The Structure of Clostridioides difficile SecA2 ATPase Exposes Regions Responsible for Differential Target Recognition of the SecA1 and SecA2-Dependent Systems


Abstract
:1. Introduction
2. Results
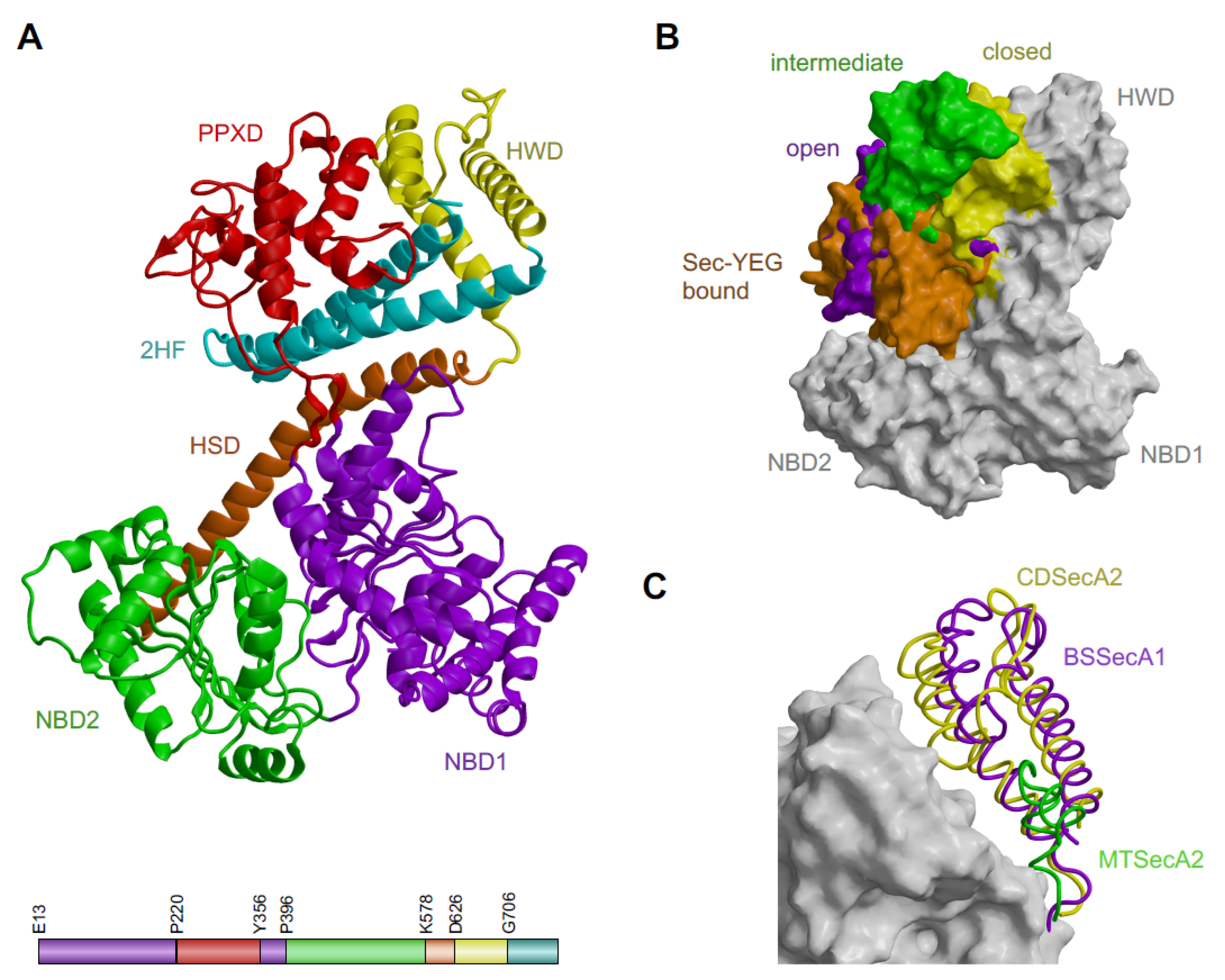
2.1. Except for the C-Terminal Tail Absence, the Structure of CDSecA2 Is More Similar to Its SecA(1) Homologues Than to M. tuberculosis SecA2
2.2. CDSecA2 Is an Active ATPase In Vitro
2.3. The Structure of CDSecA2 in Complex with ATP-γ-S Shows Conserved Regions for Nucleotide Binding
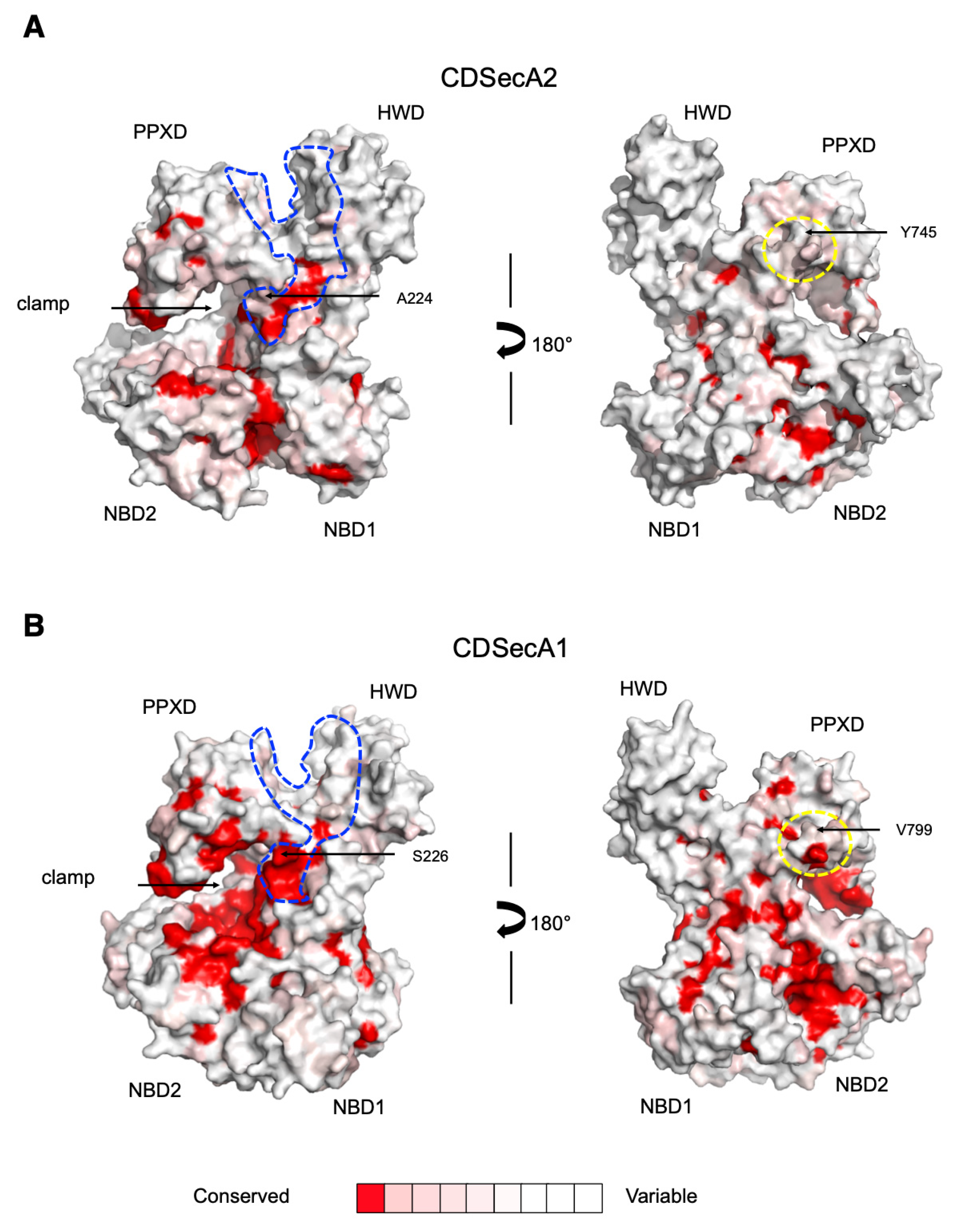
2.4. Movement of CDSecA2 Domains Affects the Conformation of the Target Protein Binding Site
3. Discussion
4. Materials and Methods
4.1. CDSecA2 Protein Expression and Purification
4.2. Crystallization and Structure Determination of CDSecA2
4.3. ATPase Activity Assay
4.4. Prediction of Evolutionary Conservation of SecA2 Proteins
4.5. Modelling of Dimer in Open and YEG-Bound Conformation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SecA2 | protein translocase subunit SecA2 |
| CDSecA2 | C. difficile SecA2 |
| CDSecA1 | C. difficile SecA1 |
| Cwps | cell wall proteins |
| PPXD | preprotein cross-linking domain |
| NBD | nucleotide binding domain |
| HSD | helical scaffold domain |
| HWD | helical wing domain |
| CTT | C-terminal tail |
References
- Feltcher, M.E.; Braunstein, M. Emerging themes in SecA2-mediated protein export. Nat. Rev. Microbiol. 2012, 10, 779–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensing, B.A.; Seepersaud, R.; Yen, Y.T.; Sullam, P.M. Selective transport by SecA2: An expanding family of customized motor proteins. Biochim. Biophys. Acta-Mol. Cell Res. 2014, 1843, 1674–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsirigotaki, A.; Chatzi, A.; Koukaki, M.; De Geyter, J.; Portaliou, A.G.; Orfanoudaki, G.; Sardis, M.F.; Trelle, M.B.; Jørgensen, T.J.; Karamanou, S.; et al. Long-Lived folding intermediates predominate the targeting-competent secretome. Structure 2018, 26, 695–707.e5. [Google Scholar] [CrossRef] [Green Version]
- De Geyter, J.; Tsirigotaki, A.; Orfanoudaki, G.; Zorzini, V.; Economou, A.; Karamanou, S. Protein folding in the cell envelope of Escherichia coli. Nat. Microbiol. 2016, 1, 16107. [Google Scholar] [CrossRef] [PubMed]
- Von Heijne, G. The signal peptide. J. Membr. Biol. 1990, 115, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Auclair, S.M.; Moses, J.P.; Musial-Siwek, M.; Kendall, D.A.; Oliver, D.B.; Mukerji, I. Mapping of the signal peptide-binding domain of Escherichia coli SecA using förster resonance energy Transfer. Biochemistry 2010, 49, 782–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelis, I.; Bonvin, A.M.J.J.; Keramisanou, D.; Koukaki, M.; Gouridis, G.; Karamanou, S.; Economou, A.; Kalodimos, C.G. Structural Basis for Signal-sequence recognition by the translocase motor SecA as determined by NMR. Cell 2007, 131, 756–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grady, L.M.; Michtavy, J.; Oliver, D. Characterization of the Escherichia coli SecA signal peptide-binding site. J. Bacteriol. 2011, 194, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Musial-Siwek, M.; Rusch, S.L.; Kendall, D.A. Probing the affinity of SecA for signal peptide in different environments. Biochemistry 2005, 44, 13987–13996. [Google Scholar] [CrossRef] [Green Version]
- Kourtz, L.; Oliver, D. Tyr-326 plays a critical role in controlling SecA-preprotein interaction. Mol. Microbiol. 2000, 37, 1342–1356. [Google Scholar] [CrossRef]
- Cranford-Smith, T.; Huber, D. The way is the goal: How SecA transports proteins across the cytoplasmic membrane in bacteria. FEMS Microbiol. Lett. 2018, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagan, R.P.; Fairweather, N.F. Clostridium difficile has two parallel and essential sec secretion systems. J. Biol. Chem. 2011, 286, 27483–27493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspers, M.; Freudl, R. Corynebacterium glutamicum possesses two secA homologous genes that are essential for viability. Arch. Microbiol. 2008, 189, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Mau, S.-M.; Oh, S.-Y.; Kern, V.J.; Missiakas, M.M.; Schneewind, O. Secretion genes as determinants of Bacillus anthracis chain length. J. Bacteriol. 2012, 194, 3841–3850. [Google Scholar] [CrossRef] [Green Version]
- Braunstein, M.; Espinosa, B.J.; Chan, J.; Belisle, J.T.; Jacobs, W.R. SecA2 functions in the secretion of superoxide dismutase A and in the virulence of Mycobacterium tuberculosis. Mol. Microbiol. 2003, 48, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, H.S.; Wolschendorf, M.; Abshire, M.; Niederweis, M.E.; Braunstein, M. Identification of two Mycobacterium smegmatis lipoproteins exported by a SecA2-dependent pathway. J. Bacteriol. 2007, 189, 5090–5100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenz, L.L.; Mohammadi, S.; Geissler, A.; Portnoy, D.A. SecA2-dependent secretion of autolytic enzymes promotes Listeria monocytogenes pathogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 12432–12437. [Google Scholar] [CrossRef] [Green Version]
- Hunt, J.F.; Weinkauf, S.; Henry, L.; Fak, J.J.; McNicholas, P.; Oliver, D.B.; Deisenhofer, J. Nucleotide control of interdomain interactions in the conformational reaction cycle of SecA. Science 2002, 297, 2018–2026. [Google Scholar] [CrossRef]
- Zimmer, J.; Rapoport, T.A. Conformational flexibility and peptide interaction of the translocation ATPase SecA. J. Mol. Biol. 2009, 394, 606–612. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Arockiasamy, A.; Ronning, D.R.; Savva, C.G.; Holzenburg, A.; Braunstein, M.; Jacobs, W.R., Jr.; Sacchettini, J.C. Crystal structure of Mycobacterium tuberculosis SecA, a preprotein translocating ATPase. Proc. Natl. Acad. Sci. USA 2003, 100, 2243–2248. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Kim, J.; Na Im, H.; Yoon, J.Y.; An, O.R.; Yoon, H.J.; Kim, J.Y.; Min, H.K.; Kim, S.-J.; Lee, J.Y.; et al. Structural basis for the inhibition of Mycobacterium tuberculosis l,d-transpeptidase by meropenem, a drug effective against extensively drug-resistant strains. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 420–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmer, J.; Li, W.; Rapoport, T.A. A Novel dimer interface and conformational changes revealed by an X-ray structure of B. subtilis SecA. J. Mol. Biol. 2006, 364, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Swanson, S.; Ioerger, T.R.; Rigel, N.W.; Miller, B.K.; Braunstein, M.; Sacchettini, J.C. Structural similarities and differences between two functionally distinct SecA proteins, Mycobacterium tuberculosis SecA1 and SecA2. J. Bacteriol. 2016, 198, 720–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmer, J.; Nam, Y.; Rapoport, T.A. Structure of a complex of the ATPase SecA and the protein-translocation channel. Nature 2008, 455, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Godzik, A. FATCAT: A web server for flexible structure comparison and structure similarity searching. Nucleic Acids Res. 2004, 32, W582–W585. [Google Scholar] [CrossRef] [Green Version]
- Osborne, A.R.; Clemons, W.M.; Rapoport, T.A. A large conformational change of the translocation ATPase SecA. Proc. Natl. Acad. Sci. USA 2004, 101, 10937–10942. [Google Scholar] [CrossRef] [Green Version]
- Papanikolau, Y.; Papadovasilaki, M.; Ravelli, R.B.G.; McCarthy, A.A.; Cusack, S.; Economou, A.; Petratos, K. Structure of dimeric SecA, the Escherichia coli preprotein translocase motor. J. Mol. Biol. 2007, 366, 1545–1557. [Google Scholar] [CrossRef]
- Wang, S.; Jomaa, A.; Jaskolowski, M.; Yang, C.-I.; Ban, N.; Shan, S.-O. The molecular mechanism of cotranslational membrane protein recognition and targeting by SecA. Nat. Struct. Mol. Biol. 2019, 26, 919–929. [Google Scholar] [CrossRef]
- Li, L.; Park, E.; Ling, J.; Ingram, J.; Ploegh, H.; Rapoport, T.A. Crystal structure of a substrate-engaged SecY protein-translocation channel. Nature 2016, 531, 395–399. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Wu, X.; Sun, D.; Park, E.; Catipovic, M.; Rapoport, T.A.; Gao, N.; Li, L. Structure of the substrate-engaged SecA-SecY protein translocation machine. Nat. Commun. 2019, 10, 2872. [Google Scholar] [CrossRef]
- Or, E.; Boyd, D.; Gon, S.; Beckwith, J.; Rapoport, T. The bacterial ATPase SecA functions as a monomer in protein translocation. J. Biol. Chem. 2004, 280, 9097–9105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabowicz, M.; Yeh, J.; Silhavy, T.J. Dominant negative LptE mutation that supports a role for LptE as a plug in the LptD barrel. J. Bacteriol. 2013, 195, 1327–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, D.; Rajagopalan, N.; Preissler, S.; Rocco, M.A.; Merz, F.; Krämer, G.; Bukau, B. SecA Interacts with ribosomes in order to facilitate posttranslational translocation in bacteria. Mol. Cell 2011, 41, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Huber, D.; Jamshad, M.; Hanmer, R.; Schibich, D.; Döring, K.; Marcomini, I.; Krämer, G.; Bukau, B. SecA cotranslationally interacts with nascent substrate proteins in vivo. J. Bacteriol. 2016, 199, e00622-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamshad, M.; Knowles, T.J.; Whit, S.A.; Ward, D.G.; Mohammed, F.; Rahman, K.F.; Wynne, M.; Hughes, G.W.; Krämer, G.; Bukau, B.; et al. The C-terminal tail of the bacterial translocation ATPase SecA modulates its activity. eLife 2019, 8, e48385. [Google Scholar] [CrossRef]
- Feltcher, M.E.; Gibbons, H.S.; Ligon, L.S.; Braunstein, M. Protein export by the mycobacterial SecA2 system is determined by the preprotein mature domain. J. Bacteriol. 2013, 195, 672–681. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.M.; D’lima, N.G.; Rigel, N.W.; Gibbons, H.S.; McCann, J.R.; Braunstein, M.; Teschke, C.M. ATPase activity of Mycobactenum tuberculosis SecA1 and SecA2 proteins and its importance for SecA2 function in macrophages. J. Bacteriol. 2008, 190, 4880–4887. [Google Scholar] [CrossRef] [Green Version]
- Pražnikar, J.; Turk, D. Free kick instead of cross-validation in maximum-likelihood refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 3124–3134. [Google Scholar] [CrossRef] [Green Version]
- Rigel, N.W.; Gibbons, H.S.; McCann, J.R.; McDonough, J.A.; Kurtz, H.S.; Braunstein, M. The Accessory SecA2 system of mycobacteria requires ATP binding and the canonical SecA1. J. Biol. Chem. 2009, 284, 9927–9936. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, C.; Oliver, D. Two distinct ATP-binding domains are needed to promote protein export by Escherichia coli SecA ATPase. Mol. Microbiol. 1993, 10, 483–497. [Google Scholar] [CrossRef]
- Robson, A.; Booth, A.E.; Gold, V.A.; Clarke, A.R.; Collinson, I. A large conformational change couples the ATP binding site of SecA to the SecY protein channel. J. Mol. Biol. 2007, 374, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Tokuda, H.; Nishiyama, K.-I. Multiple SecA molecules drive protein translocation across a single translocon with SecG inversion. J. Biol. Chem. 2011, 287, 455–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zito, C.R.; Antony, E.; Hunt, J.F.; Oliver, D.B.; Hingorani, M.M. Role of a conserved glutamate residue in the Escherichia coli SecA ATPase mechanism. J. Biol. Chem. 2005, 280, 14611–14619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, B.W.; Rapoport, T.A. Mapping polypeptide interactions of the SecA ATPase during translocation. Proc. Natl. Acad. Sci. USA 2009, 106, 20800–20805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erlandson, K.J.; Miller, S.B.M.; Nam, Y.; Osborne, A.R.; Zimmer, J.; Rapoport, T.A. A role for the two-helix finger of the SecA ATPase in protein translocation. Nature 2008, 455, 984–987. [Google Scholar] [CrossRef] [Green Version]
- Allen, W.J.; Corey, R.A.; Oatley, P.; Sessions, R.B.; Baldwin, S.A.; Radford, S.E.; Tuma, R.; Collinson, I. Two-way communication between SecY and SecA suggests a Brownian ratchet mechanism for protein translocation. eLife 2016, 5, e15598. [Google Scholar] [CrossRef]
- Catipovic, M.; Bauer, B.W.; Loparo, J.J.; Rapoport, T.A. Protein translocation by the SecA ATPase occurs by a power-stroke mechanism. EMBO J. 2019, 38, e10114. [Google Scholar] [CrossRef]
- Bhanu, M.K.; Zhao, P.; Kendall, D.A. Mapping of the SecA signal peptide binding site and dimeric interface by using the substituted cysteine accessibility method. J. Bacteriol. 2013, 195, 4709–4715. [Google Scholar] [CrossRef] [Green Version]
- De Keyzer, J.; Van Der Sluis, E.O.; Spelbrink, R.E.J.; Nijstad, N.; De Kruijff, B.; Nouwen, N.; Van Der Does, C.; Driessen, A.J.M. Covalently dimerized SecA is functional in protein translocation. J. Biol. Chem. 2005, 280, 35255–35260. [Google Scholar] [CrossRef] [Green Version]
- Jilaveanu, L.B.; Zito, C.R.; Oliver, D. Dimeric SecA is essential for protein translocation. Proc. Natl. Acad. Sci. USA 2005, 102, 7511–7516. [Google Scholar] [CrossRef] [Green Version]
- Jilaveanu, L.B.; Oliver, D. SecA dimer cross-linked at its subunit interface is functional for protein translocation. J. Bacteriol. 2006, 188, 335–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusters, I.; Bogaart, G.V.D.; Kedrov, A.; Krasnikov, V.; Fulyani, F.; Poolman, B.; Driessen, A.J.M. Quaternary structure of SecA in solution and bound to SecYEG probed at the single molecule level. Structure 2011, 19, 430–439. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, T.; Lindenthal, C.; Oliver, D. SecA functions in vivo as a discrete anti-parallel dimer to promote protein transport. Mol. Microbiol. 2017, 103, 439–451. [Google Scholar] [CrossRef]
- Ding, H.; Hunt, J.F.; Mukerji, I.; Oliver, D. Bacillus subtilis SecA ATPase exists as an antiparallel dimer in solution. Biochemistry 2003, 42, 8729–8738. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Bauer, B.W.; Rapoport, T.A.; Gumbart, J.C. Conformational changes of the clamp of the protein translocation ATPase SecA. J. Mol. Biol. 2015, 427, 2348–2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; Beer, T.A.P.D.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Vassylyev, D.G.; Mori, H.; Vassylyeva, M.N.; Tsukazaki, T.; Kimura, Y.; Tahirov, T.H.; Ito, K. Crystal Structure of the Translocation ATPase SecA from Thermus thermophilus Reveals a Parallel, Head-to-Head Dimer. J. Mol. Biol. 2006, 364, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Celniker, G.; Nimrod, G.; Ashkenazy, H.; Glaser, F.; Martz, E.; Mayrose, V.B.; Pupko, T.; Ben-Tal, N. ConSurf: Using evolutionary data to raise testable hypotheses about protein function. Isr. J. Chem. 2013, 53, 199–206. [Google Scholar] [CrossRef]
- Das, S.; Oliver, D. Mapping of the SecA·SecY and SecA·SecG interfaces by site-directed in vivo photocross-linking. J. Biol. Chem. 2011, 286, 12371–12380. [Google Scholar] [CrossRef] [Green Version]
- Stols, L.; Gu, M.; Dieckman, L.; Raffen, R.; Collart, F.R.; Donnelly, M.I. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr. Purif. 2002, 25, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Studier, F.W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turk, D. MAIN software for density averaging, model building, structure refinement and validation. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1342–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrejašič, M.; Pražnikar, J.; Turk, D. PURY: A database of geometric restraints of hetero compounds for refinement in complexes with macromolecular structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2008, 64, 1093–1109. [Google Scholar] [CrossRef]
- Merritt, E.A.; Bacon, D.J. Raster3D: Photorealistic molecular graphics. Methods Enzymol. 1997, 277, 505–524. [Google Scholar]
- Pei, J.; Kim, B.-H.; Grishin, N.V. PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008, 36, 2295–2300. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystallization and Data Collection | ||
|---|---|---|
| Crystal | CDSecA2 | CDSecA2 with ATP-γ-S |
| Unit cell | ||
| a, b, c (Å) α, β, γ (deg) | a = 84.986, b = 96.847, c = 114.739 α = β = γ = 90.0° | a = 82.940, b = 96.837, c = 114.680 α = β = γ = 90.0 |
| Space group | P21 21 21 (number 19) | P21 21 21 (number 19) |
| Molecules per au | 1 | 1 |
| Wavelength (Å) | 0.9184 | 1.0000 |
| Resolution range (Å) | 48.4–2.3 | 50–2.6 |
| No. of unique reflections | 42821 | 29038 |
| Completeness (last shell) (%) | 99.8 (98.8) | 99 (96.9) |
| Multiplicity | 7.0 | 6.32 |
| R meas (last shell) (%) | 22.0 (163.3) | 19.9 (171) |
| CC (1/2) (last shell) (%) | 99.8 (61.9) | 99.6 (80.8) |
| I/σ (last shell) | 8.0 (1.2) | 7.1 (1.33) |
| Refinement | ||
| PDB ID | 6SXH | 6T4H |
| Resolution range (Å) | 47.5–2.3 | 49.34–2.9 |
| No. of reflections in working set | 42717 | 21017 |
| No. of reflections in test set | 42717 | 21017 |
| R-work | 0.243 | 0.335 |
| R-kick | 0.272 | 0.380 |
| RMSD from ideal geometry | ||
| Bond length (Å) | 0.019 | 0.014 |
| Bond angles (°) | 2.07 | 1.78 |
| No. of atoms in au | 8578 | 7720 |
| Protein atoms | 7621 | 7635 |
| Water molecules | 315 | 42 |
| Mean B value (Å2) | 29.32 | 48.49 |
| Ramachandran plot statistics | ||
| Favored | 727 (94.8%) | 644 (83.7%) |
| Allowed | 38 (5.0%) | 88 (11.4%) |
| Outliers | 2 (0.3%) | 37 (4.8%) |
| PDB ID | Molecule-in Complex with | Species | Conformation | RMSD (Å) (Sequence Length/Identity (%)) |
|---|---|---|---|---|
| 6SXH * | SecA2 | C. difficile | Closed | |
| 6T4H | SecA2-ATP-γ-S | C. difficile | Closed | 0.85 (768/100) |
| 1NKT | SecA1-ADP-Mg | M. tuberculosis | Closed | 1.78 (621/46) |
| 1M6N * | SecA1 | B. subtilis | Closed | 2.31 (624/50) |
| 4UAQ * | SecA2 | M. tuberculosis | Intermediate | 3.03 (563/32) |
| 2IBM | SecA1-ADP | B. subtilis | Closed | 3.07 (625/49) |
| 3JV2 * | SecA1-peptide | B. subtilis | Open | 2.10 (621/50) |
| 6ITC | SecA1-SecYEG-peptide | B. subtilis | YEG-bound | 2.30 (624/50) |
| 5EUL | SecA1- SecY-peptide | B. subtilis | YEG-bound | 2.51 (572, 45) |
| 3DIN * | SecA1-SecYEG | T. maritima | YEG-bound | 3.13 (597/43) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lindič, N.; Loboda, J.; Usenik, A.; Vidmar, R.; Turk, D. The Structure of Clostridioides difficile SecA2 ATPase Exposes Regions Responsible for Differential Target Recognition of the SecA1 and SecA2-Dependent Systems. Int. J. Mol. Sci. 2020, 21, 6153. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176153
Lindič N, Loboda J, Usenik A, Vidmar R, Turk D. The Structure of Clostridioides difficile SecA2 ATPase Exposes Regions Responsible for Differential Target Recognition of the SecA1 and SecA2-Dependent Systems. International Journal of Molecular Sciences. 2020; 21(17):6153. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176153
Chicago/Turabian StyleLindič, Nataša, Jure Loboda, Aleksandra Usenik, Robert Vidmar, and Dušan Turk. 2020. "The Structure of Clostridioides difficile SecA2 ATPase Exposes Regions Responsible for Differential Target Recognition of the SecA1 and SecA2-Dependent Systems" International Journal of Molecular Sciences 21, no. 17: 6153. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176153