In Silico Structural and Biochemical Functional Analysis of a Novel CYP21A2 Pathogenic Variant

 , , ,
, , , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Case Report
2.2. DNA Studies
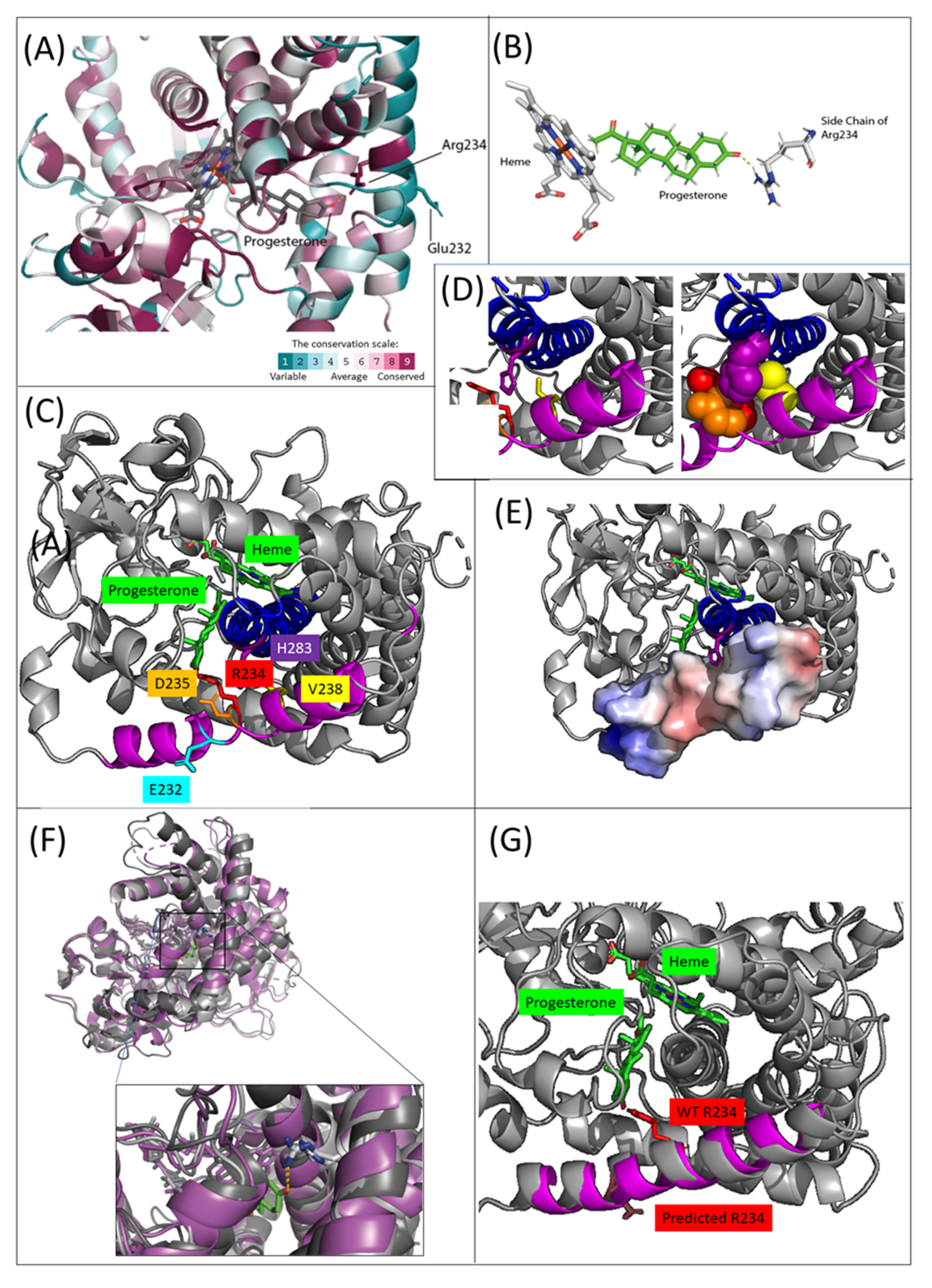
2.3. In Silico Analysis of Protein Structure and Function
2.4. In Vitro Biochemical Evaluation of Enzymatic Activity
3. Discussion
4. Materials and Methods
4.1. Genetic Analysis
4.2. In Silico Studies: Mechanistic Analysis of Protein Function with a Three-Dimensional Molecular CYP21A2 Model
4.3. Biochemical Evaluation of Enzymatic Activity
4.3.1. Cell Transfection and Steroid Profiling
4.3.2. Western Blot
4.3.3. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- White, P.C.; New, M.I.; Dupont, B. HLA-linked congenital adrenal hyperplasia results from a defective gene encoding a cytochrome P-450 specific for steroid 21-hydroxylation. Proc. Natl. Acad. Sci. USA 1984, 81, 7505–7509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, P.C.; Speiser, P.W. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr. Rev. 2000, 21, 245–291. [Google Scholar] [CrossRef] [PubMed]
- Speiser, P.W.; Arlt, W.; Auchus, R.J.; Baskin, L.S.; Conway, G.S.; Merke, D.P.; Meyer-Bahlburg, H.F.L.; Miller, W.L.; Murad, M.H.; Oberfield, S.E.; et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2018, 103, 4043–4088. [Google Scholar] [CrossRef] [PubMed]
- Sperling, M. Pediatric Endocrinology, 4th ed.; Saunders: Philadelphia, PA, USA, 2014. [Google Scholar]
- Higashi, Y.; Yoshioka, H.; Yamane, M.; Gotoh, O.; Fujii-Kuriyama, Y. Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome: A pseudogene and a genuine gene. Proc. Natl. Acad. Sci. USA 1986, 83, 2841–2845. [Google Scholar] [CrossRef] [Green Version]
- Nebert, D.W.; Nelson, D.R.; Coon, M.J.; Estabrook, R.W.; Feyereisen, R.; Fujii-Kuriyama, Y.; Gonzalez, F.J.; Guengerich, F.P.; Gunsalus, I.C.; Johnson, E.F.; et al. The P450 superfamily: Update on new sequences, gene mapping, and recommended nomenclature. DNA Cell Biol. 1991, 10, 1–14. [Google Scholar] [CrossRef]
- Pallan, P.S.; Wang, C.; Lei, L.; Yoshimoto, F.K.; Auchus, R.J.; Waterman, M.R.; Guengerich, F.P.; Egli, M. Human Cytochrome P450 21A2, the Major Steroid 21-Hydroxylase: Structure of the Enzyme.Progesterone Substrate Complex and Rate-Limiting C-H Bond Cleavage. J. Biol. Chem. 2015, 290, 13128–13143. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Pallan, P.S.; Zhang, W.; Lei, L.; Yoshimoto, F.K.; Waterman, M.R.; Egli, M.; Guengerich, F.P. Functional analysis of human cytochrome P450 21A2 variants involved in congenital adrenal hyperplasia. J. Biol. Chem. 2017, 292, 10767–10778. [Google Scholar] [CrossRef] [Green Version]
- Pharmacogene. Pharmacogene Variation (PharmVar) Consortium. Available online: https://www.pharmvar.org/gene/CYP1A2 (accessed on 20 March 2020).
- White, P.C.; New, M.I.; Dupont, B. Structure of human steroid 21-hydroxylase genes. Proc. Natl. Acad. Sci. USA 1986, 83, 5111–5115. [Google Scholar] [CrossRef] [Green Version]
- Donohoue, P.A.; Van Dop, C.; Jospe, N.; Migeon, C.J. Congenital adrenal hyperplasia: Molecular mechanisms resulting in 21-hydroxylase deficiency. Acta Endocrinol. Suppl. 1986, 279, 315–320. [Google Scholar] [CrossRef]
- Higashi, Y.; Tanae, A.; Inoue, H.; Hiromasa, T.; Fujii-Kuriyama, Y. Aberrant splicing and missense mutations cause steroid 21-hydroxylase [P-450(C21)] deficiency in humans: Possible gene conversion products. Proc. Natl. Acad. Sci. USA 1988, 85, 7486–7490. [Google Scholar] [CrossRef] [Green Version]
- Prader, A. [Genital findings in the female pseudo-hermaphroditism of the congenital adrenogenital syndrome; morphology, frequency, development and heredity of the different genital forms]. Helv. Paediatr. Acta 1954, 9, 231–248. [Google Scholar]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celniker, G.; Nimrod, G.; Ashkenazy, H.; Glaser, F.; Martz, E.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf: Using evolutionary data to raise testable hypotheses about protein function. Isr. J. Chem. 2013, 53, 199–206. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef] [Green Version]
- Glaser, F.; Pupko, T.; Paz, I.; Bell, R.E.; Bechor-Shental, D.; Martz, E.; Ben-Tal, N. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 2003, 19, 163–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armon, A.; Graur, D.; Ben-Tal, N. ConSurf: An algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J. Mol. Biol. 2001, 307, 447–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonetti, L.; Bruque, C.D.; Fernández, C.S.; Benavides-Mori, B.; Delea, M.; Kolomenski, J.E.; Espeche, L.D.; Buzzalino, N.D.; Nadra, A.D.; Dain, L. CYP21A2 mutation update: Comprehensive analysis of databases and published genetic variants. Hum. Mutat. 2018, 39, 5–22. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Barbaro, M.; Soardi, F.C.; Östberg, L.J.; Persson, B.; de Mello, M.P.; Wedell, A.; Lajic, S. In vitro functional studies of rare CYP 21A2 mutations and establishment of an activity gradient for nonclassic mutations improve phenotype predictions in congenital adrenal hyperplasia. Clin. Endocrinol. 2015, 82, 37–44. [Google Scholar] [CrossRef]
- Tardy, V.; Menassa, R.; Sulmont, V.; Lienhardt-Roussie, A.; Lecointre, C.; Brauner, R.; David, M.; Morel, Y. Phenotype-genotype correlations of 13 rare CYP21A2 mutations detected in 46 patients affected with 21-hydroxylase deficiency and in one carrier. J. Clin. Endocrinol. Metab. 2010, 95, 1288–1300. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.R.; Carmichael, H.; Andrew, S.F.; Miller, T.C.; Moon, J.E.; Derr, M.A.; Hwa, V.; Hirschhorn, J.N.; Dauber, A. Large-scale pooled next-generation sequencing of 1077 genes to identify genetic causes of short stature. J. Clin. Endocrinol. Metab. 2013, 98, E1428–E1437. [Google Scholar] [CrossRef] [Green Version]
- Hauer, N.N.; Popp, B.; Schoeller, E.; Schuhmann, S.; Heath, K.E.; Hisado-Oliva, A.; Klinger, P.; Kraus, C.; Trautmann, U.; Zenker, M.; et al. Clinical relevance of systematic phenotyping and exome sequencing in patients with short stature. Genet. Med. Off. J. Am. Coll. Med. Genet. 2018, 20, 630–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauer, N.N.; Popp, B.; Taher, L.; Vogl, C.; Dhandapany, P.S.; Buttner, C.; Uebe, S.; Sticht, H.; Ferrazzi, F.; Ekici, A.B.; et al. Evolutionary conserved networks of human height identify multiple Mendelian causes of short stature. Eur. J. Hum. Genet. EJHG 2019, 27, 1061–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.H.; Shen, Y.; Walvoord, E.C.; Miller, T.C.; Moon, J.E.; Hirschhorn, J.N.; Dauber, A. Whole exome sequencing to identify genetic causes of short stature. Horm. Res. Paediatr. 2014, 82, 44–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Takahashi, T.; Ohura, T.; Adachi, H.; Takahashi, I.; Ogawa, E.; Okuda, H.; Kobayashi, H.; Hitomi, T.; Liu, W.; et al. Combined linkage analysis and exome sequencing identifies novel genes for familial goiter. J. Hum. Genet. 2013, 58, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Abreu, A.P.; Dauber, A.; Macedo, D.B.; Noel, S.D.; Brito, V.N.; Gill, J.C.; Cukier, P.; Thompson, I.R.; Navarro, V.M.; Gagliardi, P.C.; et al. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N. Engl. J. Med. 2013, 368, 2467–2475. [Google Scholar] [CrossRef] [Green Version]
- Sabo, A.; Mishra, P.; Dugan-Perez, S.; Voruganti, V.S.; Kent, J.W., Jr.; Kalra, D.; Cole, S.A.; Comuzzie, A.G.; Muzny, D.M.; Gibbs, R.A.; et al. Exome sequencing reveals novel genetic loci influencing obesity-related traits in Hispanic children. Obesity 2017, 25, 1270–1276. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.L.; Vega-Warner, V.; Gillies, C.; Sampson, M.G.; Kher, V.; Sethi, S.K.; Otto, E.A. Whole Exome Sequencing Reveals Novel PHEX Splice Site Mutations in Patients with Hypophosphatemic Rickets. PLoS ONE 2015, 10, e0130729. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Ellard, S.; Baple, E.L.; Berry, I.; Forrester, N.; Turnbull, C.; Owens, M.; Eccles, D.M.; Abbs, S.; Scott, R.; Deans, Z.C.; et al. ACGS Best Practice Guidelines for Variant Classification. ACGS Guidelines. 2019. Available online: https://www.acgs.uk.com/ (accessed on 1 April 2020).
- Miosge, L.A.; Field, M.A.; Sontani, Y.; Cho, V.; Johnson, S.; Palkova, A.; Balakishnan, B.; Liang, R.; Zhang, Y.; Lyon, S. Comparison of predicted and actual consequences of missense mutations. Proc. Natl. Acad. Sci. USA 2015, 112, E5189–E5198. [Google Scholar] [CrossRef] [Green Version]
- Majithia, A.R.; Tsuda, B.; Agostini, M.; Gnanapradeepan, K.; Rice, R.; Peloso, G.; Patel, K.A.; Zhang, X.; Broekema, M.F.; Patterson, N.; et al. Prospective functional classification of all possible missense variants in PPARG. Nat. Genet. 2016, 48, 1570–1575. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, T.O.; Zhang, J.H.; Parra, E.; Ellis, B.K.; Simpson, C.; Lee, W.M.; Balko, J.; Fu, L.; Wong, B.Y.; Cole, D.E. Vitamin D binding protein is a key determinant of 25-hydroxyvitamin D levels in infants and toddlers. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2013, 28, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Wierbowski, S.D.; Fragoza, R.; Liang, S.; Yu, H. Extracting complementary insights from molecular phenotypes for prioritization of disease-associated mutations. Curr. Opin. Syst. Biol. 2018, 11, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Ittisoponpisan, S.; Islam, S.A.; Khanna, T.; Alhuzimi, E.; David, A.; Sternberg, M.J.E. Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated? J. Mol. Biol. 2019, 431, 2197–2212. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.; Fowler, D.M.; Hartmann-Petersen, R.; Lindorff-Larsen, K. Biophysical and mechanistic models for disease-causing protein variants. Trends Biochem. Sci. 2019, 44, 575–588. [Google Scholar]
- Keegan, C.E.; Killeen, A.A. An overview of molecular diagnosis of steroid 21-hydroxylase deficiency. J. Mol. Diagn. JMD 2001, 3, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Samandari, E.; Kempna, P.; Nuoffer, J.M.; Hofer, G.; Mullis, P.E.; Fluck, C.E. Human adrenal corticocarcinoma NCI-H295R cells produce more androgens than NCI-H295A cells and differ in 3beta-hydroxysteroid dehydrogenase type 2 and 17,20 lyase activities. J. Endocrinol. 2007, 195, 459–472. [Google Scholar] [CrossRef] [Green Version]
- Kempna, P.; Hirsch, A.; Hofer, G.; Mullis, P.E.; Fluck, C.E. Impact of differential P450c17 phosphorylation by cAMP stimulation and by starvation conditions on enzyme activities and androgen production in NCI-H295R cells. Endocrinology 2010, 151, 3686–3696. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cohen, M.; Pignatti, E.; Dines, M.; Mory, A.; Ekhilevitch, N.; Kolodny, R.; Flück, C.E.; Tiosano, D. In Silico Structural and Biochemical Functional Analysis of a Novel CYP21A2 Pathogenic Variant. Int. J. Mol. Sci. 2020, 21, 5857. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165857
Cohen M, Pignatti E, Dines M, Mory A, Ekhilevitch N, Kolodny R, Flück CE, Tiosano D. In Silico Structural and Biochemical Functional Analysis of a Novel CYP21A2 Pathogenic Variant. International Journal of Molecular Sciences. 2020; 21(16):5857. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165857
Chicago/Turabian StyleCohen, Michal, Emanuele Pignatti, Monica Dines, Adi Mory, Nina Ekhilevitch, Rachel Kolodny, Christa E. Flück, and Dov Tiosano. 2020. "In Silico Structural and Biochemical Functional Analysis of a Novel CYP21A2 Pathogenic Variant" International Journal of Molecular Sciences 21, no. 16: 5857. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165857