–3775. [Google Scholar] [CrossRef] [Green Version]
- Saravanan, R.; Joshi, M.; Mohanram, H.; Bhunia, A.; Mangoni, M.L.; Bhattacharjya, S. NMR Structure of Temporin-1 Ta in Lipopolysaccharide Micelles: Mechanistic Insight into Inactivation by Outer Membrane. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Bhunia, A.; Saravanan, R.; Mohanram, H.; Mangoni, M.L.; Bhattacharjya, S. NMR structures and interactions of temporin-1Tl and temporin-1Tb with lipopolysaccharide micelles: Mechanistic insights into outer membrane permeabilization and synergistic activity. J. Biol. Chem. 2011, 286, 24394–24406. [Google Scholar] [CrossRef] [Green Version]
- Avitabile, C.; D’Andrea, L.D.; Saviano, M.; Olivieri, M.; Cimmino, A.; Romanelli, A. Binding studies of antimicrobial peptides to Escherichia coli cells. Biochem. Biophys. Res. Commun. 2016, 478, 149–153. [Google Scholar] [CrossRef]
- Grassi, L.; Maisetta, G.; Maccari, G.; Esin, S.; Batoni, G. Analogs of the frog-skin antimicrobial peptide temporin 1Tb exhibit a wider spectrum of activity and a stronger antibiofilm potential as compared to the parental peptide. Front. Chem. 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- Manzo, G.; Ferguson, P.M.; Gustilo, V.B.; Hind, C.K.; Clifford, M.; Bui, T.T.; Drake, A.F.; Atkinson, R.A.; Sutton, J.M.; Batoni, G.; et al. Minor sequence modifications in temporin B cause drastic changes in antibacterial potency and selectivity by fundamentally altering membrane activity. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Capparelli, R.; Romanelli, A.; Iannaccone, M.; Nocerino, N.; Ripa, R.; Pensato, S.; Pedone, C.; Iannelli, D. Synergistic antibacterial and anti-inflammatory activity of temporin A and modified temporin B in vivo. PLoS ONE 2009, 4. [Google Scholar] [CrossRef]
- Grieco, P.; Luca, V.; Auriemma, L.; Carotenuto, A.; Saviello, M.R.; Campiglia, P.; Barra, D.; Novellino, E.; Mangoni, M.L. Alanine scanning analysis and structure-function relationships of the frog-skin antimicrobial peptide temporin-1Ta. J. Pept. Sci. 2011, 17, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Merlino, F.; Carotenuto, A.; Casciaro, B.; Martora, F.; Loffredo, M.R.; Di Grazia, A.; Yousif, A.M.; Brancaccio, D.; Palomba, L.; Novellino, E.; et al. Glycine-replaced derivatives of [Pro3,DLeu9]TL, a temporin L analogue: Evaluation of antimicrobial, cytotoxic and hemolytic activities. Eur. J. Med. Chem. 2017, 139, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Kumar, A.; Tripathi, A.K.; Tandon, A.; Ghosh, J.K. Modulation of anti-endotoxin property of Temporin L by minor amino acid substitution in identified phenylalanine zipper sequence. Biochem. J. 2016, 473, 4045–4062. [Google Scholar] [CrossRef] [PubMed]
- Bhunia, A.; Mohanram, H.; Domadia, P.N.; Torres, J.; Bhattacharjya, S. Designed β-boomerang antiendotoxic and antimicrobial peptides. Structures and activities in lipopolysaccharide. J. Biol. Chem. 2009, 284, 21991–22004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanram, H.; Bhattacharjya, S. Resurrecting inactive antimicrobial peptides from the lipopolysaccharide trap. Antimicrob. Agents Chemother. 2014, 58, 1987–1996. [Google Scholar] [CrossRef] [Green Version]
- Mohanram, H.; Bhattacharjya, S. ’Lollipop’-shaped helical structure of a hybrid antimicrobial peptide of temporin B-lipopolysaccharide binding motif and mapping cationic residues in antibacterial activity. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 1362–1372. [Google Scholar] [CrossRef]
- Rozek, T.; Wegener, K.L.; Bowie, J.H.; Olver, I.N.; Carver, J.A.; Wallace, J.C.; Tyler, M.J. The antibiotic and anticancer active aurein peptides from the Australian Bell Frogs Litoria aurea and Litoria raniformis: The solution structure of aurein 1.2. Eur. J. Biochem. 2000, 267, 5330–5341. [Google Scholar] [CrossRef]
- Rozek, T.; Bowie, J.H.; Wallace, J.C.; Tyler, M.J. The antibiotic and anticancer active aurein peptides from the Australian Bell FrogsLitoria aurea andLitoria raniformis. Part 2. Sequence determination using electrospray mass spectrometry. Rapid Commun. Mass Spectrom. 2000, 14, 2002–2011. [Google Scholar] [CrossRef]
- Cheng, J.T.J.; Hale, J.D.; Elliot, M.; Hancock, R.E.W.; Straus, S.K. Effect of membrane composition on antimicrobial peptides aurein 2.2 and 2.3 from australian southern bell frogs. Biophys. J. 2009, 96. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.-L.; Cheng, J.T.-J.; Hale, J.; Pan, J.; Hancock, R.E.W.; Straus, S.K. Characterization of the structure and membrane interaction of the antimicrobial peptides aurein 2.2 and 2.3 from Australian southern bell frogs. Biophys. J. 2007, 92. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.T.J.; Hale, J.D.; Elliott, M.; Hancock, R.E.W.; Straus, S.K. The importance of bacterial membrane composition in the structure and function of aurein 2.2 and selected variants. Biochim. Biophys. Acta Biomembr. 2011, 1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.T.J.; Hale, J.D.; Kindrachuk, J.; Jessen, H.; Elliott, M.; Hancock, R.E.W.; Straus, S.K. Importance of residue 13 and the C-terminus for the structure and activity of the antimicrobial peptide aurein 2.2. Biophys. J. 2010, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzo, G.; Ferguson, P.M.; Hind, C.K.; Clifford, M.; Gustilo, V.B.; Ali, H.; Bansal, S.S.; Bui, T.T.; Drake, A.F.; Atkinson, R.A.; et al. Temporin L and aurein 2.5 have identical conformations but subtly distinct membrane and antibacterial activities. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, D.I.; Le Brun, A.P.; Whitwell, T.C.; Sani, M.A.; James, M.; Separovic, F. The antimicrobial peptide aurein 1.2 disrupts model membranes via the carpet mechanism. Phys. Chem. Chem. Phys. 2012, 14, 15739–15751. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.I.; Sani, M.A.; Miles, A.J.; Wallace, B.A.; Separovic, F. Membrane defects enhance the interaction of antimicrobial peptides, aurein 1.2 versus caerin 1.1. Biochim. Biophys. Acta Biomembr. 2013, 1828, 1863–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzel, M.; Senges, C.H.R.; Zhang, J.; Suleman, S.; Nguyen, M.; Kumar, P.; Chiriac, A.I.; Stepanek, J.J.; Raatschen, N.; May, C.; et al. Antimicrobial peptides from the aurein family form ion-selective pores in Bacillus subtilis. ChemBioChem 2015, 16. [Google Scholar] [CrossRef]
- Haney, E.F.; Nguyen, L.T.; Schibli, D.J.; Vogel, H.J. Design of a novel tryptophan-rich membrane-active antimicrobial peptide from the membrane-proximal region of the HIV glycoprotein, gp41. Beilstein J. Org. Chem. 2012, 8, 1172–1184. [Google Scholar] [CrossRef]
- Chan, D.I.; Prenner, E.J.; Vogel, H.J. Tryptophan- and arginine-rich antimicrobial peptides: Structures and mechanisms of action. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1184–1202. [Google Scholar] [CrossRef] [Green Version]
- Frank, R. The SPOT-synthesis Technique. Synthethic Peptide Arrays on Membrane Support—Principles and Applications. J. Immunol. Methods 2002, 267, 13–26. [Google Scholar] [CrossRef]
- Hilpert, K.; Winkler, D.F.H.; Hancock, R.E.W. Peptide arrays on cellulose support: SPOT synthesis, a time and cost efficient method for synthesis of large numbers of peptides in a parallel and addressable fashion. Nat. Protoc. 2007, 2, 1333–1349. [Google Scholar] [CrossRef]
- Winkler, D.F.H.; Hilpert, K.; Brandt, O.; Hancock, R.E.W. Peptide Microarrays. Methods and Protocols; Cretich, M., Chiari, M., Eds.; Springer: Dordrecht, The Netherlands; Heidelberg, Germany; London, UK; New York, NY, USA, 2009; p. 570. [Google Scholar] [CrossRef]
- Haney, E.F.; Mansour, S.C.; Hilchie, A.L.; de la Fuente-Núñez, C.; Hancock, R.E.W. High Throughput Screening Methods for Assessing Antibiofilm and Immunomodulatory Activities of Synthetic Peptides. Peptides 2015, 71, 276–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherkasov, A.; Hilpert, K.; Jenssen, H.; Fjell, C.D.; Waldbrook, M.; Mullaly, S.C.; Volkmer, R.; Hancock, R.E.W. Use of Artificial Intelligence in the Design of Small Peptide Antibiotics Effective against a Broad Spectrum of Highly Antibiotic-Resistant Superbugs. ACS Chem. Biol. 2009, 4, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Winkler, D.F.H.; Hilpert, K.; Brandt, O.; Hancock, R.E.W. Synthesis of peptide arrays using SPOT-technology and the CelluSpots-method. Methods Mol. Biol. 2009, 570, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Raheem, N.; Kumar, P.; Lee, E.; Cheng, J.T.J.; Hancock, R.E.W.; Straus, S.K. Insights into the mechanism of action of two analogues of aurein 2.2. Biochim. Biophys. Acta Biomembr. 2020, 1862. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H.; Chen, C.; Jou, M.L.; Lee, A.Y.L.; Lin, Y.C.; Yu, Y.P.; Huang, W.T.; Wu, S.H. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: Evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. 2005, 33, 4053–4064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchand, C.; Krajewski, K.; Lee, H.-F.; Antony, S.; Johnson, A.A.; Amin, R.; Roller, P.; Kvaratskhelia, M.; Pommier, Y. Covalent binding of the natural antimicrobial peptide indolicidin to DNA abasic sites. Nucleic Acids Res. 2006, 34, 5157–5165. [Google Scholar] [CrossRef]
- Overhage, J.; Campisano, A.; Bains, M.; Torfs, E.C.W.; Rehm, B.H.A.; Hancock, R.E.W. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect. Immun. 2008, 76, 4176–4182. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, M.; Resende, J.M.; Moraes, C.M.; Marquette, A.; Chich, J.-F.; Metz-Boutigue, M.-H.; Bechinger, B. Membrane structure and interactions of human catestatin by multidimensional solution and solid-state NMR spectroscopy. FASEB J. 2010, 24, 1737–1746. [Google Scholar] [CrossRef] [Green Version]
- Khafagy, E.S.; Morishita, M.; Ida, N.; Nishio, R.; Isowa, K.; Takayama, K. Structural requirements of penetratin absorption enhancement efficiency for insulin delivery. J. Control Release 2010, 143, 302–310. [Google Scholar] [CrossRef]
- Vogt, T.C.B.; Bechinger, B. The interactions of histidine-containing amphipathic helical peptide antibiotics with lipid bilayers. The effects of charges and pH. J. Biol. Chem. 1999, 274, 29115–29121. [Google Scholar] [CrossRef] [Green Version]
- Kichler, A.; Leborgne, C.; März, J.; Danos, O.; Bechinger, B. Histidine-rich amphipathic peptide antibiotics promote efficient delivery of DNA into mammalian cells. Proc. Natl. Acad. Sci. USA 2003, 100, 1564–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moulay, G.; Leborgne, C.; Mason, A.J.; Aisenbrey, C.; Kichler, A.; Bechinger, B. Histidine-rich designer peptides of the LAH4 family promote cell delivery of a multitude of cargo. J. Pept. Sci. 2017, 23, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjya, S.; Domadia, P.N.; Bhunia, A.; Malladi, S.; David, S.A. High-resolution solution structure of a designed peptide bound to lipopolysaccharide: Transferred nuclear overhauser effects, micelle selectivity, and anti-endotoxic activity. Biochemistry 2007, 46, 5864–5874. [Google Scholar] [CrossRef] [PubMed]
- Mohanram, H.; Bhattacharjya, S. β-boomerang antimicrobial and antiendotoxic peptides: Lipidation and disulfide bond effects on activity and structure. Pharmaceuticals 2014, 7, 482–501. [Google Scholar] [CrossRef]
- Mathew, B.; Nagaraj, R. Variations in the interaction of human defensins with Escherichia coli: Possible implications in bacterial killing. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Shen, M.; Gohain, N.; Tolbert, W.D.; Chen, F.; Zhang, N.; Yang, K.; Wang, A.; Su, Y.; Cheng, T.; et al. Design of a Potent Antibiotic Peptide Based on the Active Region of Human Defensin 5. J. Med. Chem. 2015, 58, 3083–3093. [Google Scholar] [CrossRef]
- Du, H.; Puri, S.; McCall, A.; Norris, H.L.; Russo, T.; Edgerton, M. Human salivary protein histatin 5 has potent bactericidal activity against ESKAPE pathogens. Front. Cell. Infect. Microbiol. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Papareddy, P.; Mörgelin, M.; Walse, B.; Schmidtchen, A.; Malmsten, M. Antimicrobial activity of peptides derived from human ß-amyloid precursor protein. J. Pept. Sci. 2012, 18, 183–191. [Google Scholar] [CrossRef]
- Kasetty, G.; Papareddy, P.; Kalle, M.; Rydengrd, V.; Walse, B.; Svensson, B.; Mörgelin, M.; Malmsten, M.; Schmidtchen, A. The C-terminal sequence of several human serine proteases encodes host defense functions. J. Innate Immun. 2011, 3, 471–482. [Google Scholar] [CrossRef]
- Papareddy, P.; Rydengård, V.; Pasupuleti, M.; Walse, B.; Mörgelin, M.; Chalupka, A.; Malmsten, M.; Schmidtchen, A. Proteolysis of human thrombin generates novel host defense peptides. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef] [Green Version]
- Petrlova, J.; Hansen, F.C.; Van Der Plas, M.J.A.; Huber, R.G.; Mörgelin, M.; Malmsten, M.; Bond, P.J.; Schmidtchen, A. Aggregation of thrombin-derived C-terminal fragments as a previously undisclosed host defense mechanism. Proc. Natl. Acad. Sci. USA 2017, 114, E4213–E4222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasetty, G.; Papareddy, P.; Kalle, M.; Rydengård, V.; Mörgelin, M.; Albiger, B.; Malmsten, M.; Schmidtchen, A. Structure-activity studies and therapeutic potential of host defense peptides of human thrombin. Antimicrob. Agents Chemother. 2011, 55, 2880–2890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.B.; Kim, M.S.; Kim, S.C. A novel antimicrobial peptide from Bufo bufo gargarizans. Biochem. Biophys. Res. Commun. 1996, 218, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Iwamuro, S. Potential Roles of Histones in Host Defense as Antimicrobial Agents. Infect. Disord. Drug Targets 2012, 8, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Hoeksema, M.; Van Eijk, M.; Haagsman, H.P.; Hartshorn, K.L. Histones as mediators of host defense, inflammation and thrombosis. Future Microbiol. 2016, 11, 441–453. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.H.; Sung, B.H.; Kim, S.C. Buforins: Histone H2A-derived antimicrobial peptides from toad stomach. Biochim. Biophys. Acta Biomembr. 2009, 1788, 1564–1569. [Google Scholar] [CrossRef] [Green Version]
- Sim, S.; Wang, P.; Beyer, B.N.; Cutrona, K.J.; Radhakrishnan, M.L.; Elmore, D.E. Investigating the nucleic acid interactions of histone-derived antimicrobial peptides. FEBS Lett. 2017, 591, 706–717. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, H.R.; Thomas, U.; Pellegrini, A. A Helix-Loop-Helix Peptide at the Upper Lip of the Active Site Cleft of Lysozyme Confers Potent Antimicrobial Activity with Membrane Permeabilization Action. J. Biol. Chem. 2001, 276, 43767–43774. [Google Scholar] [CrossRef] [Green Version]
- Hunter, H.N.; Jing, W.; Schibli, D.J.; Trinh, T.; Park, I.Y.; Kim, S.C.; Vogel, H.J. The interactions of antimicrobial peptides derived from lysozyme with model membrane systems. Biochim. Biophys. Acta Biomembr. 2005, 1668, 175–189. [Google Scholar] [CrossRef] [Green Version]
- Harder, J.; Schröder, J.M. RNase 7, a novel innate immune defense antimicrobial protein of healthy human skin. J. Biol. Chem. 2002, 277, 46779–46784. [Google Scholar] [CrossRef] [Green Version]
- Pulido, D.; Moussaoui, M.; Andreu, D.; Nogués, M.V.; Torrent, M.; Boix, E. Antimicrobial action and cell agglutination by the eosinophil cationic protein are modulated by the cell wall lipopolysaccharide structure. Antimicrob. Agents Chemother. 2012, 56, 2378–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svendsen, J.S.M.; Grant, T.M.; Rennison, D.; Brimble, M.A.; Svenson, J. Very Short and Stable Lactoferricin-Derived Antimicrobial Peptides: Design Principles and Potential Uses. Acc. Chem. Res. 2019, 52, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Hossain, F.; Moghal, M.M.R.; Islam, M.Z.; Moniruzzaman, M.; Yamazaki, M. Membrane potential is vital for rapid permeabilization of plasma membranes and lipid bilayers by the antimicrobial peptide lactoferricin B. J. Biol. Chem. 2019, 294, 10449–10462. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Chau, J.K.; Perry, N.A.; de Boer, L.; Zaat, S.A.J.; Vogel, H.J. Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS ONE 2010, 5, e12684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gifford, J.L.; Hunter, H.N.; Vogel, H.J. Lactoferricin: A lactoferrin-derived peptide with antimicrobial, antiviral, antitumor and immunological properties. Cell. Mol. Life Sci. 2005, 62, 2588–2598. [Google Scholar] [CrossRef] [PubMed]
- Hunter, H.N.; Ross Demcoe, A.; Jenssen, H.; Gutteberg, T.J.; Vogel, H.J. Human lactoferricin is partially folded in aqueous solution and is better stabilized in a membrane mimetic solvent. Antimicrob. Agents Chemother. 2005, 49, 3387–3395. [Google Scholar] [CrossRef] [Green Version]
- Haney, E.F.; Nazmi, K.; Bolscher, J.G.M.; Vogel, H.J. Structural and biophysical characterization of an antimicrobial peptide chimera comprised of lactoferricin and lactoferrampin. Biochim. Biophys. Acta 2012, 1818, 762–775. [Google Scholar] [CrossRef] [Green Version]
- Arias, M.; Hilchie, A.L.; Haney, E.F.; Bolscher, J.G.M.; Hyndman, M.E.; Hancock, R.E.W.; Vogel, H.J. Anticancer activities of bovine and human lactoferricin-derived peptides. Biochem. Cell Biol. 2017, 95, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Pane, K.; Sgambati, V.; Zanfardino, A.; Smaldone, G.; Cafaro, V.; Angrisano, T.; Pedone, E.; Di Gaetano, S.; Capasso, D.; Haney, E.F.; et al. A new cryptic cationic antimicrobial peptide from human apolipoprotein E with antibacterial activity and immunomodulatory effects on human cells. FEBS J. 2016, 283, 2115–2131. [Google Scholar] [CrossRef] [Green Version]
- Thomas, G. Furin at the cutting edge: From protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 2002, 3, 753–766. [Google Scholar] [CrossRef] [Green Version]
- Seidah, N.G.; Sadr, M.S.; Chrétien, M.; Mbikay, M. The multifaceted proprotein convertases: Their unique, redundant, complementary, and opposite functions. J. Biol. Chem. 2013, 288, 21473–21481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharjya, S.; Xu, P.; Xiang, H.; Chrétien, M.; Seidah, N.G.; Ni, F. pH-induced conformational transitions of a molten-globule-like state of the inhibitory prodomain of furin: Implications for zymogen activation. Protein Sci. 2001, 10, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjya, S.; Xu, P.; Wang, P.; Osborne, M.J.; Ni, F. Conformational analyses of a partially-folded bioactive prodomain of human furin. Biopolymers 2007, 86, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Harioudh, M.K.; Dewangan, R.P.; Ng, W.J.; Ghosh, J.K.; Bhattacharjya, S. Cell-Selective Pore Forming Antimicrobial Peptides of the Prodomain of Human Furin: A Conserved Aromatic/Cationic Sequence Mapping, Membrane Disruption, and Atomic-Resolution Structure and Dynamics. ACS Omega 2018, 3, 14650–14664. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | Net Charge |
|---|---|---|
| Temporin A (TA) | FLPLIGRVLSGIL-NH2 | +2 |
| Temporin B (TB) | LLPIVGNLLKSLL-NH2 | +2 |
| Temporin L (TL) | FVQWFSKFLGRIL-NH2 | +3 |
| TB_KKG6A | KKLLPIVANLLKSLL-NH2 | +4 |
| TB_L1FK | FLPIVGLLKSLLK-NH2 | +3 |
| TB-YK | KKYLLPIVGNLLKSLL-NH2 | +3 |
| TL (P3, D-P10) | FVPWFSKFLpRIL-NH2 | +2 |
| TL analog 9 | FVPWFSKFlkRIL-NH2 | +4 |
| TL analog 10 | FVPWFSKFlWRIL-NH2 | +3 |
| TA-β-boomerang (FG21) | FLPLIGRVLSGILGWKRKRFG-NH2 | +6 |
| TB-β-boomerang (LG21) | LLPIVGNLLKSLLGWKRKRFG-NH2 | +6 |
| Name | Sequence | Net Charge |
|---|---|---|
| aurein 2.2 | GLFDIVKKVVGALGSL-NH2 | +2 |
| aurein 2.3 | GLFDIVKKVVGAIGSL-NH2 | +2 |
| aurein 2.2-Δ3 | GLFDIVKKVVGAL-NH2 | +2 |
| peptide 73 | RLWDIVRRWVGWL-NH2 | +3 |
| peptide 77 | RLWDIVRRVWGWL-NH2 | +3 |
| Name | Sequence | Net Charge |
|---|---|---|
| YW12 | YVLWKRKRMIFI-OH | +4 |
| YI12WF 1 | YVLWKRKRFIFI-NH2 | +5 |
| YI12WY | YVLWKRKRYIFI-NH2 | +5 |
| YI12WW | YVLWKRKRWIFI-NH2 | +5 |
| YI12FF | YVLFKRKRFIFI-NH2 | +5 |
| C4/C8-YI13C | C4/C8-YVLWKRKRKFCFI-NH2 | +6 |
| Name | Sequence | Net Charge |
|---|---|---|
| TCP (C-terminal of thrombin) | GKYGFYTHVFRLKKWIQKVIDQFGE | +5 |
| Buforin II | TRSSRAGLQFPVGRVHRLLRK | +8 |
| Human lysozyme | DNIADAVACAKRVVRDPQGIRAWVAWRNR | +4 |
| Lactoferricin | KCFQWQRNMRKVRGPPVSCIKRDS | +6 |
| ApoE | LRVRLASHLRKLRKRLLR | +10 |
| Prodomain of Furin (YR26) | YYHFWHRGVTKRSLSPHRPRHSRLQR | +12 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattacharjya, S.; Straus, S.K. Design, Engineering and Discovery of Novel α-Helical and β-Boomerang Antimicrobial Peptides against Drug Resistant Bacteria. Int. J. Mol. Sci. 2020, 21, 5773. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165773
Bhattacharjya S, Straus SK. Design, Engineering and Discovery of Novel α-Helical and β-Boomerang Antimicrobial Peptides against Drug Resistant Bacteria. International Journal of Molecular Sciences. 2020; 21(16):5773. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165773
Chicago/Turabian StyleBhattacharjya, Surajit, and Suzana K. Straus. 2020. "Design, Engineering and Discovery of Novel α-Helical and β-Boomerang Antimicrobial Peptides against Drug Resistant Bacteria" International Journal of Molecular Sciences 21, no. 16: 5773. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165773