Aberrant CXCR4 Signaling at Crossroad of WHIM Syndrome and Waldenstrom’s Macroglobulinemia


1
Humanitas Clinical and Research Center-IRCCS, Via Manzoni 56, I-20089 Rozzano (Milan), Italy
2
Department of Medical Biotechnologies and Translational Medicine, University of Milan, Via Fratelli Cervi, I-20090 Segrate (Milan), Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(16), 5696; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165696
Submission received: 30 June 2020
/
Revised: 4 August 2020
/
Accepted: 6 August 2020
/
Published: 8 August 2020
(This article belongs to the Special Issue Cancer Immunoediting and Beyond)
Abstract
:Given its pleiotropic functions, including its prominent role in inflammation, immune responses and cancer, the C-X-C chemokine receptor type 4 (CXCR4) has gained significant attention in recent years and has become a relevant target in drug development. Although the signaling properties of CXCR4 have been extensively studied, several aspects deserve deeper investigations. Mutations in the C-term tail of the CXCR4 gene cause WHIM syndrome, a rare congenital immunodeficiency associated by chronic leukopenia. Similar mutations have also been recently identified in 30% of patients affected by Waldenstrom’s macroglobulinaemia, a B-cell neoplasia with bone marrow accumulation of malignant cells. An ample body of work has been generated to define the impact of WHIM mutations on CXCR4 signaling properties and evaluate their role on pathogenesis, diagnosis, and response to therapy, although the identity of disease-causing signaling pathways and their relevance for disease development in different genetic variants are still open questions. This review discusses the current knowledge on biochemical properties of CXCR4 mutations to identify their prototypic signaling profile potentially useful to highlighting novel opportunities for therapeutic intervention.
1. Introduction
The CXCL12 receptor CXCR4 is a G protein-coupled receptor (GPCR) identified for the first time in peripheral blood leukocytes [1], and initially described because of its role as a co-receptor for HIV [2]. CXCR4 is endowed with potent chemotactic properties for leukocytes but is also highly expressed in a variety of cell types, including endothelial and epithelial cells, hematopoietic stem cells, stromal fibroblasts, and cancer cells [3]. In addition to its well-established functions in hematopoiesis and immune responses, the CXCL12/CXCR4 axis plays a pivotal role in a plethora of physiological processes, including neurogenesis, cardiogenesis and neovascular formation. Receptor dysfunctions have been associated with several pathological processes, including immunodeficiencies, autoimmune diseases and cancer [4]. In particular, aberrant CXCR4 signaling caused by heterogeneous mutations affecting the C-terminal (C-ter) region of the receptor has been reported in a rare primary immunodeficiency, the Warts, Hypogammaglobulinaemia, Infections and Myelokathexis (WHIM) syndrome [5], and more recently in Waldenstrom’s macroglobulinaemia (WM), an indolent form of B-cell non-Hodgkin lymphoma [6]. These mutations act as gain-of-function mutations that turn CXCR4 into a truncated receptor with impaired internalization/desensitization and amplified signaling properties that dramatically impact on its biological functions, resulting in a “high-performance“ receptor associated with aberrant expression and activity, and with chemotherapy resistance in WM patients [7]. Since CXCR4 is an attractive target for diverse diseases and is considered as one of the best potential targets in hematological tumors, several efforts are currently ongoing aimed at developing specific inhibitors including small molecules, peptides, antibodies, and siRNA that abrogate receptor activity [8]. Of note, several clinical trials proposed CXCR4 inhibitors for treatment of hematological tumors alone or in combination with chemotherapy or biological agents [9]. Although the CXCR4 antagonist Plerixafor (also known as AMD3100) is an Food and Drug Administration (FDA)-approved drug and represents a valid therapeutic approach in both WHIM and WM, the complexity of its use in clinical practice indicates the need to develop new strategies. Other CXCR4 antagonists such as Mavorixafor (also known as AMD11070) the humanized IgG blocking monoclonal antibody BMS-936564 (also known as Ulocuplumab) are currently tested in clinical trials for WHIM and WM and showed promising results [9]. Targeting the other part of the axis, CXCL12, also represents an attractive alternative approach that may facilitate regulation rather than the abrogation of receptor activity, although the extremely high evolutionary conservation of this chemokine gets the generation of efficacious antibodies complicated. However, a selective inhibitor of CXCL12, the oligonucleotide NOX-A12, has been recently generated by Spiegelmer technology [10], and tested with promising results in clinical trials for chronic lymphocytic leukemia (#NCT01486797) [11,12] and multiple myeloma (#NCT01521533) [13], opening a new scenario in treatment of WHIM and WM. Nevertheless, only an accurate knowledge of the molecular mechanisms defining CXCR4 signaling properties will allow for developing innovative, and more targeted, therapeutic interventions. Thus, CXCR4 genetic is a crucial hub for the pathogenesis of both WHIM syndrome and WM, and deciphering the aberrant signaling profile represents a fundamental element for the identification of relevant biomarkers and the development of precision therapy in both diseases. In this review, we attempt a systematic description of WHIM mutations aimed at outlying the global organization of this group of mutations, drawing up a prototypic WHIM-mutated CXCR4 profile and identifying aspects deserving further investigation.
1.1. WHIM Syndrome
The CXCL12/CXCR4 axis has profound influences on immune system homeostasis, exerting a fundamental role in bone marrow (BM) colonization during ontogenesis, as well as in hematopoietic stem cells homeostasis [14]. Besides regulating BM homeostasis, CXCR4 plays a prominent function in orchestrating both innate and adaptive immune responses: it regulates leukocyte trafficking and distribution in peripheral tissues, participates in lymph node organization, and contributes to the formation and stabilization of the immunological synapse thus sustaining B cell functions as well as T cell priming [7]. Consistent with this, the prominent role of CXCR4 in immunodeficiencies has been largely documented. WHIM syndrome is a rare immunodeficiency disease caused by the combination of four main clinical manifestations: Warts, Hypogammaglobulinaemia, Infections and Myelokathexis. The first case report described a 9-year-old girl who had severe neutropenia with retention of fully mature and apoptotic neutrophils in the BM, a picture indicated as myelokathexis [15]. Several years later, the reason behind neutrophil retention/apoptosis in the BM has been linked to heterozygous mutations in the CXCR4 gene [16]. To date, 9 heterozygous mutations have been described causing WHIM syndrome [5]. All but one mutation span in the C-ter of CXCR4, thereby generating an amiss truncated receptor with delayed internalization and prolonged signaling. CXCR4 is physiologically involved in retention of neutrophils and other leukocyte subtypes in the BM, and WHIM mutations exaggerate this process causing a delay in neutrophil egress from BM to blood and enhancing neutrophil homing from blood to BM, resulting in myelokathectic neutropenia. This, in turn, increases susceptibility to bacterial and viral infections. Consistent with the gain-of-function profile of WHIM mutations, inhibitors of CXCR4 offer an opportunity to provide more specific and targeted therapy. However, this type of treatment will need to be lifelong. Plerixafor is a specific CXCR4 antagonist [17] approved by the FDA in 2008 in combination with Granulocyte Colony-Stimulating Factor (G-CSF) for hematopoietic stem cell mobilization and transplantation in patients receiving cytoreductive treatment for multiple myeloma or non-Hodgkins lymphoma (nHL) [18,19]. Efforts to repurpose Plerixafor in WHIM syndrome are underway, and to date several small studies have demonstrated its ability to reverse panleukopenia in WHIM patients.
1.2. Waldenstrom’s Macroglobulinaemia
The CXCL12/CXCR4 axis represents a promising prognostic marker and a potential target for therapy in oncology, as it has been shown to be operative in about 20 different cancer types where it promotes cancer cell survival and proliferation, tumor angiogenesis, and cancer cells migration to metastatic sites. It also affects chemosensitivity and disease progression by directing CXCR4-expressing tumor cells through concentration gradients of CXCL12 to reside in protective niches [19]. Consistent with this, in recent years genomic findings have provided important insights into the relevance of the CXCL12/CXCR4 axis for pathogenesis, prognostication, and treatment outcome of cancer. As CXCL12 regulates hematopoietic cell trafficking to positioning in the BM [20], it is not surprising that aberrant CXCR4 expression on cancer cells from several hematopoietic malignancies influences neoplasia progression by controlling cancer cell migration to BM as well as to lymph nodes, suggesting CXCR4 as a novel, reliable prognostic biomarker [21]. In particular, somatic CXCR4 mutations have been reported in indolent forms of B-cell nHL, follicular lymphoma, and WM [22,23]. WM is a rare form of nHL or lymphoplasmacytic lymphoma [24], characterized by lymphoplasmacytic infiltrate in BM, lymph nodes and spleen, frequently associated with the presence of monoclonal immunoglobulin M (IgM) protein in the blood. Approximately 30–40% WM patients carry mutations in heterozygosis of CXCR4 [25]. These somatic mutations are primarily subclonal, and almost always associated with the MYD88L265P mutation, the first identified recurring mutation in almost 67–90% of non-IgM secreting WM patients [6]. All 17 CXCR4 heterozygous mutations identified so far in WM span in the C-ter of the receptor, and closely resemble the already documented germline mutations of CXCR4 C-ter occurring in heterozygosis in WHIM syndrome [9]. Although their relevance for clinical presentation and overall survival, as well as their relationship with resistance to chemotherapy are still unsolved issues [26,27,28], several studies reported that patients with CXCR4 mutations present a significantly lower rate of adenopathy, and those with CXCR4 nonsense mutations have an increased BM disease burden, serum IgM levels, and/or risk of symptomatic hyperviscosity [29]. Moreover, Plerixafor inhibition of CXCL12/CXCR4 axis can reverse the tumor-promoting signals of stromal cells, increasing the spontaneous apoptosis rate of tumor cells and enhancing their response to chemotherapy [30,31]. Taken together, these observations clearly demonstrate that blocking the CXCL12/CXCR4 axis might be a promising approach for potentiating the effects of currently used therapeutic regimen in WM.
2. Genetic Barcode of WHIM Mutations
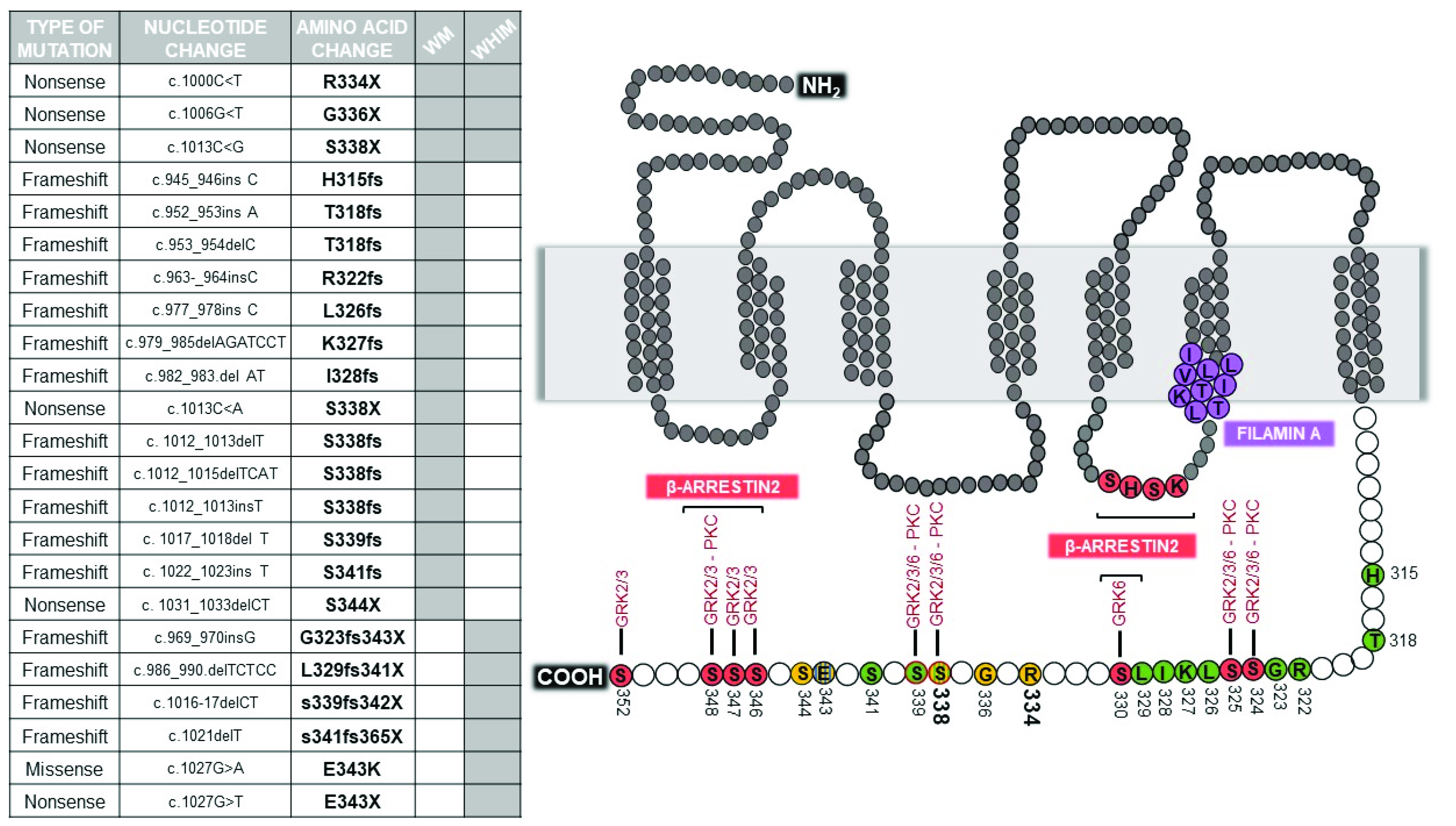
At least 20 different WHIM/WHIM-like mutations have been described so far in the genomic region of chromosome 2q21 that encodes the C-ter of CXCR4 [9,29] (Figure 1). Interestingly, these mutations occur in the context of a gene that is highly conserved across species and span in a genomic region that is even more highly conserved than the gene as a whole [32].
In WHIM syndrome, all but one of the CXCR4 germline mutations truncate receptor C-ter by premature termination (4 NS) or by frame-shifts (4 FS) that introduce from 3 to 24 additional new amino acids [5]. The only missense mutation (MS) is E343K, which involves a single amino acid substitution and a charge change [32]. By far the most common WHIM mutation is R334X, which accounts for half of genotyped cases in the literature, including both de novo and familial cases. Conversely, CXCR4 S338X accounts for 15 of the genotyped cases [33]. The remaining 29 genotyped cases are distributed among the remaining mutations, 8 of which were observed in only 1–3 cases each.
Acquired CXCR4 WHIM-like mutations, are present in up to 40% of patients with WM and are nearly always observed in conjunction with the MYD88L265P mutation [29]. CXCR4 mutations are essentially unique to WM, as they have not been described so far in other malignancies, except for several cases of patients affected by follicular lymphoma [9,22]. Among the different CXCR4 mutations observed, 5 are NS and 12 FS [34]. The most frequent mutations are the S338X (C1013G) followed by S338X (C1013A), which generate a stop codon in place of a serine at amino acid position 338. Several other mutations to S338 has been described, suggesting a hotspot locus in the CXCR4 gene. The S338 hotspot mutation has been identified in half of CXCR4 mutated cases in WM. Interestingly, WM acquired exact the same R334X mutation that has been also previously described in WHIM syndrome. Differently from MYD88L265P, CXCR4 mutant clonality is highly variable, and multiple CXCR4 mutations are present within individual patients in separate clones or are present as heterozygous events [29]. The subclonal nature suggests that these mutations are acquired after MYD88L265P, although this could occur early in WM pathogenesis.
3. Signaling of WHIM Mutations
Like all GPCRs, the structure of CXCR4 contains 7 transmembrane helices, an extracellular N-terminal domain and an intracellular C-ter domain. CXCR4 activity, trafficking, and signaling properties are finely coordinated by physical interaction with several canonical (i.e., G proteins, GPCR kinases (GRKs), and β-arrestins) and non-canonical proteins (i.e., Filamin A (FLNA), Atrophin–Interacting Protein 4 (AIP4), CD74 and CD164) [35]. CXCR4 is known to couple to the G protein αi (Gαi) that mediates most of receptor signaling pathways, and to Gα12–13 and Gαq [36]. These signaling pathways include the activation of Src and PI3K kinases, and Phospholipase C (PLC) that in turn activates Protein Kinase B (PKB also known as AKT) and Protein Kinase C (PKC)/ Mitogen-Activated Protein Kinase (MAPK), respectively. AKT and PKC are also activated by the interaction of CXCR4 with the Human Leukocyte Antigen (HLA) class II histocompatibility antigen gamma chain (CD74) [37] and Endolyn (CD164) [38]. After CXCL12 activation, Gαi is negatively regulated by powerful desensitization mechanisms that involve GRKs, which phosphorylates specific serines and threonines distributed along the C-ter to which β-arrestins are recruited to drive receptor internalization [39,40]. CXCR4/β-arrestins interaction is finely tuned by FLNA which stabilizes the receptor at the plasma membrane by blocking receptor endocytosis [41], unlike AIP4 which promotes receptors ubiquitination and targeting to multivescicular bodies for degradation [42]. By linking CXCR4 to actin, FLNA also orchestrates the deep cytoskeletal rearrangements required to sustain cell migration, like other non-canonical constitutively interacting proteins such as Drebrin [43], diaphanous-related formin-2 (mDIA2) [44], PI3-kinase isoform p110γ [45] and the motor protein NonMuscle Myosin H Chain (NMMHC) that selectively bind the receptor C-ter domain [46].
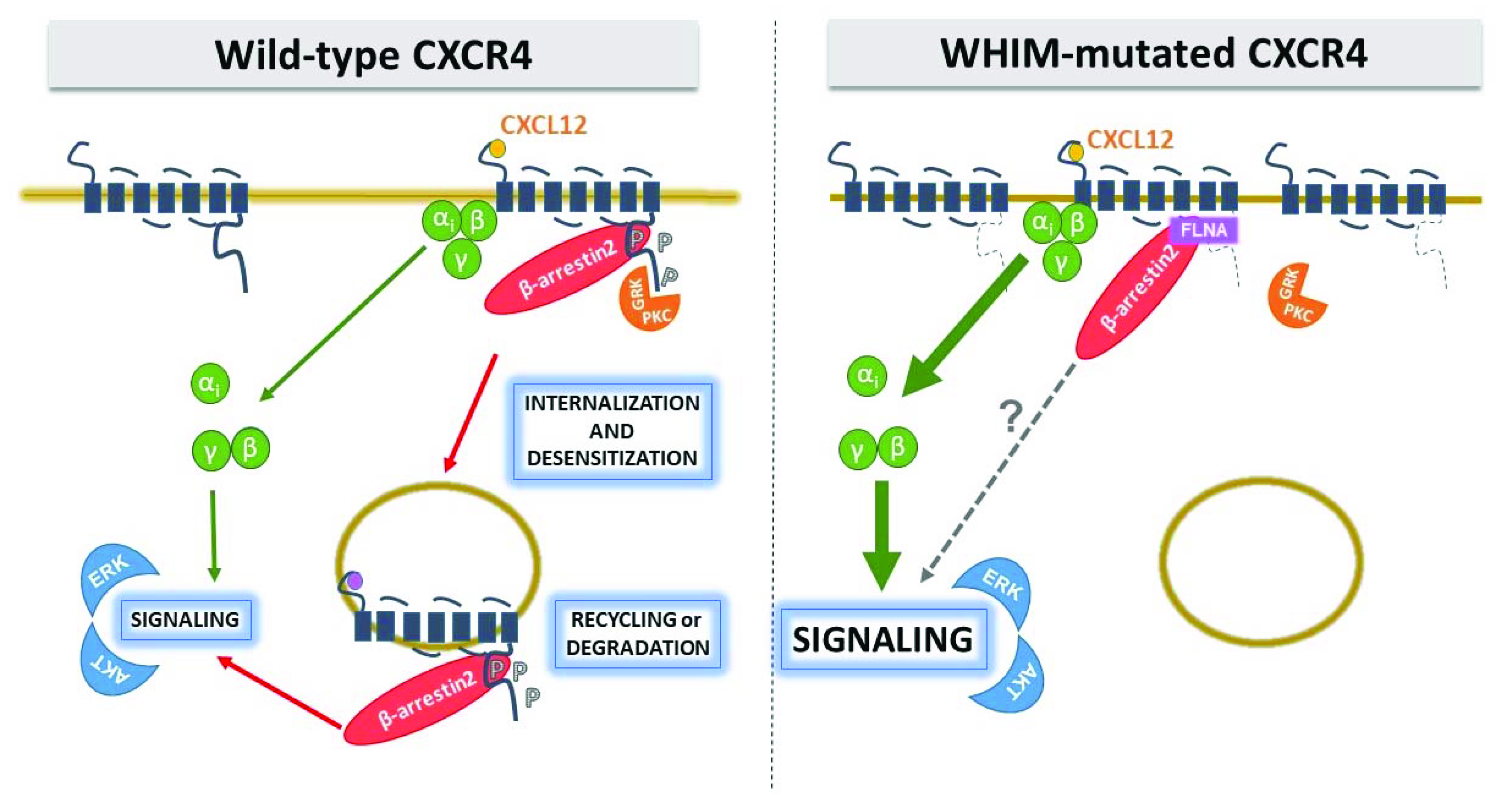
To date, all but one WHIM CXCR4 variant show impaired internalization, with prolonged receptor residence time on the cell surface which is thought to contribute to amplification and prolongation of receptor signaling activity [47]. Thus, paradoxically, a loss of structure from WHIM mutations leads to a gain-of-function by stabilizing the mutant receptor on the cell surface (Figure 2).
The signaling profile of WHIM-mutated CXCR4 has been currently explored for 5 mutations (the NS mutations R334X, G336X, S338X; the MS mutation E343K; the FS mutation L329fs341X) (Table 1), whereas signaling properties of remaining variants are presently largely unknown. To date, R334X and S338X are the best characterized mutations, and S338X is the mutation whose signaling properties has been investigated for both WHIM and WM diseases. Studies have been conducted either in ex vivo setting using freshly isolated leukocytes (PBMC) or CD34+ cells from patients, and immortalized cells (EBV [48,49], BCWM.1 [50], MWCL-1 [51] for WHIM and WM, respectively), and in in vitro experimental settings using several cell lines (Table 1).
3.1. The Gain-of-Function Nature of WHIM Mutations
The binding of CXCL12 to CXCR4 takes place through a two-step mechanism [64]. CXCL12 contact at the extracellular domain causes a first conformational change which strengthens chemokine binding to a receptor pocket. Next, a second conformational change activates the intracellular trimeric G protein by the dissociation of Gαi subunit from the Gβ/Gγ dimer at the third receptor intracellular loop [65,66]. Once activated, the Gαi subunit inhibits production of cAMP by adenyl cyclases and stimulates the activity of Src family tyrosine kinases that modulate cell cycle progression by activating the Ras/Raf/MEK/ERK pathway. Conversely, Gβγ and Gα subunits stimulate the activity of PI3Ks, which mediate gene transcription, cell adhesion and migration, and cell survival by phosphorylating AKT and several focal adhesion components. AKT also triggers the activity of PLC that hydrolyses PIP2 into IP3 and DiAcylGlycerol (DAG), which promote Ca2+ mobilization from the intracellular stores and PKC/MAPK activation, respectively.
With the rest of GPCR structure left intact, WHIM-mutated CXCR4 remains fully competent in downstream signaling via G protein. Impaired internalization prolongs receptor residence time on the cell surface and contributes to amplification and prolongation of receptor signaling that have been observed using multiple signaling assays in different cellular contexts [47]. To date, increased ERK and AKT activation have been described for R334X and S338X, and amplified calcium flux and chemotaxis has been proven for all mutations but not yet reported for S338X and L329fs341X, respectively (Table 1). Interestingly, polymerization of actin monomers into F-actin filaments has been also evaluated in T lymphocytes of R334X and S338X WHIM patients, with results showing a protracted rise of F-actin after CXCL12 stimulation [33]. These observations are consistent with the “hyperactive” signaling nature of WHIM-mutated CXCR4 and might reflect an increased ability of WHIM variants to activate G proteins. However, effect of WHIM mutations on G protein recruitment and activation deserves further investigation. Lagane and colleagues measured CXCL12-induced 35S-GTPγS binding to activated Gαi-containing membranes from HEK293 cells expressing similar amounts of CXCR4 WT and S338X, and reported that the truncation of the C-ter improved CXCL12-induced coupling efficiency and potency of the receptor [61], confirming previous observations by Balabanian and colleagues on S338X and R334X using the same in vitro assay [33]. Thus, the enhanced responsiveness of WHIM-mutated CXCR4 to CXCL12 is likely the consequence of an improved activation of receptor-associated G proteins.
3.2. Hyperactivity of WHIM-Mutated CXCR4: A Question of TAIL or deTAILs?
Upon activation, GPCRs are rapidly phosphorylated, typically by members of the GRK family. This triggers the recruitment of β-arrestins at receptor C-ter, which prevents further activation of their cognate G protein, initiates β-arrestin-dependent signaling, and leads to receptor desensitization and internalization [39,40]. The CXCR4 C-ter contains 18 potential phosphorylation sites: 15 serines and 3 threonines. The “phosphorylation barcode” of the CXCR4 C-ter includes 7 serine residues that are phosphorylated by specific GRKs and PKC isoforms following CXCL12 stimulation: S321, S324, S325, S330, S339, a residue between S346 and S348, and either S351 or S352 (Figure 1) [67]. CXCL12 induces long-lasting GRK2/3-dependent phosphorylation of a S346/347 phosphosite which precedes less stable phosphorylation at S324/325 and S338/339 phosphosites [68]. S330 and S339 are phosphorylated by GRK6, as well as S324/325 that are additionally phosphorylated by PKC [67]. The S346/347 residues have been shown to be rapidly phosphorylated by GRK2, GRK3 and PKC(ɑ) following CXCL12 stimulation and together with the SHSK motif in intracellular loop 3 (ICL3) are involved in the recruitment of β-arrestin2 [69], which initiates the process of receptor desensitization [68,70]. Of note, β-arrestin2 binding to these two sites has distinct functional consequences; whereas β-arrestin2 interaction with the C-ter mediates receptor desensitization and endocytosis, its binding to ICL3 is important for β-arrestin2-mediated signaling [69].
The hierarchy of CXCR4 phosphorylation events may partially explain how small structural changes caused by WHIM mutations are leveraged into large functional effects in CXCR4. All WHIM mutants lacking the S346/347 phosphosite have impaired phosphorylation at the intact proximal S324/325 and S338/339 sites, and this is consistent with impaired CXCL12-induced receptor downregulation from the cell surface (Table 1). However, although the MS E343Kmutation shows impaired phosphorylation at serine phosphosites and results in increased receptor signaling, the effect on blocking normal receptor downregulation is impaired [32]. Similarly, the NS S336X resulted in enhanced migration, but calcium flux and receptor downregulation were unaffected [56]. Phosphorylation of CXCR4 C-ter is mandatory for docking of β-arrestins, which also serves as scaffold for a number of downstream effectors involved in G protein-independent signal pathways, enhancing receptor-mediated ERK and p38 activation which promote cell migration [71]. However, the role of β-arrestins and GRKs in orchestrating the puzzling behavior of WHIM mutations is still an unsolved and controversial issue. McCormick and colleagues demonstrated in HeLa cells that R334X is defective at recruiting β-arrestin2 and GRK6 proteins after exposure to CXCL12, and displays a delay in receptor downregulation, signaling and trafficking in comparison to WT [55]. These authors proposed that truncated residues of CXCR4 C-ter are important for the interaction with GRK6 but not GRK3, providing evidence that different GRKs may require distinct CXCR4 structural elements for interaction. This was consistent with GRK6 depletion results that showed a delayed CXCL12-induced ERK activation, similarly to that seen downstream R334X. Conversely, a study by Lagane and colleagues in leukocytes isolated from WHIM patients carrying the S338X mutation demonstrated that β-arrestin2 interaction with CXCR4 is preserved, despite that the receptor is not internalized, and results in an enhanced ERK activation and consequent chemotaxis to CXCL12 [61]. Furthermore, the authors demonstrated that β-arrestin2 also bind the SHSK motif of ICL3, with prolonged signaling downstream WHIM receptor mutants. Of note, the observation that CXCR4 C-ter and ICL3 (LKTTVILIL) interact with FLNA (Figure 1), an important regulatory element of the cytoskeleton known to act as a platform for receptor signaling and intracellular trafficking [72], that not only links CXCR4 to the actin cytoskeleton, but also suggests its role in shaping receptor signaling by regulating β-arrestin2/ICL3 interaction [41]. Gomez-Mouton and colleagues demonstrated that FLNA also binds the ICL3 of R334X and suggested a role both in receptor stabilization at the cell surface and in increased β-arrestin2 binding to the mutant, providing a new molecular mechanism for hyperactivation of WHIM receptor signaling [41]. Interestingly, a detailed study of two unrelated patients with a full clinical form of WHIM syndrome but lacking detectable mutations in CXCR4 showed hyperactive responses to CXCL12 [33]. Balabanian and colleagues identified a defective activity of GRK3 as a consequence of decreased GRK3 levels, likely resulted from defective mRNA synthesis [62]. As GRK3 specifically regulates CXCL12-mediated desensitization and internalization of CXCR4, its defective activation possibly contributes to the amplification of the G protein-dependent responses in WHIM cells as shown by gene transfer mediated expression of GRK3 in this patient’s fibroblasts or leukocytes that recovered normal chemotactic response and CXCR4 internalization in response to CXCL12 [62]. These results underline the complexity of CXCR4 modulation by WHIM mutations, and clearly demonstrate that the critical biochemical feature shared by WHIM patients carrying C-ter truncating mutations of CXCR4 and those who lack detectable mutations in CXCR4 is the gain in functional hyperactivity of CXCR4 in response to CXCL12. This further supports the general understanding of WHIM syndrome as an immunodeficiency caused by functional hyperactivity of CXCR4. Thus, while investigating the molecular basis accounting for altered CXCR4 signaling in WHIM patients, possible functional alterations of components normally involved in the CXCR4 signaling machinery have emerged.
3.3. Effect of Homo or Heterodimerization on WHIM-Mutated CXCR4 Signaling: A Still Unanswered Question
In the past two decades, an increasing amount of evidence suggested that beyond monomeric entities, GPCRs could also exist and function as oligomeric complexes [73], thus further adding complexity to the signaling basis of chemokine-mediated responses. CXCR4 was shown to constitutively dimerize, with CXCL12 binding further enhancing receptor homodimerization [4]. Furthermore, CXCR4 form heterodimers with other chemokine receptors, including its atypical chemokine receptor counterpart ACKR3 (also known as CXCR7). Although the biological significance of CXCR4/ACKR3 heterodimerization remains poorly understood, it has been shown that by this mechanism ACKR3 acts as key regulator of CXCR4 signaling properties, including its ability to mobilize intracellular Ca2+ [74,75,76,77]. Once homodimerized, CXCR4 becomes phosphorylated through the rapid binding and activation of Janus Kinase 2 (JAK2) and 3 (JAK3) mediated by a G protein-independent pathway [78]. JAK proteins in turn activate the STAT signaling involved in regulation of various cellular processes, including Ca2+ mobilization from intracellular stores and the transcription of target genes [4]. However, despite the emerging interest in this pathway, the role of JAK/STAT in CXCR4 signal transduction is still matter of debate.
As WHIM mutations occur in heterozygous dominant condition, the WT and WHIM-mutated CXCR4 alleles are likely co-expressed. The crystal structure of CXCR4 revealed that the receptor is a homodimer [79,80], thus both the WT and WHIM-mutated forms could coexist as independent monomers and/or as homodimers and/or as heterodimers in patient cells. Although at present the biochemical evidence that WHIM-mutated variants can heterodimerize with the WT receptor is limited [61], this element has potential unpredictable effects on receptor expression levels, G protein/β-arrestin coupling and signaling properties. The stoichiometry of the different forms could also vary according to cell type and among different patients, possibly contributing to the phenotypic heterogeneity observed in different patients with the same pedigree. Moreover, it has recently been reported that in addition to the ligand-mediated conformational change of the receptor that activates G proteins, CXCR4 undergoes changes associated with receptor nanoclustering that are necessary for signaling and could establish a new target for potential intervention in WHIM and WM patients [81]. A deeper understanding of the functional and pharmacological properties of CXCR4 heterodimers may therefore indicate more specific targets for drug design and development.
4. Concluding Remarks
After the initial description of the presence of mutations in the CXCR4 gene in both WHIM and WM diseases, several studies have revealed a striking concordance in the molecular and biochemical properties of the underlying mutations, suggesting that hyperactivation of receptor signaling by truncation of the C-ter is an essential aspect in pathogenesis. Biochemical studies have provided support for the model that impaired CXCR4 downregulation and desensitization leads to the characteristic immunologic and hematologic alterations, although the broad heterogeneity in clinical manifestations and disease severity and different abnormalities in immunological parameters in the two diseases tempt us to speculate that they are likely supported by different underlying mechanisms. Since genetic studies have also identified patients with the same clinical features but without mutation of CXCR4, uncharacterized downstream regulators of the receptor may also be involved in a proportion of cases. Though the CXCR4 inhibitor AMD3100 is an FDA-approved drug and represents a valid therapeutic approach in WHIM and WM, issues related to the complexity of its use in clinical practice indicate the need to develop new therapies. These will require a more precise understanding of signaling properties of CXCR4 mutants and the role they may play in WHIM and WM pathogenesis.
Author Contributions
E.M.B. and S.M. designed the work and wrote the manuscript; M.L. supervised and revised the manuscript; E.M.B. funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a research grant of E.M.B. from the Italian Ministry of Health (Project for Young Researcher GR-2013-02356521).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Loetscher, M.; Geiser, T.; O’Reilly, T.; Zwahlen, R.; Baggiolini, M.; Moser, B. Cloning of a human seven-transmembrane domain receptor, LESTR, that is highly expressed in leukocytes. J. Biol. Chem. 1994, 269, 232–237. [Google Scholar] [PubMed]
- Chen, B. Molecular Mechanism of HIV-1 Entry. Trends Microbiol. 2019, 27, 878–891. [Google Scholar] [CrossRef] [PubMed]
- Teixido, J.; Martinez-Moreno, M.; Diaz-Martinez, M.; Sevilla-Movilla, S. The good and bad faces of the CXCR4 chemokine receptor. Int. J. Biochem. Cell Biol. 2018, 95, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Pawig, L.; Klasen, C.; Weber, C.; Bernhagen, J.; Noels, H. Diversity and Inter-Connections in the CXCR4 Chemokine Receptor/Ligand Family: Molecular Perspectives. Front. Immunol. 2015, 6, 429. [Google Scholar] [CrossRef] [Green Version]
- McDermott, D.H.; Murphy, P.M. WHIM syndrome: Immunopathogenesis, treatment and cure strategies. Immunol. Rev. 2019, 287, 91–102. [Google Scholar] [CrossRef]
- Kapoor, P.; Ansell, S.M.; Braggio, E. Waldenstrom Macroglobulinemia: Genomic Aberrations and Treatment. Cancer Treat. Res. 2016, 169, 321–361. [Google Scholar] [CrossRef]
- Pozzobon, T.; Goldoni, G.; Viola, A.; Molon, B. CXCR4 signaling in health and disease. Immunol. Lett. 2016, 177, 6–15. [Google Scholar] [CrossRef]
- Tsou, L.K.; Huang, Y.H.; Song, J.S.; Ke, Y.Y.; Huang, J.K.; Shia, K.S. Harnessing CXCR4 antagonists in stem cell mobilization, HIV infection, ischemic diseases, and oncology. Med. Res. Rev. 2018, 38, 1188–1234. [Google Scholar] [CrossRef]
- Scala, S.; D’Alterio, C.; Milanesi, S.; Castagna, A.; Carriero, R.; Farina, F.M.; Locati, M.; Borroni, E.M. New Insights on the Emerging Genomic Landscape of CXCR4 in Cancer: A Lesson from WHIM. Vaccines 2020, 8, 164. [Google Scholar] [CrossRef] [Green Version]
- Vater, A.; Klussmann, S. Toward third-generation aptamers: Spiegelmers and their therapeutic prospects. Curr. Opin. Drug Discov. Dev. 2003, 6, 253–261. [Google Scholar]
- Hoellenriegel, J.; Zboralski, D.; Maasch, C.; Rosin, N.Y.; Wierda, W.G.; Keating, M.J.; Kruschinski, A.; Burger, J.A. The Spiegelmer NOX-A12, a novel CXCL12 inhibitor, interferes with chronic lymphocytic leukemia cell motility and causes chemosensitization. Blood 2014, 123, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Steurer, M.; Montillo, M.; Scarfo, L.; Mauro, F.R.; Andel, J.; Wildner, S.; Trentin, L.; Janssens, A.; Burgstaller, S.; Fromming, A.; et al. Olaptesed pegol (NOX-A12) with bendamustine and rituximab: A phase IIa study in patients with relapsed/refractory chronic lymphocytic leukemia. Haematologica 2019, 104, 2053–2060. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.L.; Sattler, M.; Azab, A.K.; Eulberg, D.; Kruschinski, A.; Manley, P.W.; Stone, R.; Griffin, J.D. Inhibition of SDF-1-induced migration of oncogene-driven myeloid leukemia by the L-RNA aptamer (Spiegelmer), NOX-A12, and potentiation of tyrosine kinase inhibition. Oncotarget 2017, 8, 109973–109984. [Google Scholar] [CrossRef] [Green Version]
- Karpova, D.; Bonig, H. Concise Review: CXCR4/CXCL12 Signaling in Immature Hematopoiesis--Lessons From Pharmacological and Genetic Models. Stem Cells 2015, 33, 2391–2399. [Google Scholar] [CrossRef] [PubMed]
- Zuelzer, W.W. “Myelokathexis”—A New Form of Chronic Granulocytopenia. Report of a Case. N. Engl. J. Med. 1964, 270, 699–704. [Google Scholar] [CrossRef]
- Hernandez, P.A.; Gorlin, R.J.; Lukens, J.N.; Taniuchi, S.; Bohinjec, J.; Francois, F.; Klotman, M.E.; Diaz, G.A. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat. Genet. 2003, 34, 70–74. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Mozobil(R) (Plerixafor, AMD3100), 10 years after its approval by the US Food and Drug Administration. Antivir. Chem. Chemother. 2019, 27, 2040206619829382. [Google Scholar] [CrossRef] [Green Version]
- De Clercq, E. AMD3100/CXCR4 Inhibitor. Front. Immunol. 2015, 6, 276. [Google Scholar] [CrossRef]
- Scala, S. Molecular Pathways: Targeting the CXCR4-CXCL12 Axis—Untapped Potential in the Tumor Microenvironment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 4278–4285. [Google Scholar] [CrossRef] [Green Version]
- Lim, V.Y.; Zehentmeier, S.; Fistonich, C.; Pereira, J.P. A Chemoattractant-Guided Walk Through Lymphopoiesis: From Hematopoietic Stem Cells to Mature B Lymphocytes. Adv. Immunol. 2017, 134, 47–88. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Gao, L.; Luan, J.; Zhang, H.; Xiao, T. C-X-C Chemokine Receptor 4 in Diffuse Large B Cell Lymphoma: Achievements and Challenges. Acta Haematol. 2019, 142, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Krysiak, K.; Gomez, F.; White, B.S.; Matlock, M.; Miller, C.A.; Trani, L.; Fronick, C.C.; Fulton, R.S.; Kreisel, F.; Cashen, A.F.; et al. Recurrent somatic mutations affecting B-cell receptor signaling pathway genes in follicular lymphoma. Blood 2017, 129, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Hunter, Z.R.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Manning, R.J.; Tripsas, C.; Patterson, C.J.; Sheehy, P.; et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood 2014, 123, 1637–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, M.Y.; Radojcic, V.; Maillard, I. Oncogenic Notch signaling in T-cell and B-cell lymphoproliferative disorders. Curr. Opin. Hematol. 2016, 23, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Passaro, D.; Irigoyen, M.; Catherinet, C.; Gachet, S.; Da Costa De Jesus, C.; Lasgi, C.; Tran Quang, C.; Ghysdael, J. CXCR4 Is Required for Leukemia-Initiating Cell Activity in T Cell Acute Lymphoblastic Leukemia. Cancer Cell 2015, 27, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Paulus, A.; Akhtar, S.; Yousaf, H.; Manna, A.; Paulus, S.M.; Bashir, Y.; Caulfield, T.R.; Kuranz-Blake, M.; Chitta, K.; Wang, X.; et al. Waldenstrom macroglobulinemia cells devoid of BTK(C481S) or CXCR4(WHIM-like) mutations acquire resistance to ibrutinib through upregulation of Bcl-2 and AKT resulting in vulnerability towards venetoclax or MK2206 treatment. Blood Cancer J. 2017, 7, e565. [Google Scholar] [CrossRef]
- Castillo, J.J.; Xu, L.; Gustine, J.N.; Keezer, A.; Meid, K.; Dubeau, T.E.; Liu, X.; Demos, M.G.; Kofides, A.; Tsakmaklis, N.; et al. CXCR4 mutation subtypes impact response and survival outcomes in patients with Waldenstrom macroglobulinaemia treated with ibrutinib. Br. J. Haematol. 2019, 187, 356–363. [Google Scholar] [CrossRef]
- Castillo, J.J.; Moreno, D.F.; Arbelaez, M.I.; Hunter, Z.R.; Treon, S.P. CXCR4 mutations affect presentation and outcomes in patients with Waldenstrom macroglobulinemia: A systematic review. Expert Rev. Hematol. 2019, 12, 873–881. [Google Scholar] [CrossRef]
- Hunter, Z.R.; Yang, G.; Xu, L.; Liu, X.; Castillo, J.J.; Treon, S.P. Genomics, Signaling, and Treatment of Waldenstrom Macroglobulinemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 994–1001. [Google Scholar] [CrossRef]
- Shen, Z.H.; Zeng, D.F.; Ma, Y.Y.; Zhang, X.; Zhang, C.; Kong, P.Y. Are there any new insights for G-CSF and/or AMD3100 in chemotherapy of haematological malignants? Med. Oncol. 2015, 32, 262. [Google Scholar] [CrossRef]
- Shen, Z.H.; Zeng, D.F.; Kong, P.Y.; Ma, Y.Y.; Zhang, X. AMD3100 and G-CSF disrupt the cross-talk between leukemia cells and the endosteal niche and enhance their sensitivity to chemotherapeutic drugs in biomimetic polystyrene scaffolds. Blood Cells Mol. Dis. 2016, 59, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chen, H.; Ojode, T.; Gao, X.; Anaya-O’Brien, S.; Turner, N.A.; Ulrick, J.; DeCastro, R.; Kelly, C.; Cardones, A.R.; et al. WHIM syndrome caused by a single amino acid substitution in the carboxy-tail of chemokine receptor CXCR4. Blood 2012, 120, 181–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balabanian, K.; Lagane, B.; Pablos, J.L.; Laurent, L.; Planchenault, T.; Verola, O.; Lebbe, C.; Kerob, D.; Dupuy, A.; Hermine, O.; et al. WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood 2005, 105, 2449–2457. [Google Scholar] [CrossRef] [PubMed]
- Poulain, S.; Roumier, C.; Venet-Caillault, A.; Figeac, M.; Herbaux, C.; Marot, G.; Doye, E.; Bertrand, E.; Geffroy, S.; Lepretre, F.; et al. Genomic Landscape of CXCR4 Mutations in Waldenstrom Macroglobulinemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 1480–1488. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, A.; Zarca, A.; Neves, M.; Caspar, B.; Hill, S.J.; Mayor, F., Jr.; Smit, M.J.; Marin, P. CXCR4/ACKR3 Phosphorylation and Recruitment of Interacting Proteins: Key Mechanisms Regulating Their Functional Status. Mol. Pharmacol. 2019, 96, 794–808. [Google Scholar] [CrossRef] [Green Version]
- Busillo, J.M.; Benovic, J.L. Regulation of CXCR4 signaling. Biochim. Et Biophys. Acta 2007, 1768, 952–963. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, V.; Lue, H.; Kraemer, S.; Korbiel, J.; Krohn, R.; Ohl, K.; Bucala, R.; Weber, C.; Bernhagen, J. A functional heteromeric MIF receptor formed by CD74 and CXCR4. FEBS Lett. 2009, 583, 2749–2757. [Google Scholar] [CrossRef] [Green Version]
- Forde, S.; Tye, B.J.; Newey, S.E.; Roubelakis, M.; Smythe, J.; McGuckin, C.P.; Pettengell, R.; Watt, S.M. Endolyn (CD164) modulates the CXCL12-mediated migration of umbilical cord blood CD133+ cells. Blood 2007, 109, 1825–1833. [Google Scholar] [CrossRef] [Green Version]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. Beta-arrestins and cell signaling. Annu. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef] [Green Version]
- Moore, C.A.; Milano, S.K.; Benovic, J.L. Regulation of receptor trafficking by GRKs and arrestins. Annu. Rev. Physiol. 2007, 69, 451–482. [Google Scholar] [CrossRef]
- Gomez-Mouton, C.; Fischer, T.; Peregil, R.M.; Jimenez-Baranda, S.; Stossel, T.P.; Nakamura, F.; Manes, S. Filamin A interaction with the CXCR4 third intracellular loop regulates endocytosis and signaling of WT and WHIM-like receptors. Blood 2015, 125, 1116–1125. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, D.; Robia, S.L.; Marchese, A. The E3 ubiquitin ligase atrophin interacting protein 4 binds directly to the chemokine receptor CXCR4 via a novel WW domain-mediated interaction. Mol. Biol. Cell 2009, 20, 1324–1339. [Google Scholar] [CrossRef] [PubMed]
- Perez-Martinez, M.; Gordon-Alonso, M.; Cabrero, J.R.; Barrero-Villar, M.; Rey, M.; Mittelbrunn, M.; Lamana, A.; Morlino, G.; Calabia, C.; Yamazaki, H.; et al. F-actin-binding protein drebrin regulates CXCR4 recruitment to the immune synapse. J. Cell Sci. 2010, 123, 1160–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyse, M.M.; Goicoechea, S.; Garcia-Mata, R.; Nestor-Kalinoski, A.L.; Eisenmann, K.M. mDia2 and CXCL12/CXCR4 chemokine signaling intersect to drive tumor cell amoeboid morphological transitions. Biochem. Biophys. Res. Commun. 2017, 484, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Schmid, M.C.; Avraamides, C.J.; Dippold, H.C.; Franco, I.; Foubert, P.; Ellies, L.G.; Acevedo, L.M.; Manglicmot, J.R.; Song, X.; Wrasidlo, W.; et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer Cell 2011, 19, 715–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rey, M.; Vicente-Manzanares, M.; Viedma, F.; Yanez-Mo, M.; Urzainqui, A.; Barreiro, O.; Vazquez, J.; Sanchez-Madrid, F. Cutting edge: Association of the motor protein nonmuscle myosin heavy chain-IIA with the C terminus of the chemokine receptor CXCR4 in T lymphocytes. J Immunol. 2002, 169, 5410–5414. [Google Scholar] [CrossRef] [Green Version]
- Heusinkveld, L.E.; Majumdar, S.; Gao, J.L.; McDermott, D.H.; Murphy, P.M. WHIM Syndrome: From Pathogenesis Towards Personalized Medicine and Cure. J. Clin. Immunol. 2019, 39, 532–556. [Google Scholar] [CrossRef] [PubMed]
- Chae, K.M.; Ertle, J.O.; Tharp, M.D. B-cell lymphoma in a patient with WHIM syndrome. J. Am. Acad. Dermatol. 2001, 44, 124–128. [Google Scholar] [CrossRef]
- Imashuku, S.; Miyagawa, A.; Chiyonobu, T.; Ishida, H.; Yoshihara, T.; Teramura, T.; Kuriyama, K.; Imamura, T.; Hibi, S.; Morimoto, A.; et al. Epstein-Barr virus-associated T-lymphoproliferative disease with hemophagocytic syndrome, followed by fatal intestinal B lymphoma in a young adult female with WHIM syndrome. Warts, hypogammaglobulinemia, infections, and myelokathexis. Ann. Hematol. 2002, 81, 470–473. [Google Scholar] [CrossRef]
- Ditzel Santos, D.; Ho, A.W.; Tournilhac, O.; Hatjiharissi, E.; Leleu, X.; Xu, L.; Tassone, P.; Neri, P.; Hunter, Z.R.; Chemaly, M.A.; et al. Establishment of BCWM.1 cell line for Waldenstrom’s macroglobulinemia with productive in vivo engraftment in SCID-hu mice. Exp. Hematol. 2007, 35, 1366–1375. [Google Scholar] [CrossRef]
- Hodge, L.S.; Novak, A.J.; Grote, D.M.; Braggio, E.; Ketterling, R.P.; Manske, M.K.; Price Troska, T.L.; Ziesmer, S.C.; Fonseca, R.; Witzig, T.E.; et al. Establishment and characterization of a novel Waldenstrom macroglobulinemia cell line, MWCL-1. Blood 2011, 117, e190–e197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, D.H.; Lopez, J.; Deng, F.; Liu, Q.; Ojode, T.; Chen, H.; Ulrick, J.; Kwatemaa, N.; Kelly, C.; Anaya-O’Brien, S.; et al. AMD3100 is a potent antagonist at CXCR4(R334X), a hyperfunctional mutant chemokine receptor and cause of WHIM syndrome. J. Cell. Mol. Med. 2011, 15, 2071–2081. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Choi, U.; Whiting-Theobald, N.L.; Linton, G.F.; Brenner, S.; Sechler, J.M.; Murphy, P.M.; Malech, H.L. Enhanced function with decreased internalization of carboxy-terminus truncated CXCR4 responsible for WHIM syndrome. Exp. Hematol. 2005, 33, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Reger, R.; Segerberg, F.; Lambert, M.; Leijonhufvud, C.; Baumer, Y.; Carlsten, M.; Childs, R. Enhanced Bone Marrow Homing of Natural Killer Cells Following mRNA Transfection With Gain-of-Function Variant CXCR4(R334X). Front. Immunol. 2019, 10, 1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormick, P.J.; Segarra, M.; Gasperini, P.; Gulino, A.V.; Tosato, G. Impaired recruitment of Grk6 and beta-Arrestin 2 causes delayed internalization and desensitization of a WHIM syndrome-associated CXCR4 mutant receptor. PLoS ONE 2009, 4, e8102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulino, A.V.; Moratto, D.; Sozzani, S.; Cavadini, P.; Otero, K.; Tassone, L.; Imberti, L.; Pirovano, S.; Notarangelo, L.D.; Soresina, R.; et al. Altered leukocyte response to CXCL12 in patients with warts hypogammaglobulinemia, infections, myelokathexis (WHIM) syndrome. Blood 2004, 104, 444–452. [Google Scholar] [CrossRef]
- Cao, Y.; Hunter, Z.R.; Liu, X.; Xu, L.; Yang, G.; Chen, J.; Patterson, C.J.; Tsakmaklis, N.; Kanan, S.; Rodig, S.; et al. The WHIM-like CXCR4(S338X) somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom’s Macroglobulinemia. Leukemia 2015, 29, 169–176. [Google Scholar] [CrossRef]
- Cao, Y.; Hunter, Z.R.; Liu, X.; Xu, L.; Yang, G.; Chen, J.; Tsakmaklis, N.; Kanan, S.; Castillo, J.J.; Treon, S.P. CXCR4 WHIM-like frameshift and nonsense mutations promote ibrutinib resistance but do not supplant MYD88(L265P)-directed survival signalling in Waldenstrom macroglobulinaemia cells. Br. J. Haematol. 2015, 168, 701–707. [Google Scholar] [CrossRef]
- Gustine, J.N.; Xu, L.; Tsakmaklis, N.; Demos, M.G.; Kofides, A.; Chen, J.G.; Liu, X.; Munshi, M.; Guerrera, M.L.; Chan, G.G.; et al. CXCR4 (S338X) clonality is an important determinant of ibrutinib outcomes in patients with Waldenstrom macroglobulinemia. Blood Adv. 2019, 3, 2800–2803. [Google Scholar] [CrossRef] [Green Version]
- Castellani, F.; Visentin, A.; Campagnolo, M.; Salvalaggio, A.; Cacciavillani, M.; Candiotto, C.; Bertorelle, R.; Trentin, L.; Briani, C. The Bruton tyrosine kinase inhibitor ibrutinib improves anti-MAG antibody polyneuropathy. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7. [Google Scholar] [CrossRef] [Green Version]
- Lagane, B.; Chow, K.Y.; Balabanian, K.; Levoye, A.; Harriague, J.; Planchenault, T.; Baleux, F.; Gunera-Saad, N.; Arenzana-Seisdedos, F.; Bachelerie, F. CXCR4 dimerization and beta-arrestin-mediated signaling account for the enhanced chemotaxis to CXCL12 in WHIM syndrome. Blood 2008, 112, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Balabanian, K.; Levoye, A.; Klemm, L.; Lagane, B.; Hermine, O.; Harriague, J.; Baleux, F.; Arenzana-Seisdedos, F.; Bachelerie, F. Leukocyte analysis from WHIM syndrome patients reveals a pivotal role for GRK3 in CXCR4 signaling. J. Clin. Investig. 2008, 118, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Pan, C.; Lopez, L.; Gao, J.; Velez, D.; Anaya-O’Brien, S.; Ulrick, J.; Littel, P.; Corns, J.S.; Ellenburg, D.T.; et al. WHIM Syndrome Caused by Waldenstrom’s Macroglobulinemia-Associated Mutation CXCR4 (L329fs). J. Clin. Immunol. 2016, 36, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Crump, M.P.; Gong, J.H.; Loetscher, P.; Rajarathnam, K.; Amara, A.; Arenzana-Seisdedos, F.; Virelizier, J.L.; Baggiolini, M.; Sykes, B.D.; Clark-Lewis, I. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 1997, 16, 6996–7007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kehrl, J.H. The impact of RGS and other G-protein regulatory proteins on Galphai-mediated signaling in immunity. Biochem. Pharmacol. 2016, 114, 40–52. [Google Scholar] [CrossRef] [Green Version]
- Oldham, W.M.; Hamm, H.E. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2008, 9, 60–71. [Google Scholar] [CrossRef]
- Busillo, J.M.; Armando, S.; Sengupta, R.; Meucci, O.; Bouvier, M.; Benovic, J.L. Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J. Biol. Chem. 2010, 285, 7805–7817. [Google Scholar] [CrossRef] [Green Version]
- Mueller, W.; Schutz, D.; Nagel, F.; Schulz, S.; Stumm, R. Hierarchical organization of multi-site phosphorylation at the CXCR4 C terminus. PLoS ONE 2013, 8, e64975. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.J.; Zhao, J.; Sun, Y.; Hu, W.; Wu, Y.L.; Cen, B.; Wu, G.X.; Pei, G. beta-arrestin differentially regulates the chemokine receptor CXCR4-mediated signaling and receptor internalization, and this implicates multiple interaction sites between beta-arrestin and CXCR4. J. Biol. Chem. 2000, 275, 2479–2485. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Busillo, J.M.; Stumm, R.; Benovic, J.L. G Protein-Coupled Receptor Kinase 3 and Protein Kinase C Phosphorylate the Distal C-Terminal Tail of the Chemokine Receptor CXCR4 and Mediate Recruitment of beta-Arrestin. Mol. Pharmacol. 2017, 91, 554–566. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Hu, Y.; Zhou, Q. The CXCL12-CXCR4 signaling axis plays a key role in cancer metastasis and is a potential target for developing novel therapeutics against metastatic cancer. Curr. Med. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Razinia, Z.; Makela, T.; Ylanne, J.; Calderwood, D.A. Filamins in mechanosensing and signaling. Annu. Rev. Biophys. 2012, 41, 227–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angers, S.; Salahpour, A.; Bouvier, M. Dimerization: An emerging concept for G protein-coupled receptor ontogeny and function. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 409–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doijen, J.; Van Loy, T.; De Haes, W.; Landuyt, B.; Luyten, W.; Schoofs, L.; Schols, D. Signaling properties of the human chemokine receptors CXCR4 and CXCR7 by cellular electric impedance measurements. PLoS ONE 2017, 12, e0185354. [Google Scholar] [CrossRef] [PubMed]
- Levoye, A.; Balabanian, K.; Baleux, F.; Bachelerie, F.; Lagane, B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood 2009, 113, 6085–6093. [Google Scholar] [CrossRef] [Green Version]
- Luker, K.E.; Gupta, M.; Luker, G.D. Imaging chemokine receptor dimerization with firefly luciferase complementation. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 823–834. [Google Scholar] [CrossRef]
- Sierro, F.; Biben, C.; Martinez-Munoz, L.; Mellado, M.; Ransohoff, R.M.; Li, M.; Woehl, B.; Leung, H.; Groom, J.; Batten, M.; et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc. Natl. Acad. Sci. USA 2007, 104, 14759–14764. [Google Scholar] [CrossRef] [Green Version]
- Mellado, M.; Rodriguez-Frade, J.M.; Vila-Coro, A.J.; Fernandez, S.; Martin de Ana, A.; Jones, D.R.; Toran, J.L.; Martinez, A.C. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. 2001, 20, 2497–2507. [Google Scholar] [CrossRef] [Green Version]
- Handel, T.M. The Structure of a CXCR4:Chemokine Complex. Front. Immunol. 2015, 6, 282. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Chien, E.Y.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Munoz, L.; Rodriguez-Frade, J.M.; Barroso, R.; Sorzano, C.O.S.; Torreno-Pina, J.A.; Santiago, C.A.; Manzo, C.; Lucas, P.; Garcia-Cuesta, E.M.; Gutierrez, E.; et al. Separating Actin-Dependent Chemokine Receptor Nanoclustering from Dimerization Indicates a Role for Clustering in CXCR4 Signaling and Function. Mol. Cell 2018, 70, 106–119 e110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Mutations in C-ter of CXCR4. Mutations identified in WHIM and WM patients are listed in the table. The schematic structure of CXCR4 reports sites of mutation, with frameshift mutations highlighted in green, nonsense mutations in yellow, and missense mutations in blue. Phosphorylation sites for GRKs and PKC, and docking sites for β-arrestin2 are filled in red. The most frequent S338 and R334 mutation sites are represented in bold.
Figure 1.
Mutations in C-ter of CXCR4. Mutations identified in WHIM and WM patients are listed in the table. The schematic structure of CXCR4 reports sites of mutation, with frameshift mutations highlighted in green, nonsense mutations in yellow, and missense mutations in blue. Phosphorylation sites for GRKs and PKC, and docking sites for β-arrestin2 are filled in red. The most frequent S338 and R334 mutation sites are represented in bold.

Figure 2.
Gain-of-function nature of WHIM-mutated CXCR4. WHIM mutations cause a gain of G protein-dependent functions, at least in part by abrogating normal β-arrestin-mediated receptor downregulation. The C-ter is the site of receptor phosphorylation sites by GRKs and PKC and their loss affects β-arrestin binding and receptor internalization. P, phosphate.
Figure 2.
Gain-of-function nature of WHIM-mutated CXCR4. WHIM mutations cause a gain of G protein-dependent functions, at least in part by abrogating normal β-arrestin-mediated receptor downregulation. The C-ter is the site of receptor phosphorylation sites by GRKs and PKC and their loss affects β-arrestin binding and receptor internalization. P, phosphate.


{kind=link}
{kind=link}
Table 1.
Biological, biochemical and signaling properties of WHIM-mutated CXCR4 variants.
| Type of Mutation | G PROTEIN | β-ARRESTIN | Calcium | ERK | AKT | CXCR4 Internalization | CXCR4 Membrane Expression | CXCR4 Desensitization | CXCR4 Eterodimerization | Cell Migration | Ibrutinib Response | Disease and Cell Type | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R334X | WHIM: K562, CHO | [52] | |||||||||||
| WHIM: CD34+, K562 | [53] | ||||||||||||
| WHIM: EBV | [16] | ||||||||||||
| WHIM: NK | [54] | ||||||||||||
| WHIM: HeLa, HEK293 | [55] | ||||||||||||
| G336X | WHIM: PMBC | [56] | |||||||||||
| S338X | R | WM: BCWM.1, MWCL-1 | [57] | ||||||||||
| R | WM: BCWM.1 | [58] | |||||||||||
| R | WM: Patients (undefined) | [59] | |||||||||||
| R | WM: Patients (undefined) | [60] | |||||||||||
| WHIM: PBMC, HEK293 | [61] | ||||||||||||
| WHIM: PBMC, CHO | [33] | ||||||||||||
| WHIM: EBV | [62] | ||||||||||||
| E343K | WHIM: K562, PBMC | [32] | |||||||||||
| L329fs | WHIM: K562, PBMC, HEK293 | [63] |
Evidence for unmodified, increased or impaired activity of WHIM-mutated CXCR4 compared to WT are shown in blue, green, and red, respectively. Weak impairment is indicated as light red. White boxes indicates absence of data. Properties reported have been evaluated upon CXCL12 stimulation in the indicated cellular contexts of WHIM and WM diseases. R, resistant.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Milanesi, S.; Locati, M.; Borroni, E.M. Aberrant CXCR4 Signaling at Crossroad of WHIM Syndrome and Waldenstrom’s Macroglobulinemia. Int. J. Mol. Sci. 2020, 21, 5696. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165696
AMA Style
Milanesi S, Locati M, Borroni EM. Aberrant CXCR4 Signaling at Crossroad of WHIM Syndrome and Waldenstrom’s Macroglobulinemia. International Journal of Molecular Sciences. 2020; 21(16):5696. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165696
Chicago/Turabian StyleMilanesi, Samantha, Massimo Locati, and Elena Monica Borroni. 2020. "Aberrant CXCR4 Signaling at Crossroad of WHIM Syndrome and Waldenstrom’s Macroglobulinemia" International Journal of Molecular Sciences 21, no. 16: 5696. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165696
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.