Predicting the Development of Anti-Drug Antibodies against Recombinant alpha-Galactosidase A in Male Patients with Classical Fabry Disease

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
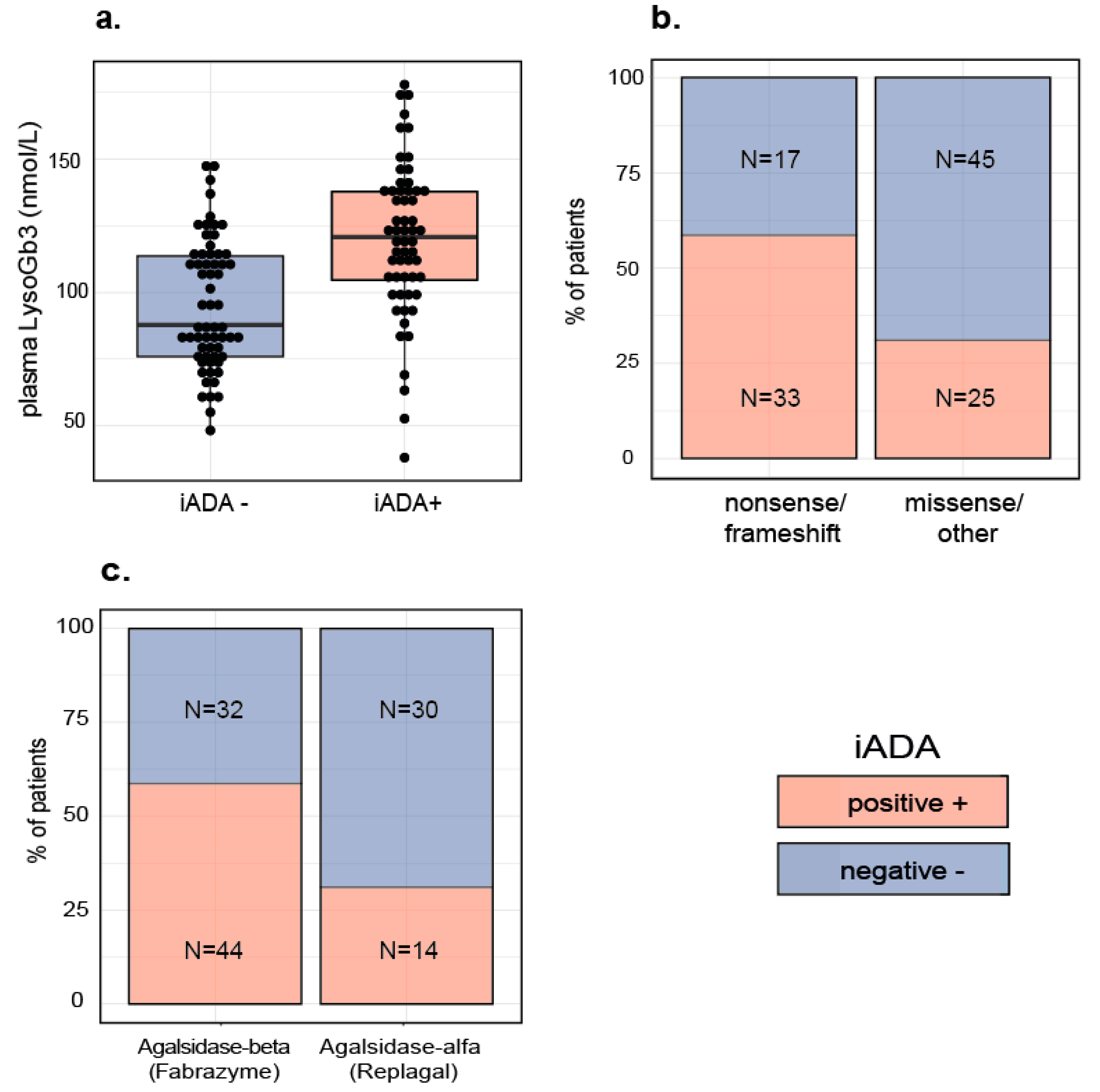
2.1. Patient Characteristics
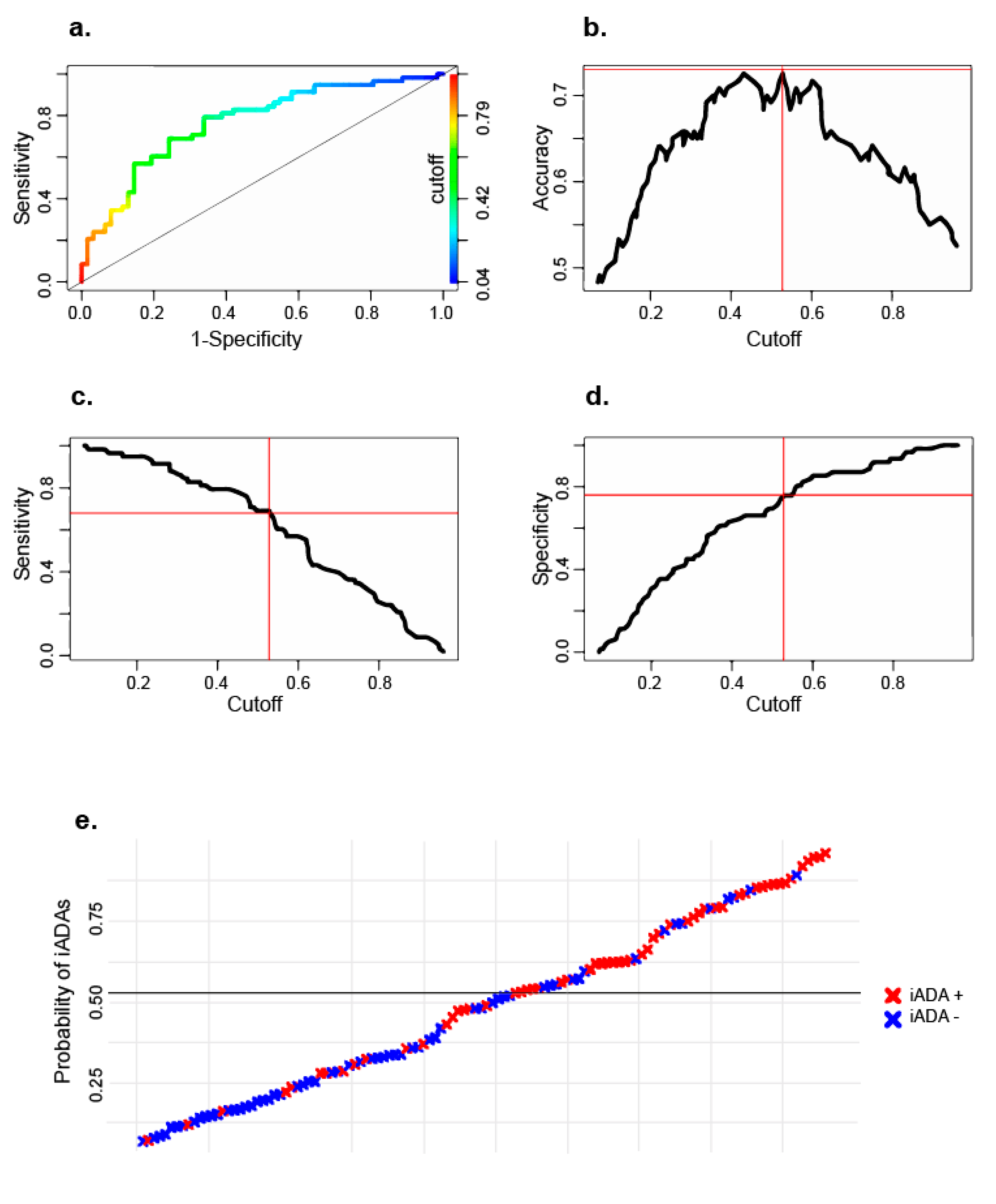
2.2. Logistic Regression Model
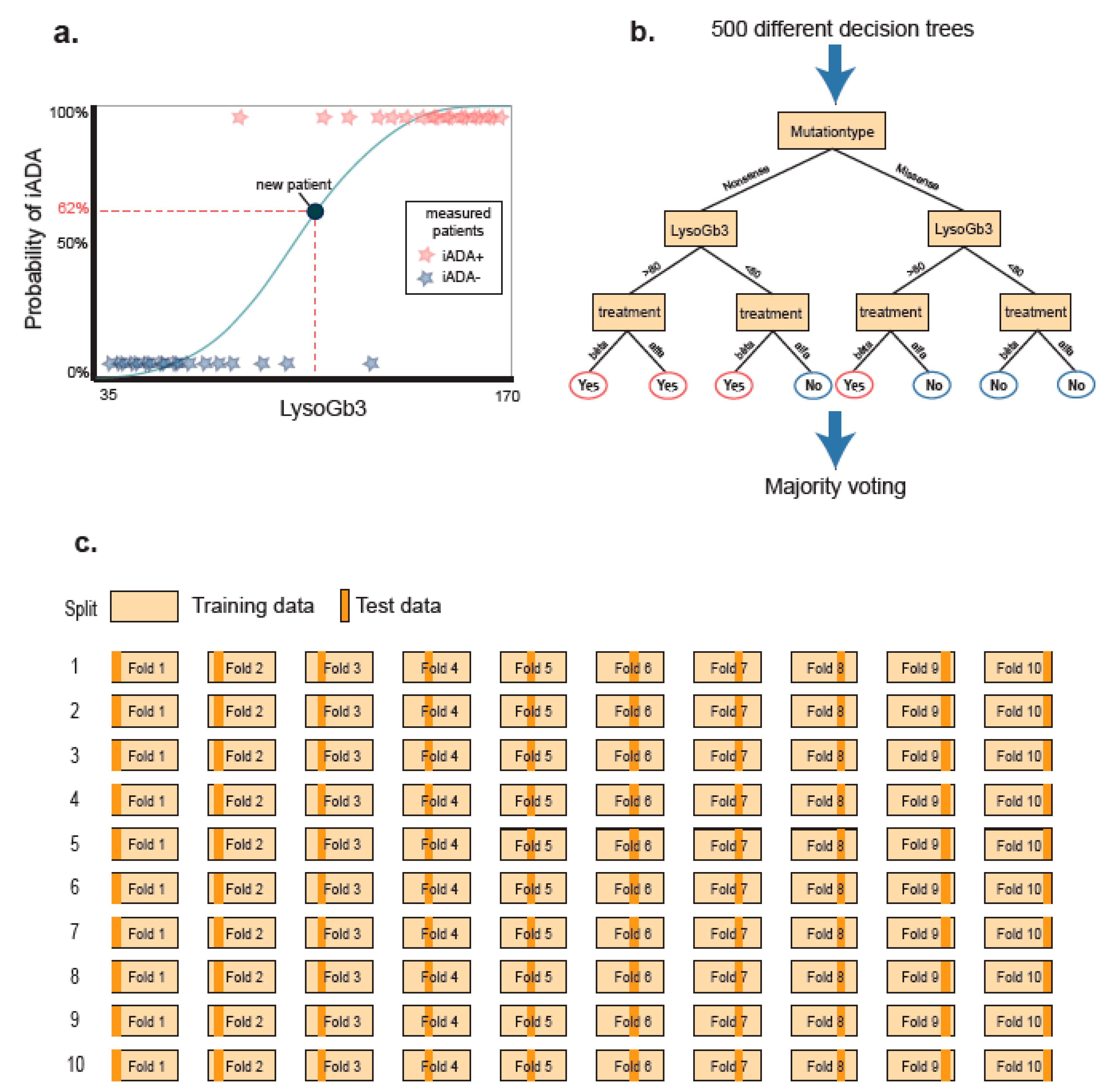
2.3. Random Forest Model
2.4. Post Hoc Analyses
2.5. Second Cohort
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Variables and Development of the Prediction Models
4.3. Laboratory Measurements
4.4. Statistics
4.4.1. Algorithms
4.4.2. Handling of Missing Data
4.4.3. Experiment and Intrinsic Validation
4.5. External Cohort
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADA | Anti-drug antibodies |
| αGAL A | Alpha-Galactosidase A (enzyme) |
| ERT | Enzyme replacement therapy |
| FD | Fabry disease |
| GB3 | Globotriaosylceramide |
| GLA | Alpha-Galactosidase A (gene) |
| iADA | Inhibiting anti-drug antibodies |
| LR | Logistic regression |
| LysoGb3 | Globotriaosylsphingosine |
| r- αGAL A | Recombinant alpha-galactosidase A (enzyme) |
| RF | Random forest |
| ROC-AUC | Area under the receiver operating characteristic curve |
References
- Desnick, R.J.; Ioannou, Y.A.; Eng, C.M. α-Galactosidase A Deficiency: Fabry Disease. In The Online Metabolic and Molecular Bases of Inherited Disease; Valle, D.L., Antonarakis, S., Ballabio, A., Beaudet, A.L., Mitchell, G.A., Eds.; OMMBID: New York, NY, USA, 2016. [Google Scholar] [CrossRef]
- Tesmoingt, C.; Lidove, O.; Reberga, A.; Thetis, M.; Ackaert, C.; Nicaise, P.; Arnaud, P.; Papo, T. Enzyme therapy in Fabry disease: Severe adverse events associated with anti-agalsidase cross-reactive IgG antibodies. Br. J. Clin. Pharm. 2009, 68, 765–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, K.; Bleasel, K.; Becker, G. Severe infusion reactions to fabry enzyme replacement therapy: Rechallenge after tracheostomy. JIMD Rep. 2012, 5, 109–112. [Google Scholar] [PubMed] [Green Version]
- Schiffmann, R.; Kopp, J.B.; Austin, H.A., 3rd; Sabnis, S.; Moore, D.F.; Weibel, T.; Balow, J.E.; Brady, R.O. Enzyme replacement therapy in Fabry disease: A randomized controlled trial. JAMA 2001, 285, 2743–2749. [Google Scholar] [CrossRef] [PubMed]
- Linthorst, G.E.; Hollak, C.E.; Donker-Koopman, W.E.; Strijland, A.; Aerts, J.M. Enzyme therapy for Fabry disease: Neutralizing antibodies toward agalsidase alpha and beta. Kidney Int. 2004, 66, 1589–1595. [Google Scholar] [CrossRef] [Green Version]
- van der Veen, S.J.; van Kuilenburg, A.B.P.; Hollak, C.E.M.; Kaijen, P.H.P.; Voorberg, J.; Langeveld, M. Antibodies against recombinant alpha-galactosidase A in Fabry disease: Subclass analysis and impact on response to treatment. Mol. Genet. Metab. 2019, 126, 162–168. [Google Scholar] [CrossRef]
- Stappers, F.; Scharnetzki, D.; Schmitz, B.; Manikowski, D.; Brand, S.M.; Grobe, K.; Lenders, M.; Brand, E. Neutralizing anti-drug antibodies in Fabry disease can inhibit endothelial enzyme uptake and activity. J. Inherit. Metab. Dis. 2020, 43, 334–347. [Google Scholar] [CrossRef]
- Ries, M.; Clarke, J.T.; Whybra, C.; Mehta, A.; Loveday, K.S.; Brady, R.O.; Beck, M.; Schiffmann, R. Enzyme replacement in Fabry disease: Pharmacokinetics and pharmacodynamics of agalsidase alpha in children and adolescents. J. Clin. Pharm. 2007, 47, 1222–1230. [Google Scholar] [CrossRef]
- Schiffmann, R.; Goker-Alpan, O.; Holida, M.; Giraldo, P.; Barisoni, L.; Colvin, R.B.; Jennette, C.J.; Maegawa, G.; Boyadjiev, S.A.; Gonzalez, D.; et al. Pegunigalsidase alfa, a novel PEGylated enzyme replacement therapy for Fabry disease, provides sustained plasma concentrations and favorable pharmacodynamics: A 1-year Phase 1/2 clinical trial. J. Inherit. Metab. Dis. 2019, 42, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, U.; Bichet, D.G.; Clarke, L.A.; Dostalova, G.; Fainboim, A.; Fellgiebel, A.; Forcelini, C.M.; An Haack, K.; Hopkin, R.J.; Mauer, M.; et al. Low-dose agalsidase beta treatment in male pediatric patients with Fabry disease: A 5-year randomized controlled trial. Mol. Genet. Metab. 2019, 127, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Benichou, B.; Goyal, S.; Sung, C.; Norfleet, A.M.; O’Brien, F. A retrospective analysis of the potential impact of IgG antibodies to agalsidase beta on efficacy during enzyme replacement therapy for Fabry disease. Mol. Genet. Metab. 2009, 96, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Lenders, M.; Stypmann, J.; Duning, T.; Schmitz, B.; Brand, S.M.; Brand, E. Serum-Mediated Inhibition of Enzyme Replacement Therapy in Fabry Disease. J. Am. Soc. Nephrol. 2016, 27, 256–264. [Google Scholar] [CrossRef]
- Walsh, C.E.; Jimenez-Yuste, V.; Auerswald, G.; Grancha, S. The burden of inhibitors in haemophilia patients. Thromb. Haemost. 2016, 116, S10–S17. [Google Scholar] [CrossRef]
- Banugaria, S.G.; Prater, S.N.; Ng, Y.K.; Kobori, J.A.; Finkel, R.S.; Ladda, R.L.; Chen, Y.T.; Rosenberg, A.S.; Kishnani, P.S. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: Lessons learned from infantile Pompe disease. Genet. Med. 2011, 13, 729–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Gelder, C.M.; Hoogeveen-Westerveld, M.; Kroos, M.A.; Plug, I.; van der Ploeg, A.T.; Reuser, A.J. Enzyme therapy and immune response in relation to CRIM status: The Dutch experience in classic infantile Pompe disease. J. Inherit. Metab. Dis. 2015, 38, 305–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langereis, E.J.; van Vlies, N.; Church, H.J.; Geskus, R.B.; Hollak, C.E.; Jones, S.A.; Kulik, W.; van Lenthe, H.; Mercer, J.; Schreider, L.; et al. Biomarker responses correlate with antibody status in mucopolysaccharidosis type I patients on long-term enzyme replacement therapy. Mol. Genet. Metab. 2015, 114, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Poelman, E.; Hoogeveen-Westerveld, M.; van den Hout, J.M.P.; Bredius, R.G.M.; Lankester, A.C.; Driessen, G.J.A.; Kamphuis, S.S.M.; Pijnappel, W.W.M.; van der Ploeg, A.T. Effects of immunomodulation in classic infantile Pompe patients with high antibody titers. Orphanet. J. Rare Dis. 2019, 14, 71. [Google Scholar] [CrossRef]
- Banugaria, S.G.; Prater, S.N.; McGann, J.K.; Feldman, J.D.; Tannenbaum, J.A.; Bailey, C.; Gera, R.; Conway, R.L.; Viskochil, D.; Kobori, J.A.; et al. Bortezomib in the rapid reduction of high sustained antibody titers in disorders treated with therapeutic protein: Lessons learned from Pompe disease. Genet. Med. 2013, 15, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Schep, S.J.; Schutgens, R.E.G.; Fischer, K.; Boes, M.L. Review of immune tolerance induction in hemophilia A. Blood Rev. 2018, 32, 326–338. [Google Scholar] [CrossRef]
- Ghosh, A.; Liao, A.; O’Leary, C.; Mercer, J.; Tylee, K.; Goenka, A.; Holley, R.; Jones, S.A.; Bigger, B.W. Strategies for the induction of immune tolerance to enzyme replacement therapy in Mucopolysaccharidosis Type I. Mol. Ther. Methods Clin. Dev. 2019, 13, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Gouw, S.C.; van der Bom, J.G.; Marijke van den Berg, H. Treatment-related risk factors of inhibitor development in previously untreated patients with hemophilia A: The CANAL cohort study. Blood 2007, 109, 4648–4654. [Google Scholar] [CrossRef]
- Singh, S.K. Impact of product-related factors on immunogenicity of biotherapeutics. J. Pharm. Sci. 2011, 100, 354–387. [Google Scholar] [CrossRef] [PubMed]
- Hermeling, S.; Crommelin, D.J.; Schellekens, H.; Jiskoot, W. Structure-immunogenicity relationships of therapeutic proteins. Pharm. Res. 2004, 21, 897–903. [Google Scholar] [CrossRef] [PubMed]
- LeMaoult, J.; Szabo, P.; Weksler, M.E. Effect of age on humoral immunity, selection of the B-cell repertoire and B-cell development. Immunol. Rev. 1997, 160, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lozier, J.; Johnson, G.; Kirshner, S.; Verthelyi, D.; Pariser, A.; Shores, E.; Rosenberg, A. Neutralizing antibodies to therapeutic enzymes: Considerations for testing, prevention and treatment. Nat. Biotechnol. 2008, 26, 901–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauhin, W.; Lidove, O.; Amelin, D.; Lamari, F.; Caillaud, C.; Mingozzi, F.; Dzangue-Tchoupou, G.; Arouche-Delaperche, L.; Douillard, C.; Dussol, B.; et al. Deep characterization of the anti-drug antibodies developed in Fabry disease patients, a prospective analysis from the French multicenter cohort FFABRY. Orphanet J. Rare Dis. 2018, 13, 127. [Google Scholar] [CrossRef] [Green Version]
- Garagiola, I.; Palla, R.; Peyvandi, F. Risk factors for inhibitor development in severe hemophilia a. Thromb. Res. 2018, 168, 20–27. [Google Scholar] [CrossRef]
- Bali, D.S.; Goldstein, J.L.; Banugaria, S.; Dai, J.; Mackey, J.; Rehder, C.; Kishnani, P.S. Predicting cross-reactive immunological material (CRIM) status in Pompe disease using GAA mutations: Lessons learned from 10 years of clinical laboratory testing experience. Am. J. Med. Genet. C Semin. Med. Genet. 2012, 160C, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Mauhin, W.; Lidove, O.; Masat, E.; Mingozzi, F.; Mariampillai, K.; Ziza, J.M.; Benveniste, O. Innate and adaptive immune response in Fabry Disease. JIMD Rep. 2015, 22, 1–10. [Google Scholar]
- Sherman, A.; Biswas, M.; Herzog, R.W. Tolerance induction in hemophilia: Innovation and accomplishments. Curr. Opin. Hematol. 2018, 25, 365–372. [Google Scholar] [CrossRef] [Green Version]
- Lenders, M.; Oder, D.; Nowak, A.; Canaan-Kuhl, S.; Arash-Kaps, L.; Drechsler, C.; Schmitz, B.; Nordbeck, P.; Hennermann, J.B.; Kampmann, C.; et al. Impact of immunosuppressive therapy on therapy-neutralizing antibodies in transplanted patients with Fabry disease. J. Intern. Med. 2017, 282, 241–253. [Google Scholar] [CrossRef] [Green Version]
- Poelman, E.; Hoogeveen-Westerveld, M.; Kroos-de Haan, M.A.; van den Hout, J.M.P.; Bronsema, K.J.; van de Merbel, N.C.; van der Ploeg, A.T.; Pijnappel, W. High sustained antibody titers in patients with classic infantile pompe disease following immunomodulation at Start of Enzyme Replacement Therapy. J. Pediatr. 2018, 195, 236–243.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenger, E.O.; Kazi, Z.; Lisi, E.; Gambello, M.J.; Kishnani, P. Immune tolerance strategies in siblings with infantile pompe disease-advantages for a preemptive approach to high-sustained antibody titers. Mol. Genet. Metab. Rep. 2015, 4, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Gouw, S.C.; van den Berg, H.M.; le Cessie, S.; van der Bom, J.G. Treatment characteristics and the risk of inhibitor development: A multicenter cohort study among previously untreated patients with severe hemophilia A. J. Thromb. Haemost. 2007, 5, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Reipert, B.M. Risky business of inhibitors: HLA haplotypes, gene polymorphisms, and immune responses. Hematology Am. Soc. Hematol. Educ. Program. 2014, 2014, 372–378. [Google Scholar] [CrossRef]
- Kempton, C.L.; Payne, A.B. HLA-DRB1-factor VIII binding is a risk factor for inhibitor development in nonsevere hemophilia: A case-control study. Blood Adv. 2018, 2, 1750–1755. [Google Scholar] [CrossRef] [Green Version]
- De Groot, A.S.; Kazi, Z.B.; Martin, R.F.; Terry, F.E.; Desai, A.K.; Martin, W.D.; Kishnani, P.S. HLA- and genotype-based risk assessment model to identify infantile onset pompe disease patients at high-risk of developing significant anti-drug antibodies (ADA). Clin. Immunol. 2019, 200, 66–70. [Google Scholar] [CrossRef]
- Lovgren, K.M.; Sondergaard, H.; Skov, S.; Wiinberg, B. Non-genetic risk factors in haemophilia A inhibitor management - the danger theory and the use of animal models. Haemophilia 2016, 22, 657–666. [Google Scholar] [CrossRef]
- Arends, M.; Biegstraaten, M.; Hughes, D.A.; Mehta, A.; Elliott, P.M.; Oder, D.; Watkinson, O.T.; Vaz, F.M.; van Kuilenburg, A.B.P.; Wanner, C.; et al. Retrospective study of long-term outcomes of enzyme replacement therapy in Fabry disease: Analysis of prognostic factors. PLoS ONE 2017, 12, e0182379. [Google Scholar] [CrossRef]
- Arends, M.; Wanner, C.; Hughes, D.; Mehta, A.; Oder, D.; Watkinson, O.T.; Elliott, P.M.; Linthorst, G.E.; Wijburg, F.A.; Biegstraaten, M.; et al. Characterization of classical and nonclassical fabry disease: A multicenter study. J. Am. Soc. Nephrol. 2017, 28, 1631–1641. [Google Scholar] [CrossRef] [Green Version]
- Collins, G.S.; Reitsma, J.B.; Altman, D.G.; Moons, K.G. Transparent Reporting of a multivariable prediction model for Individual Prognosis or Diagnosis (TRIPOD): The TRIPOD statement. Ann. Intern. Med. 2015, 162, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Gold, H.; Mirzaian, M.; Dekker, N.; Joao Ferraz, M.; Lugtenburg, J.; Codee, J.D.; van der Marel, G.A.; Overkleeft, H.S.; Linthorst, G.E.; Groener, J.E.; et al. Quantification of globotriaosylsphingosine in plasma and urine of fabry patients by stable isotope ultraperformance liquid chromatography-tandem mass spectrometry. Clin. Chem. 2013, 59, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruger, R.; Tholey, A.; Jakoby, T.; Vogelsberger, R.; Monnikes, R.; Rossmann, H.; Beck, M.; Lackner, K.J. Quantification of the Fabry marker lysoGb3 in human plasma by tandem mass spectrometry. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2012, 883–884, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Kirasich, K.S.; Trace, S.; Bivin, S. Random Forest vs. Logistic Regression: Binary Classification for Heterogeneous Datasets. SMU Data Sci. Rev. 2018, 1. Available online: https://scholar.smu.edu/datasciencereview/vol1/iss3/9 (accessed on 11 July 2020).
- Azur, M.J.; Stuart, E.A.; Frangakis, C.; Leaf, P.J. Multiple imputation by chained equations: What is it and how does it work? Int. J. Methods Psychiatr. Res. 2011, 20, 40–49. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| iADA+ | iADA− | |
|---|---|---|
| Site (N, % of total) | ||
| 23 (40%) | 16 (26%) |
| 24 (41%) | 26 (42%) |
| 11 (19%) | 20 (32%) |
| Mutation type (N, % of total) | ||
| 33 (57%) | 17 (27%) |
| 21 (36%) | 37 (60%) |
| 4 (7%) | 8 (13%) |
| Age at ERT start (years, median, range) | 37 (9–58) | 35 (13–63) |
| LysoGb3 (nmol/L, median, range) | 123 (38–178) | 96 (48–149) |
| First treatment (N, % of total) | ||
| 14 (24%) | 31 (50%) |
| 4 (7%) | 2 (3%) |
| 2 (3%) | 2 (3%) |
| 38 (66%) | 27 (44%) |
| Inhibition titer (median, range) | 113 (7–32645) | 0 (0–5) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Veen, S.J.; Vlietstra, W.J.; van Dussen, L.; van Kuilenburg, A.B.P.; Dijkgraaf, M.G.W.; Lenders, M.; Brand, E.; Wanner, C.; Hughes, D.; Elliott, P.M.; et al. Predicting the Development of Anti-Drug Antibodies against Recombinant alpha-Galactosidase A in Male Patients with Classical Fabry Disease. Int. J. Mol. Sci. 2020, 21, 5784. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165784
van der Veen SJ, Vlietstra WJ, van Dussen L, van Kuilenburg ABP, Dijkgraaf MGW, Lenders M, Brand E, Wanner C, Hughes D, Elliott PM, et al. Predicting the Development of Anti-Drug Antibodies against Recombinant alpha-Galactosidase A in Male Patients with Classical Fabry Disease. International Journal of Molecular Sciences. 2020; 21(16):5784. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165784
Chicago/Turabian Stylevan der Veen, Sanne J., Wytze J. Vlietstra, Laura van Dussen, André B.P. van Kuilenburg, Marcel G. W. Dijkgraaf, Malte Lenders, Eva Brand, Christoph Wanner, Derralynn Hughes, Perry M. Elliott, and et al. 2020. "Predicting the Development of Anti-Drug Antibodies against Recombinant alpha-Galactosidase A in Male Patients with Classical Fabry Disease" International Journal of Molecular Sciences 21, no. 16: 5784. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165784