Asymmetric Conservation within Pairs of Co-Occurred Motifs Mediates Weak Direct Binding of Transcription Factors in ChIP-Seq Data


Abstract
:
1. Introduction
2. Results
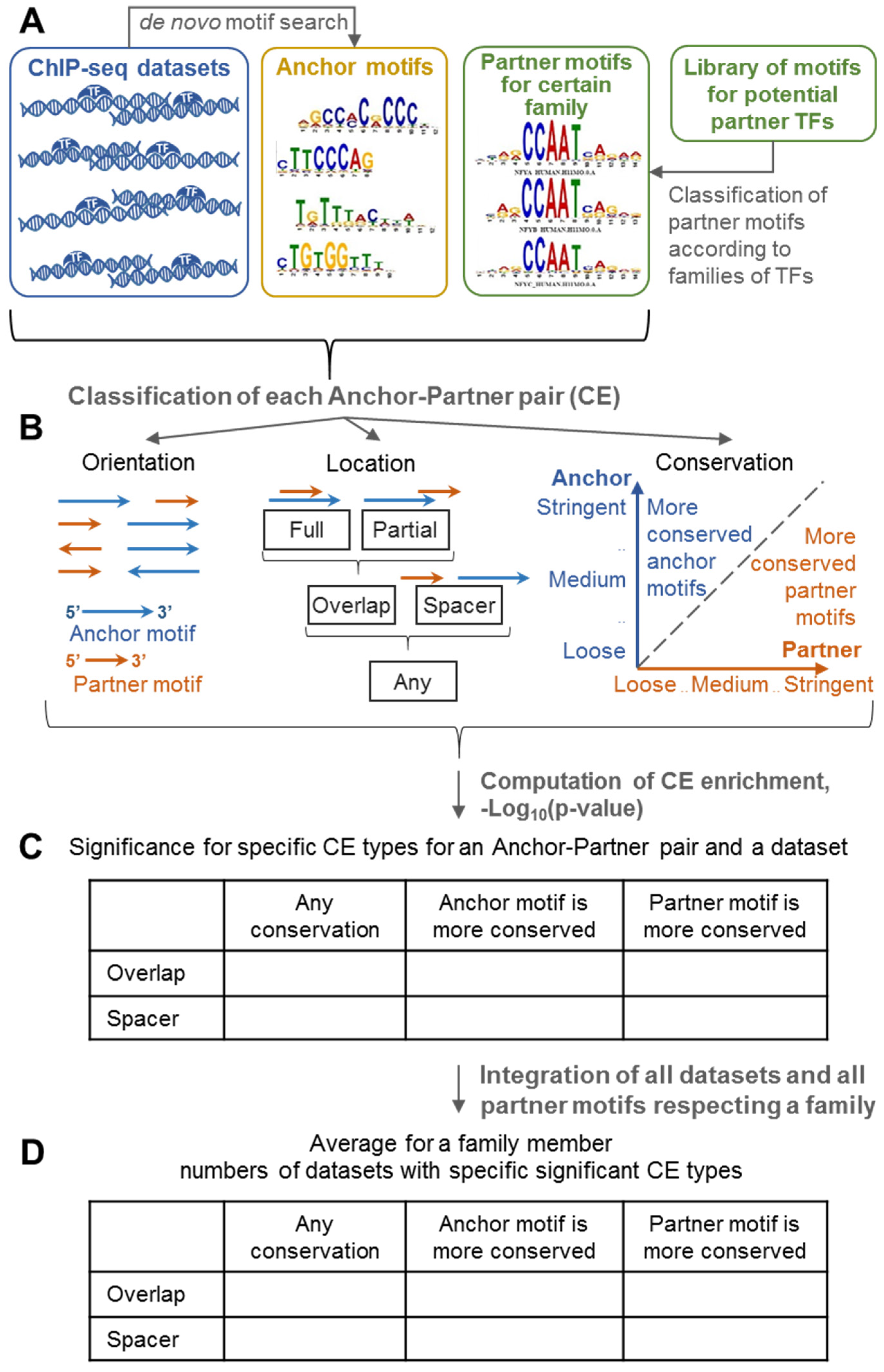
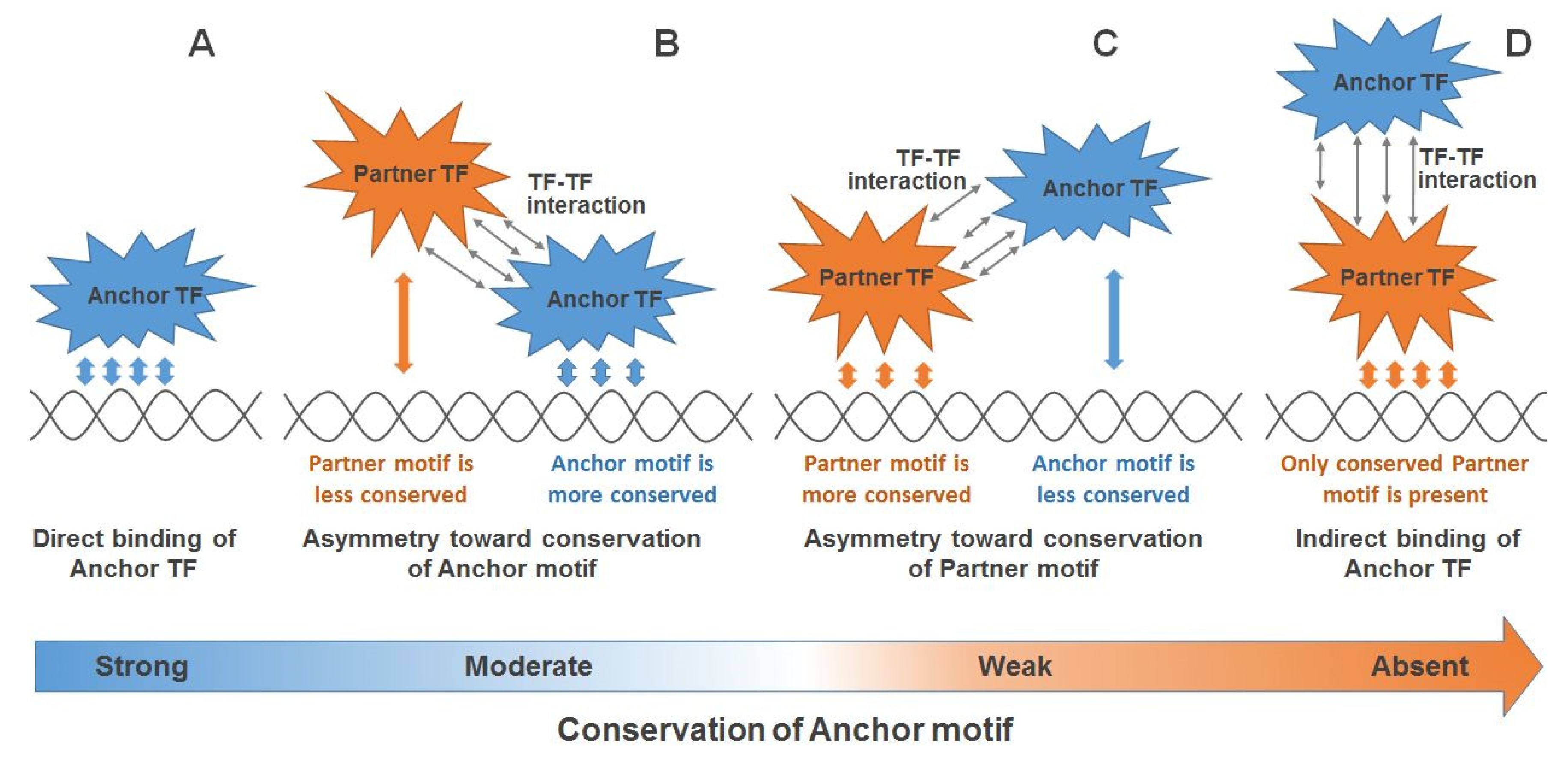
2.1. Integration of CE Significance and CE Asymmetry in the MCOT Analysis
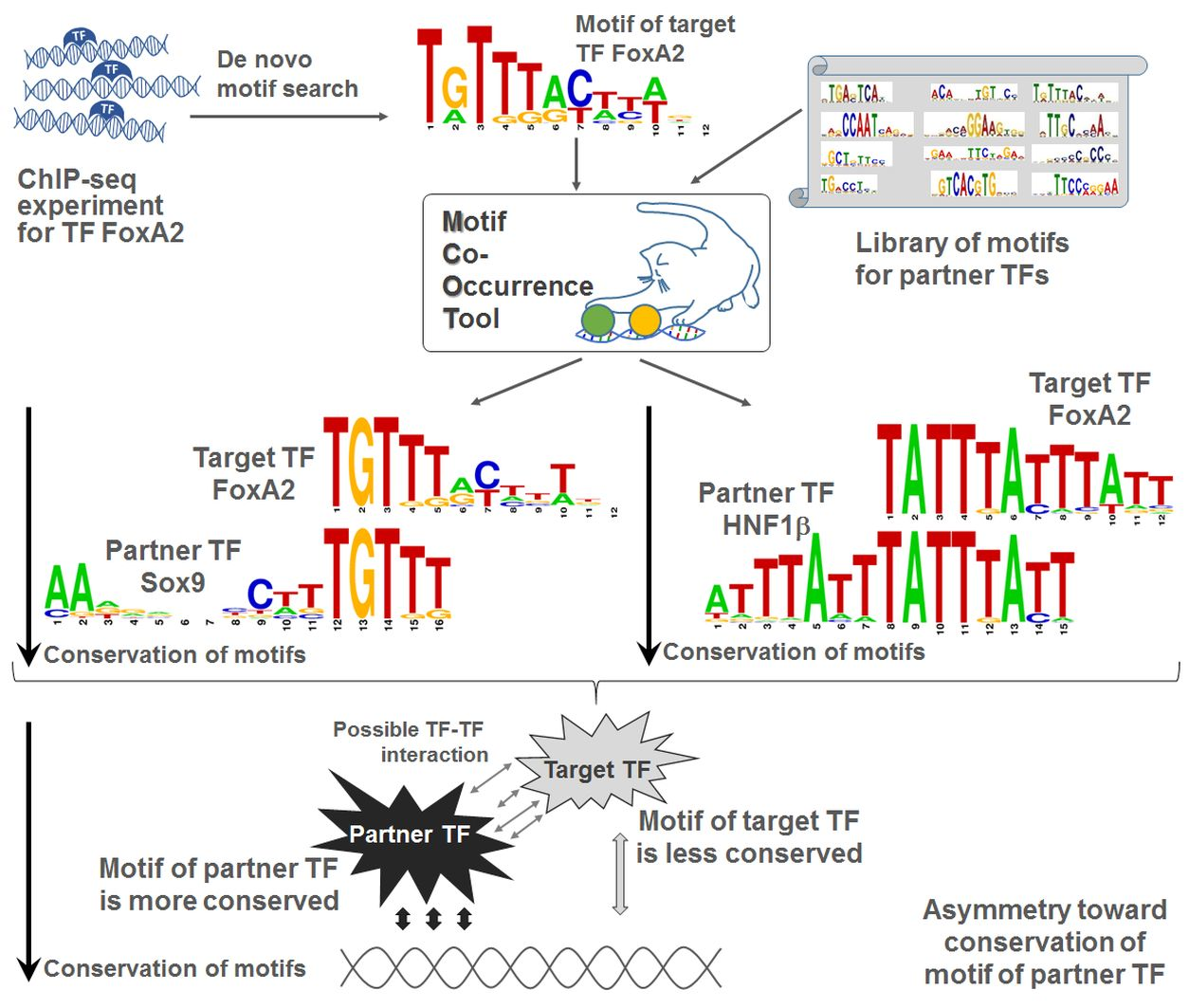
2.2. Single ChIP-Seq Dataset: Example of Significant Asymmetry within CE
2.3. Single ChIP-Seq Dataset: Multiple Partner TFs Support Binding of Anchor TF
- Thyroid hormone receptor-related factors (NR1){2.1.2} (NR1H3_MOUSE.H11MO.0.A),
- POU domain factors {3.1.10} (HNF1A_MOUSE.H11MO.0.A and HNF1B_MOUSE.H11MO.0.A),
- HD-CUT factors {3.1.9} (HNF6_MOUSE.H11MO.0.A and CUX2_MOUSE.H11MO.0.C),
- C/EBP-related {1.1.8} (NFIL3_MOUSE.H11MO.0.C),
- SOX-related factors {4.1.1} (SOX9_MOUSE.H11MO.0.A and SOX10_MOUSE.H11MO.0.B).
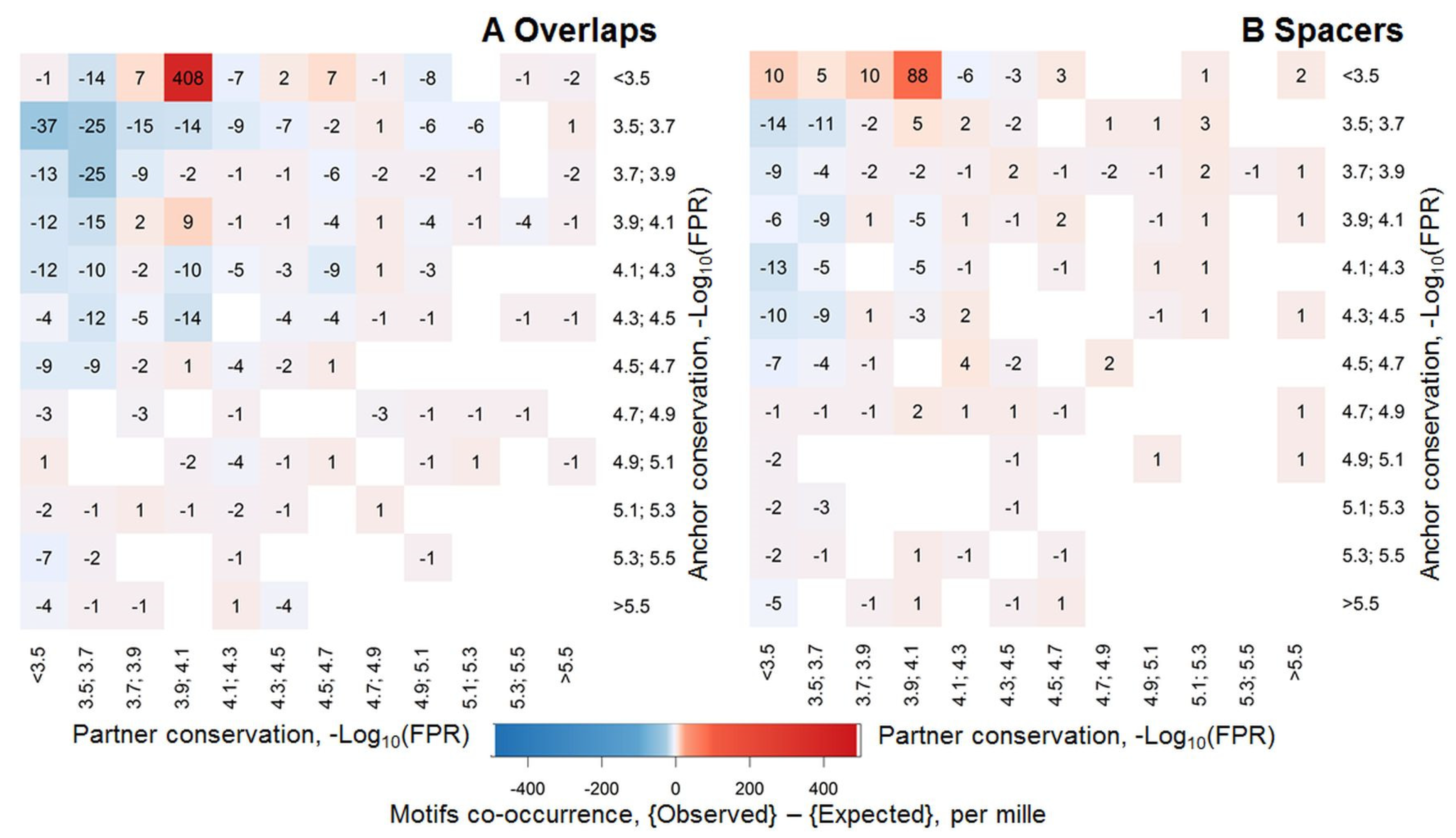
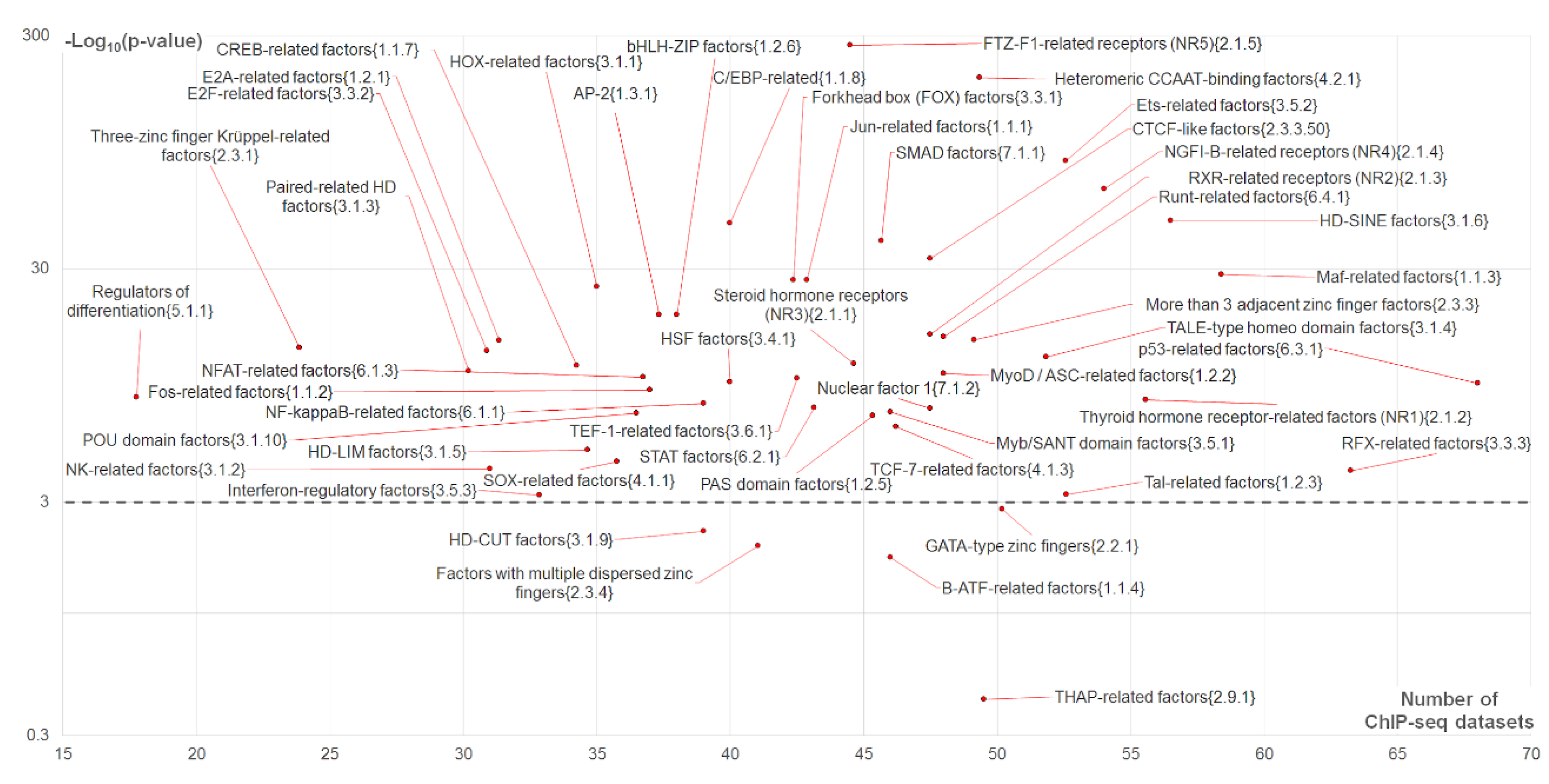
2.4. Massive Analysis of Asymmetric CEs
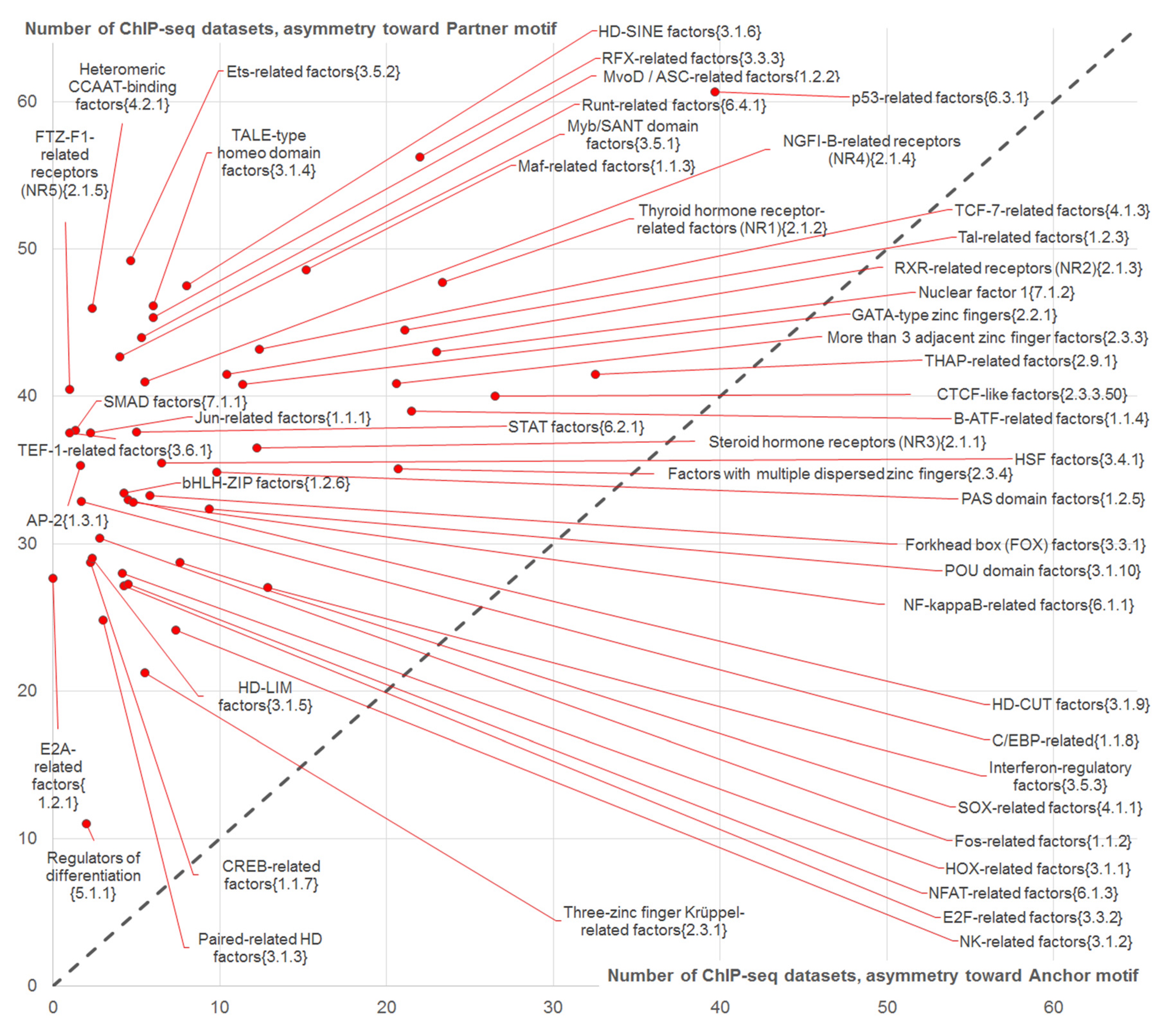
2.4.1. Analysis of Partner Motifs Classified According to the TFs Families
- High abundance of CEs (axis X in Figure 5),
- Significant enrichment of asymmetric CEs toward partner motifs vs. those asymmetric toward anchor motifs (axis Y in Figure 5);
- High abundance of asymmetric CEs toward partner motifs in comparison with that for asymmetric CEs toward anchor motifs (Figure 4).
2.4.2. Analysis of Top-Ranked Partner Motifs Classified According to TFs Families
3. Discussion
4. Materials and Methods
4.1. MCOT: Classification of Co-Occurred Motifs
4.2. Composite Elements Search and Annotation
4.3. Significances for Asymmetric CEs and for Asymmetry within CEs
- {−Log10[FPR(Anchor)] > −Log10[FPR(Partner)]} and,
- {−Log10[FPR(Anchor)] ≤ −Log10[FPR(Partner)]}.
4.4. Bonferroni Correction for Significance
- Significance of CEs regardless motifs conservation, 0.05/(NFOR × NBACK × NFLOW × NTHR × NTHR);
- Significance of asymmetric CEs toward one of motifs, 0.05/(NFOR × NBACK × NFLOW × 2);
- CE asymmetry, 0.05/(NFOR × NBACK × NFLOW).
4.5. Massive Analysis of the ChIP-Seq Data
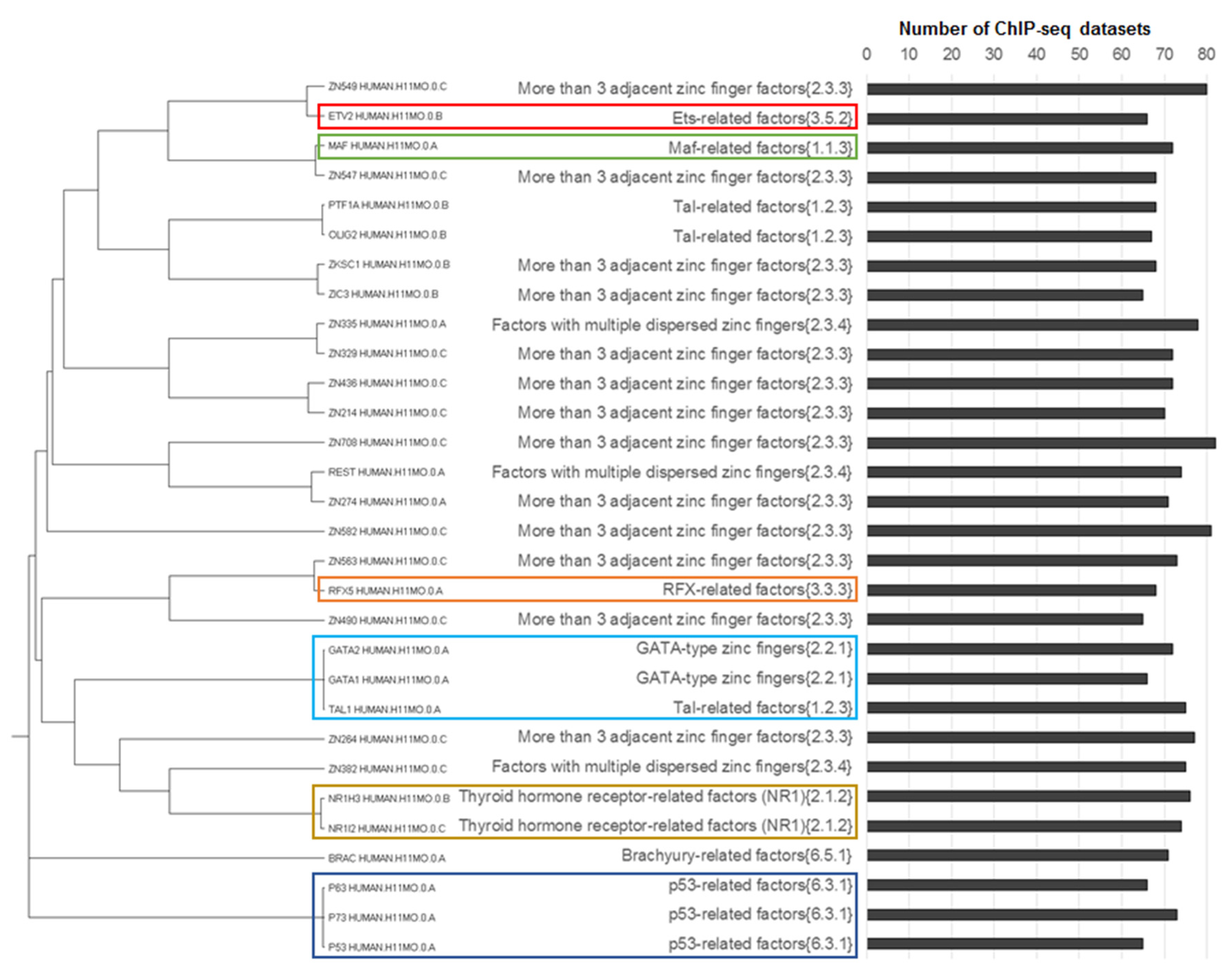
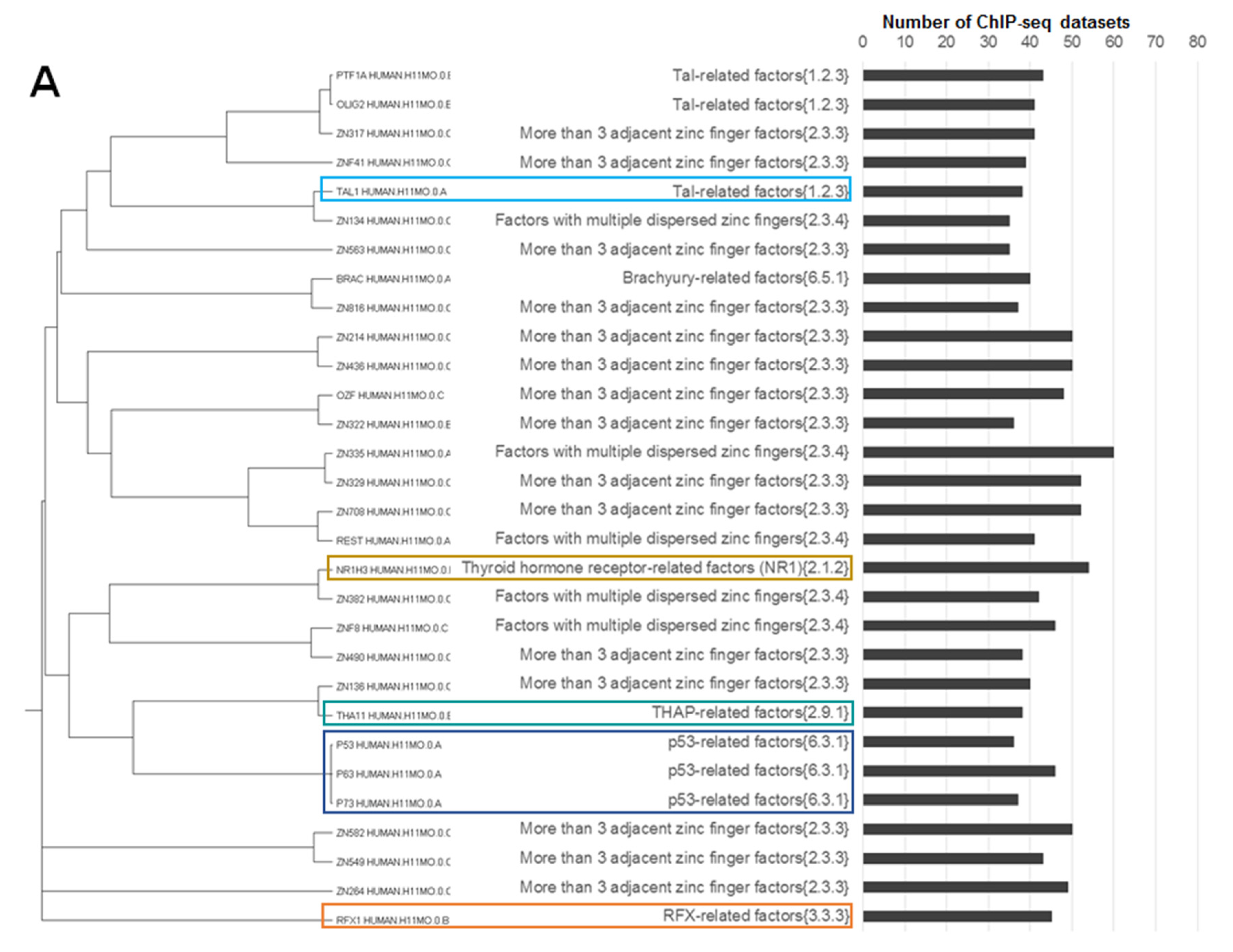
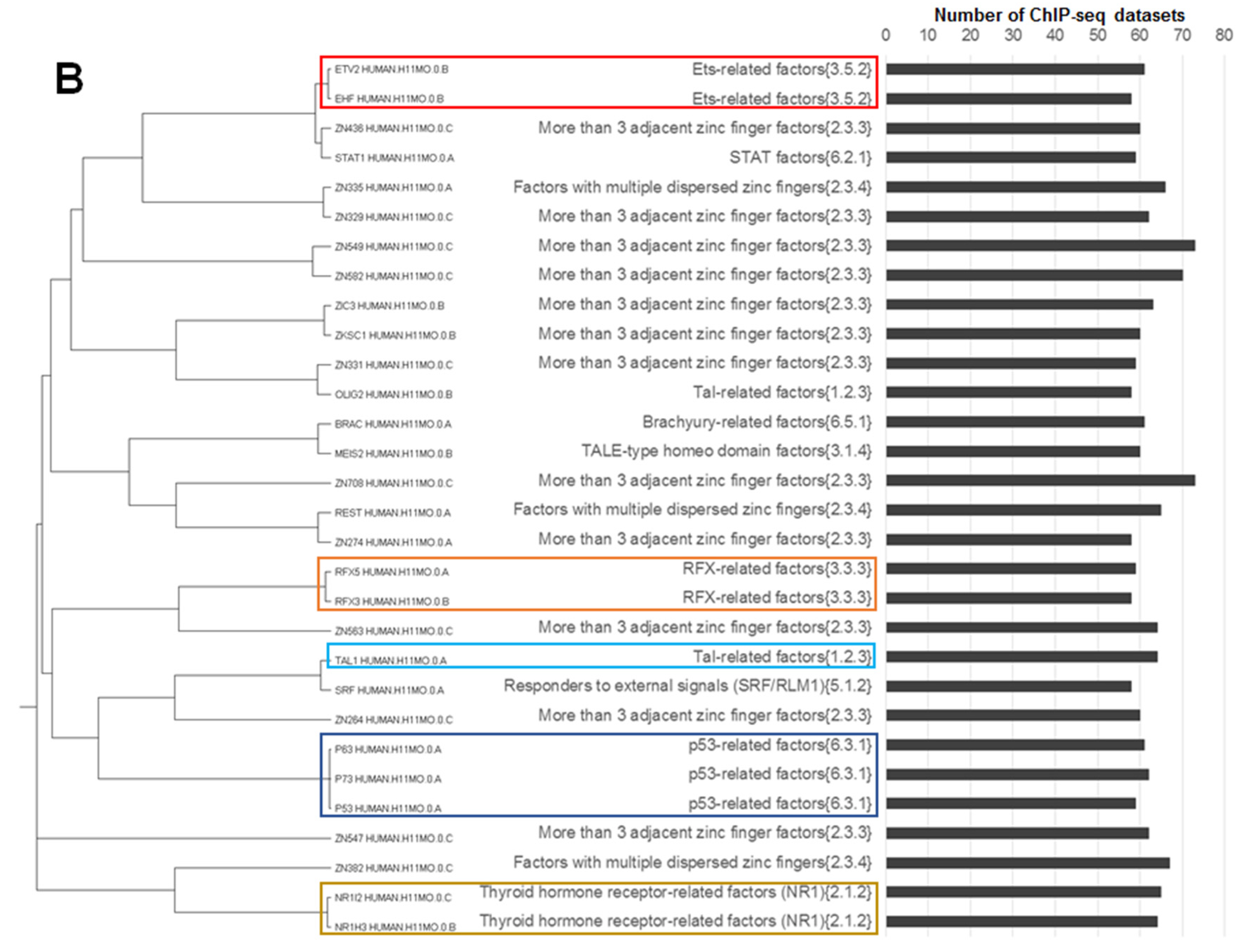
- Jun-like, out of a total 18 motifs of Jun-related {1.1.1}, Fos-related {1.1.2} and Maf-related {1.1.3} families 15 were homologous;
- ETS-like, out of a total 19 motifs of the ETS-related factors {3.5.2} family 14 were homologous;
- CTCF-like, two homologous motifs constituted the subfamily CTCF-like factors {2.3.3.50} of the largest family More than three adjacent zinc finger factors {2.3.3} consisting of 76 motifs;
- Two non-homologous motifs THA11_HUMAN.H11MO.0.B and THAP1_HUMAN.H11MO.0.C constituted the THAP-related factors {2.9.1} family.
- p53-like, all three motifs from family p53-related factors {6.3.1} were homologous;
- RFX-like, all four motifs from family RFX-related factors {3.3.3} were homologous;
- GATA-like, all five motifs from family GATA-type zinc fingers {2.2.1} were homologous, we added them to their homologue TAL1_HUMAN.H11MO.0.A from the Tal-related factors {1.2.3} family (the rest of the participants of this family were not homologous to GATA-like motifs);
- NR1H3-like motifs, four motifs from the thyroid hormone receptor-related factors (NR1) {2.1.2} family (NR1H3_HUMAN.H11MO.0.B, THA_HUMAN.H11MO.0.C, NR1I3_HUMAN.H11MO.0.C and NR1I2_HUMAN.H11MO.0.C) were homologous, this family consisted of 14 motifs; NR1H3-like motifs had close homologous motifs in families of steroid hormone receptors (NR3) {2.1.1} (e.g., ERR1_HUMAN.H11MO.0.A) and RXR-related receptors (NR2) {2.1.3} (e.g., COT2_HUMAN.H11MO.0.A);
- NFYA-like, all three motifs from the family heteromeric CCAAT-binding factors {4.2.1} were homologous.
5. Conclusions
- We proposed the approach for the computation of the significance of co-occurrence of asymmetric CEs anchor–partner with one of the participant motifs more conservative than another one, and for asymmetry within pairs of co-occurred motifs;
- We applied our approach for motifs of partner TFs from various families over-represented near motifs of anchor TFs in ChIP-seq data;
- We demonstrated that for partner motifs of almost all families of TFs only for overlapping anchor–partner pairs but not for pairs with a spacer, pairs with a higher conservation of partner motifs were significantly more abundant than those with higher conservation of anchor motifs. This observation explained a substantial portion of ChIP-seq data lacking conserved anchor motifs;
- We found that the asymmetric CEs toward partner motifs were the most reliable for partner motifs of TFs from ETS family. Hence, motifs of TFs from the ETS family tended to mediate the interaction of anchor TFs with genomic DNA.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BS | Binding Site |
| CE | Composite Element |
| DBD | DNA Binding Domain |
| FPR | False Positive Rate |
| ETS | E26 Transformation Specific |
| IDR | Intrinsically Disordered Region |
| MCOT | Motifs Co-Occurrence Tool |
| PWM | Position Weight Matrix |
| TF | Transcription Factor |
| TFBS | Transcription Factor Binding Site |
References
- Morgunova, E.; Taipale, J. Structural perspective of cooperative transcription factor binding. Curr. Opin. Struct. Biol. 2017, 47, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Reiter, F.; Wienerroither, S.; Stark, A. Combinatorial function of transcription factors and cofactors. Curr. Opin. Genet. Dev. 2017, 43, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Mayran, A.; Drouin, J. Pioneer transcription factors shape the epigenetic landscape. J. Biol. Chem. 2018, 293, 13795–13804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, X.; Verhage, L.; Hugouvieux, V.; Zubieta, C. Pioneer factors in animals and plants-colonizing chromatin for gene regulation. Molecules 2018, 23, 1914. [Google Scholar] [CrossRef] [Green Version]
- Zaret, K.S.; Carroll, J.S. Pioneer transcription factors: Establishing competence for gene expression. Genes Dev. 2011, 25, 2227–2241. [Google Scholar] [CrossRef] [Green Version]
- Nagy, G.; Nagy, L. Motif grammar: The basis of the language of gene expression. Comput. Struct. Biotechnol. [CrossRef]
- Lloyd, S.M.; Bao, X. Pinpointing the genomic localizations of chromatin-associated proteins: The yesterday, today, and tomorrow of ChIP-seq. Curr. Protoc. Cell Biol. 2019, 84, e89. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010, 38, 576–589. [Google Scholar] [CrossRef] [Green Version]
- Whitington, T.; Frith, M.C.; Johnson, J.; Bailey, T.L. Inferring transcription factor complexes from ChIP-seq data. Nucleic Acids Res. 2011, 39, 98. [Google Scholar] [CrossRef] [Green Version]
- Jankowski, A.; Prabhakar, S.; Tiuryn, J. TACO: A general-purpose tool for predicting cell-type-specific transcription factor dimers. BMC Genom. 2014, 15, 208. [Google Scholar] [CrossRef] [Green Version]
- Levitsky, V.; Zemlyanskaya, E.; Oshchepkov, D.; Podkolodnaya, O.; Ignatieva, E.; Grosse, I.; Mironova, V.; Merkulova, T. A single ChIP-seq dataset is sufficient for comprehensive analysis of motifs co-occurrence with MCOT package. Nucleic Acids Res. 2019, 47, e139. [Google Scholar] [CrossRef] [PubMed]
- Worsley Hunt, R.; Wasserman, W.W. Non-targeted transcription factors motifs are a systemic component of ChIP-seq datasets. Genome Biol. 2014, 15, 412. [Google Scholar] [CrossRef] [PubMed]
- Wingender, E.; Schoeps, T.; Haubrock, M.; Krull, M.; Dönitz, J. TFClass: Expanding the classification of human transcription factors to their mammalian orthologs. Nucleic Acids Res. 2018, 46, D343–D347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulakovskiy, I.V.; Vorontsov, I.E.; Yevshin, I.S.; Sharipov, R.N.; Fedorova, A.D.; Rumynskiy, E.I.; Medvedeva, Y.A.; Magana-Mora, A.; Bajic, V.B.; Papatsenko, D.A.; et al. HOCOMOCO: Expansion and enhancement of the collection of transcription factor binding sites models. Nucleic Acids Res. 2018, 46, D252–D259. [Google Scholar] [CrossRef]
- Wederell, E.D.; Bilenky, M.; Cullum, R.; Thiessen, N.; Dagpinar, M.; Delaney, A.; Varhol, R.; Zhao, Y.; Zeng, T.; Bernier, B.; et al. Global analysis of in vivo Foxa2-binding sites in mouse adult liver using massively parallel sequencing. Nucleic Acids Res. 2008, 36, 4549–4564. [Google Scholar] [CrossRef] [Green Version]
- Wallerman, O.; Motallebipour, M.; Enroth, S.; Patra, K.; Bysani, M.S.; Komorowski, J.; Wadelius, C. Molecular interactions between HNF4a, FOXA2 and GABP identified at regulatory DNA elements through ChIP-sequencing. Nucleic Acids Res. 2009, 37, 7498–7508. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Garcia-Bassets, I.; Benner, C.; Li, W.; Su, X.; Zhou, Y.; Qiu, J.; Liu, W.; Kaikkonen, M.U.; Ohgi, K.A.; et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 2011, 474, 390–394. [Google Scholar] [CrossRef] [Green Version]
- Tsankov, A.M.; Gu, H.; Akopian, V.; Ziller, M.J.; Donaghey, J.; Amit, I.; Gnirke, A.; Meissner, A. Transcription factor binding dynamics during human ES cell differentiation. Nature 2015, 518, 344–349. [Google Scholar] [CrossRef] [Green Version]
- Gheorghe, M.; Sandve, G.K.; Khan, A.; Chèneby, J.; Ballester, B.; Mathelier, A. A map of direct TF-DNA interactions in the human genome. Nucleic Acids Res. 2019, 47, e21. [Google Scholar] [CrossRef] [Green Version]
- Levitsky, V.G.; Kulakovskiy, I.V.; Ershov, N.I.; Oshchepkov, D.Y.; Makeev, V.J.; Hodgman, T.C.; Merkulova, T.I. Application of experimentally verified transcription factor binding sites models for computational analysis of ChIP-Seq data. BMC Genom. 2014, 15, 80. [Google Scholar] [CrossRef] [Green Version]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human transcription factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, G.; Vorontsov, I.; Penzar, D.; Groux, R.; Fornes, O.; Nikolaeva, D.D.; Ballester, B.; Grau, J.; Grosse, I.; Makeev, V.; et al. Insights gained from a comprehensive all-against-all transcription factor binding motif benchmarking study. Genome Biol. 2020, 21, 114. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.H.; Badis, G.; Berger, M.F.; Kivioja, T.; Palin, K.; Enge, M.; Bonke, M.; Jolma, A.; Varjosalo, M.; Gehrke, A.R.; et al. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J. 2010, 29, 2147–2160. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; He, Z.Y.; You, P.; Han, Q.W.; Xiang, D.; Chen, F.; Wang, M.J.; Liu, C.C.; Lin, X.W.; Borjigin, U.; et al. Reprogramming fibroblasts into bipotential hepatic stem cells by defined factors. Cell Stem Cell 2013, 13, 328–340. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Mahony, S.; Gifford, D.K. High resolution genome wide binding event finding and motif discovery reveals transcription factor spatial binding constraints. PLoS Comput. Biol. 2012, 8, e1002638. [Google Scholar] [CrossRef] [Green Version]
- Kazemian, M.; Pham, H.; Wolfe, S.A.; Brodsky, M.H.; Sinha, S. Widespread evidence of cooperative DNA binding by transcription factors in Drosophila development. Nucleic Acids Res. 2013, 41, 8237–8352. [Google Scholar] [CrossRef]
- Jankowski, A.; Szczurek, E.; Jauch, R.; Tiuryn, J.; Prabhakar, S. Comprehensive prediction in 78 human cell lines reveals rigidity and compactness of transcription factor dimers. Genome Res. 2013, 23, 1307–1318. [Google Scholar] [CrossRef]
- Richardson, L.; Venkataraman, S.; Stevenson, P.; Yang, Y.; Burton, N.; Rao, J.; Fisher, M.; Baldock, R.A.; Davidson, D.R.; Christiansen, J.H. EMAGE mouse embryo spatial gene expression database: 2010 update. Nucleic Acids Res. 2010, 38, D703–D709. [Google Scholar] [CrossRef] [Green Version]
- Dahl, R.; Ramirez-Bergeron, D.L.; Rao, S.; Simon, M.C. Spi-B can functionally replace PU.1 in myeloid but not lymphoid development. EMBO J. 2002, 21, 2220–2230. [Google Scholar] [CrossRef] [Green Version]
- DeKoter, R.P.; Lee, H.J.; Singh, H. PU.1 regulates expression of the interleukin-7 receptor in lymphoid progenitors. Immunity 2002, 16, 297–309. [Google Scholar] [CrossRef] [Green Version]
- Verger, A.; Duterque-Coquillaud, M. When Ets transcription factors meet their partners. BioEssays 2002, 24, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Hollenhorst, P.C.; McIntosh, L.P.; Graves, B.J. Genomic and biochemical insights into the specificity of ETS transcription factors. Annu. Rev. Biochem. 2011, 80, 437–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, C.D.; Newman, J.A.; Gileadi, O. Recent advances in the structural molecular biology of Ets transcription factors: Interactions, interfaces and inhibition. Biochem. Soc. Trans. 2014, 42, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Coyne, H.J., III; De, S.; Okon, M.; Green, S.M.; Bhachech, N.; Graves, B.J.; McIntosh, L.P. Autoinhibition of ETV6 (TEL) DNA binding: Appended helices sterically block the ETS domain. J. Mol. Biol. 2012, 421, 67–84. [Google Scholar] [CrossRef] [Green Version]
- Regan, M.C.; Horanyi, P.S.; Pryor, E.E., Jr.; Sarver, J.L.; Cafiso, D.S.; Bushweller, J.H. Structural and dynamic studies of the transcription factor ERG reveal DNA binding is allosterically autoinhibited. Proc. Natl. Acad. Sci. USA 2013, 110, 13374–13379. [Google Scholar] [CrossRef] [Green Version]
- Newman, J.A.; Cooper, C.D.; Aitkenhead, H.; Gileadi, O. Structural insights into the autoregulation and cooperativity of the human transcription factor ETS-2. J. Biol. Chem. 2015, 290, 8539–8549. [Google Scholar] [CrossRef] [Green Version]
- Currie, S.L.; Lau, D.; Doane, J.J.; Whitby, F.G.; Okon, M.; McIntosh, L.P.; Graves, B.J. Structured and disordered regions cooperatively mediate DNA-binding autoinhibition of ETS factors ETV1, ETV4 and ETV5. Nucleic Acids Res. 2017, 45, 2223–2241. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.M.; Donaldson, L.W.; Pufall, M.A.; Kang, H.S.; Pot, I.; Graves, B.J.; McIntosh, L.P. The structural and dynamic basis of Ets-1 DNA binding autoinhibition. J. Biol. Chem. 2005, 280, 7088–7099. [Google Scholar] [CrossRef] [Green Version]
- Pufall, M.A.; Lee, G.M.; Nelson, M.L.; Kang, H.S.; Velyvis, A.; Kay, L.E.; McIntosh, L.P.; Graves, B.J. Variable control of Ets-1 DNA binding by multiple phosphates in an unstructured region. Science 2005, 309, 142–145. [Google Scholar] [CrossRef] [Green Version]
- Green, S.M.; Coyne, H.J., III; McIntosh, L.P.; Graves, B.J. DNA binding by the ETS protein TEL (ETV6) is regulated by autoinhibition and self-association. J. Biol. Chem. 2010, 285, 18496–18504. [Google Scholar] [CrossRef] [Green Version]
- De, S.; Chan, A.C.; Coyne, H.J., III; Bhachech, N.; Hermsdorf, U.; Okon, M.; Murphy, M.E.; Graves, B.J.; McIntosh, L.P. Steric mechanism of auto-inhibitory regulation of specific and non-specific DNA binding by the ETS transcriptional repressor ETV6. J. Mol. Biol. 2014, 426, 1390–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Borrajero, C.; Lin, C.S.; Okon, M.; Scheu, K.; Graves, B.J.; Murphy, M.; McIntosh, L.P. The biophysical basis for phosphorylation-enhanced DNA-binding autoinhibition of the ETS1 transcription factor. J. Mol. Biol. 2019, 431, 593–614. [Google Scholar] [CrossRef] [PubMed]
- Xhani, S.; Lee, S.; Kim, H.M.; Wang, S.; Esaki, S.; Ha, V.; Khanezarrin, M.; Fernandez, G.L.; Albrecht, A.V.; Aramini, J.M.; et al. Intrinsic disorder controls two functionally distinct dimers of the master transcription factor PU.1. Sci. Adv. 2020, 6, eaay3178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Perumal, N.B.; Oldfield, C.J.; Su, E.W.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in transcription factors. Biochemistry 2006, 45, 6873–6888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Bulyk, M.L.; Hartemink, A.J. Intrinsic disorder within and flanking the DNA-binding domains of human transcription factors. In Proceedings of the Pacific Symposium on Biocomputing 2012, Kohala Coast, HI, USA, 3–7 January 2012; Altman, R.B., Dunker, A.K., Hunter, L., Murray, T., Klein, T.E., Eds.; World Scientific: Singapore, 2011; pp. 104–115. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, M.; Pan, G.; Nord, H.; Wallerman, O.; Wallén Arzt, E.; Berggren, O.; Elvers, I.; Eloranta, M.L.; Rönnblom, L.; Lindblad Toh, K.; et al. Allele-specific transcription factor binding to common and rare variants associated with disease and gene expression. Hum. Genet. 2016, 135, 485–497. [Google Scholar] [CrossRef]
- Cavalli, M.; Baltzer, N.; Pan, G.; Bárcenas Walls, J.R.; Smolinska Garbulowska, K.; Kumar, C.; Skrtic, S.; Komorowski, J.; Wadelius, C. Studies of liver tissue identify functional gene regulatory elements associated to gene expression, type 2 diabetes, and other metabolic diseases. Hum. Genom. 2019, 13, 20. [Google Scholar] [CrossRef]
- Li, S.; Li, Y.; Li, X.; Liu, J.; Huo, Y.; Wang, J.; Liu, Z.; Li, M.; Luo, X.-J. Regulatory mechanisms of major depressive disorder risk variants. Mol. Psychiatry. [CrossRef]
- Deplancke, B.; Alpern, D.; Gardeux, V. The Genetics of Transcription Factor DNA Binding Variation. Cell 2016, 166, 538–554. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Stamatoyannopolous, J.A.; Bailey, T.L.; Noble, W.S. Quantifying similarity between motifs. Genome Biol. 2007, 8, R24. [Google Scholar] [CrossRef] [Green Version]
- MCOT. Available online: https://gitlab.sysbio.cytogen.ru/academiq/mcot-kernel (accessed on 20 August 2020).
- HOCOMOCO. Available online: https://hocomoco11.autosome.ru/ (accessed on 20 August 2020).
- Classification of Transcription Factors in Mammalia. Available online: http://tfclass.bioinf.med.uni-goettingen.de/ (accessed on 20 August 2020).
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- MEGA. Available online: https://www.megasoftware.net/ (accessed on 20 August 2020).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Categories of Sequences | Count of Sequences | |
|---|---|---|
| With CE | Without CE | |
| Foreground | ObsCE+ | ObsCE- |
| Background | ExpCE+ | ExpCE- |
| Categories of CEs | Count of CEs with More Conserved | |
|---|---|---|
| Anchor Motif | Partner Motif | |
| Foreground | ObsCE,Anchor | ObsCE,Partner |
| Background | ExpCE,Anchor | ExpCE,Partner |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levitsky, V.; Oshchepkov, D.; Zemlyanskaya, E.; Merkulova, T. Asymmetric Conservation within Pairs of Co-Occurred Motifs Mediates Weak Direct Binding of Transcription Factors in ChIP-Seq Data. Int. J. Mol. Sci. 2020, 21, 6023. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176023
Levitsky V, Oshchepkov D, Zemlyanskaya E, Merkulova T. Asymmetric Conservation within Pairs of Co-Occurred Motifs Mediates Weak Direct Binding of Transcription Factors in ChIP-Seq Data. International Journal of Molecular Sciences. 2020; 21(17):6023. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176023
Chicago/Turabian StyleLevitsky, Victor, Dmitry Oshchepkov, Elena Zemlyanskaya, and Tatyana Merkulova. 2020. "Asymmetric Conservation within Pairs of Co-Occurred Motifs Mediates Weak Direct Binding of Transcription Factors in ChIP-Seq Data" International Journal of Molecular Sciences 21, no. 17: 6023. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176023