Insights on the Interplay between Cells Metabolism and Signaling: A Therapeutic Perspective in Pediatric Acute Leukemias

 ,
,  and
and
Abstract
:1. Introduction
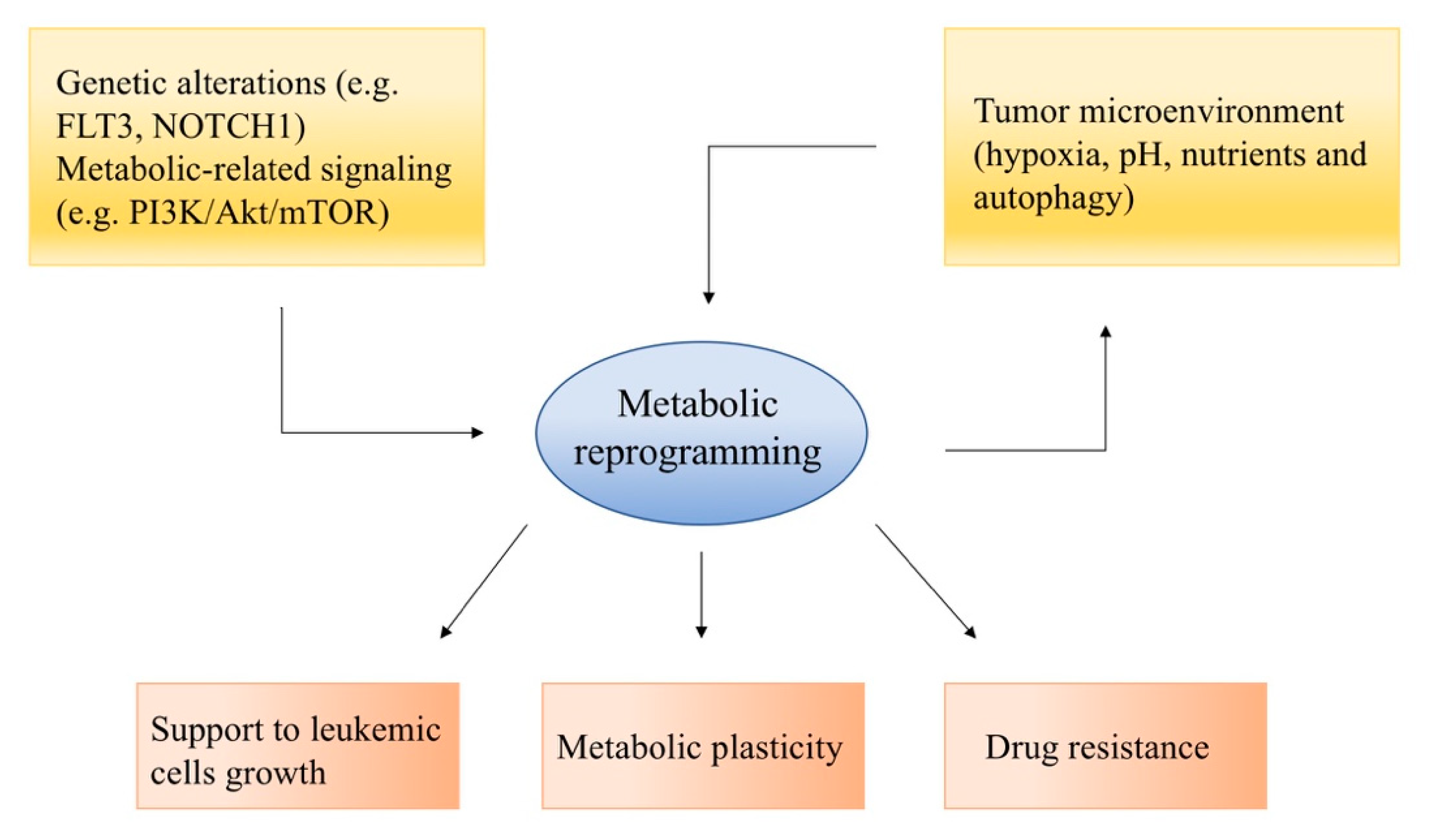
2. Metabolic Reprogramming in Acute Leukemias
3. Current Knowledge on Pediatric Acute Leukemia
3.1. Metabolic Hallmarks of Pediatric ALL
3.1.1. Treatment Efficacy Prediction
3.1.2. Metabolic Inhibitors
3.1.3. Metabolic-Related Signaling Pathways
3.2. Metabolic Hallmarks of Pediatric AML
3.2.1. From Cytogenetic Aberrations to Metabolic Response
3.2.2. Metabolic-Related Signaling Pathways
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALL | Acute Lymphoblastic Leukemia |
| AML | Acute Myeloid Leukemia |
| AMPK | AMP-activated protein kinase |
| ASNase | L-asparaginase |
| ASNS | Asparagine Synthetase |
| FAO | Fatty Acid Oxidation |
| FLT-3 | FMS-like Tyrosine Kinase 3 |
| GC | Glucocorticoids |
| HIF-1 | Hypoxia Inducible Factors |
| HSC | Hematopoietic Stem Cell |
| LIC | Leukemia-Initiating Cells |
| LKB1 | Liver Kinase B1 |
| LSC | Leukemic Stem Cells |
| mtDNA | mitochondrial DNA |
| mTOR | Mammalian Target Of Rapamycin |
| OXPHOS | Oxidative Phosphorylation |
| PDK1 | Phosphoinositide-dependent Kinase 1 |
| Ph+ | Philadelphia Chromosome-positive |
| PKB | Protein Kinase B |
| PH | Pleckstrin Homology |
| RTK | Receptor Tyrosine Kinase |
| TCA | Tricarboxylic Acid |
| 2-DG | 2-deoxy-d-glucose |
References
- Pui, C.-H.; Yang, J.J.; Hunger, S.P.; Pieters, R.; Schrappe, M.; Biondi, A.; Vora, A.; Baruchel, A.; Silverman, L.B.; Schmiegelow, K.; et al. Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. J. Clin. Oncol. 2015, 33, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Rasche, M.; Zimmermann, M.; Borschel, L.; Bourquin, J.-P.; Dworzak, M.; Klingebiel, T.; Lehrnbecher, T.; Creutzig, U.; Klusmann, J.-H.; Reinhardt, D. Successes and challenges in the treatment of pediatric acute myeloid leukemia: A retrospective analysis of the AML-BFM trials from 1987 to 2012. Leukemia 2018, 32, 2167–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masetti, R.; Rondelli, R.; Fagioli, F.; Mastronuzzi, A.; Pierani, P.; Togni, M.; Menna, G.; Pigazzi, M.; Putti, M.; Basso, G. Infants with acute myeloid leukemia treated according to the Associazione Italiana di Ematologia e Oncologia Pediatrica 2002/01 protocol have an outcome comparable to that of older children. Haematologica 2014, 99, e127–e129. [Google Scholar] [CrossRef]
- Masetti, R.; Guidi, V.; Ronchini, L.; Bertuccio, N.S.; Locatelli, F.; Pession, A. The changing scenario of non-Down syndrome acute megakaryoblastic leukemia in children. Crit. Rev. Oncol. 2019, 138, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Masetti, R.; Vendemini, F.; Zama, D.; Biagi, C.; Gasperini, P.; Pession, A. All-transretinoic acid in the treatment of pediatric acute promyelocytic leukemia. Expert Rev. Anticancer Ther. 2012, 12, 1191–1204. [Google Scholar] [CrossRef]
- Helsmoortel, H.H.; Bresolin, S.; Lammens, T.; Cave, H.; Noellke, P.; Caye, A.; Ghazavi, F.; De Vries, A.; Hasle, H.; Labarque, V.; et al. LIN28B overexpression defines a novel fetal-like subgroup of juvenile myelomonocytic leukemia. Blood 2016, 127, 1163–1172. [Google Scholar] [CrossRef] [Green Version]
- Masetti, R.; Bertuccio, S.N.; Astolfi, A.; Chiarini, F.; Lonetti, A.; Indio, V.; De Luca, M.; Bandini, J.; Serravalle, S.; Franzoni, M.; et al. Hh/Gli antagonist in acute myeloid leukemia with CBFA2T3-GLIS2 fusion gene. J. Hematol. Oncol. 2017, 10, 26. [Google Scholar] [CrossRef]
- Yu, L.; Lu, M.; Jia, D.; Ma, J.; Ben-Jacob, E.; Levine, H.; Kaipparettu, B.A.; Onuchic, J.N. Modeling the Genetic Regulation of Cancer Metabolism: Interplay between Glycolysis and Oxidative Phosphorylation. Cancer Res. 2017, 77, 1564–1574. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Suganuma, K.; Miwa, H.; Imai, N.; Shikami, M.; Gotou, M.; Goto, M.; Mizuno, S.; Takahashi, M.; Yamamoto, H.; Hiramatsu, A.; et al. Energy metabolism of leukemia cells: Glycolysis versus oxidative phosphorylation. Leuk. Lymphoma 2010, 51, 2112–2119. [Google Scholar] [CrossRef]
- Han, L.; Cavazos, A.; Baran, N.; Zhang, Q.; Kuruvilla, V.M.; Gay, J.P.; Feng, N.; Battula, V.L.; Kantarjian, H.M.; Daver, N.G.; et al. Mitochondrial Oxphos as Survival Mechanism of Minimal Residual AML Cells after Induction Chemotherapy: Survival Benefit by Complex I Inhibition with Iacs-010759. Blood 2019, 134, 5161. [Google Scholar] [CrossRef]
- Tabe, Y.; Konopleva, M.; Andreeff, M. Fatty Acid Metabolism, Bone Marrow Adipocytes, and AML. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashkovan, M.; Ferrando, A. Metabolic dependencies and vulnerabilities in leukemia. Genes Dev. 2019, 33, 1460–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Boil. 2014, 15, 243–256. [Google Scholar] [CrossRef] [Green Version]
- Kishton, R.J.; Barnes, C.E.; Nichols, A.G.; Cohen, S.; Gerriets, V.A.; Siska, P.J.; MacIntyre, A.; Goraksha-Hicks, P.; De Cubas, A.A.; Liu, T.; et al. AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival. Cell Metab. 2016, 23, 649–662. [Google Scholar] [CrossRef] [Green Version]
- De Grandis, M.; Mancini, S.J.; Aurrand-Lions, M. In quest for leukemia initiating cells in AML. Oncoscience 2018, 5, 9–10. [Google Scholar] [CrossRef]
- Hao, X.; Gu, H.; Chen, C.; Huang, D.; Zhao, Y.; Xie, L.; Zou, Y.; Shu, H.S.; Zhang, Y.; He, X.; et al. Metabolic Imaging Reveals a Unique Preference of Symmetric Cell Division and Homing of Leukemia-Initiating Cells in an Endosteal Niche. Cell Metab. 2019, 29, 950–965.e6. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-H.; Israelsen, W.J.; Lee, D.; Yu, V.W.C.; Jeanson, N.T.; Clish, C.B.; Cantley, L.C.; Heiden, M.G.V.; Scadden, D.T. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 2014, 158, 1309–1323. [Google Scholar] [CrossRef] [Green Version]
- Cilloni, D.; Saglio, G. Molecular Pathways: BCR-ABL. Clin. Cancer Res. 2011, 18, 930–937. [Google Scholar] [CrossRef] [Green Version]
- Stavropoulou, V.; Peters, A.H.F.M.; Schwaller, J. Aggressive leukemia driven by MLL-AF9. Mol. Cell. Oncol. 2017, 5, e1241854. [Google Scholar] [CrossRef] [Green Version]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias—An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irigoyen, M.; García, J.; Berra, E. The hypoxia signalling pathway in haematological malignancies. Oncotarget 2017, 8, 36832–36844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nepstad, I.; Hatfield, K.J.; Grønningsæter, I.S.; Reikvam, H. The PI3K-Akt-mTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. Int. J. Mol. Sci. 2020, 21, 2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slaninova, V.; Krafcikova, M.; Perez-Gomez, R.; Steffal, P.; Trantirek, L.; Bray, S.J.; Krejci, A. Notch stimulates growth by direct regulation of genes involved in the control of glycolysis and the tricarboxylic acid cycle. Open Boil. 2016, 6, 150155. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Farge, T.; Saland, E.; De Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef]
- Pierro, J.; Hogan, L.E.; Bhatla, T.; Carroll, W.L. New targeted therapies for relapsed pediatric acute lymphoblastic leukemia. Expert Rev. Anticancer. Ther. 2017, 17, 725–736. [Google Scholar] [CrossRef]
- Boag, J.M.; Beesley, A.H.; Firth, M.J.; Freitas, J.R.; Ford, J.; Hoffmann, K.; Cummings, A.J.; De Klerk, N.; Kees, U.R. Altered glucose metabolism in childhood pre-B acute lymphoblastic leukaemia. Leukemia 2006, 20, 1731–1737. [Google Scholar] [CrossRef]
- Järviaho, T.; Hurme-Niiranen, A.; Soini, H.; Niinimäki, R.; Möttönen, M.; Savolainen, E.-R.; Hinttala, R.; Harila-Saari, A.; Uusima, J. Novel non-neutral mitochondrial DNA mutations found in childhood acute lymphoblastic leukemia. Clin. Genet. 2017, 93, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Jerchel, I.S.; Hoogkamer, A.Q.; AriëS, I.M.; Steeghs, E.M.P.; Boer, J.M.; Besselink, N.J.M.; van de Ven, C.; de Groot-Kruseman, H.A.; de Haas, V.; Horstmann, M.A. RAS pathway mutations as a predictive biomarker for treatment adaptation in pediatric B-cell precursor acute lymphoblastic leukemia. Leukemia 2018, 32, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Sbirkov, Y.; Burnusuzov, H.; Sarafian, V. Metabolic reprogramming in childhood acute lymphoblastic leukemia. Pediatr. Blood Cancer 2020, 67, e28255. [Google Scholar] [CrossRef] [PubMed]
- Manara, E.; Baron, E.; Tregnago, C.; Aveic, S.; Bisio, V.; Bresolin, S.; Masetti, R.; Locatelli, F.; Basso, G.; Pigazzi, M. MLL-AF6 fusion oncogene sequesters AF6 into the nucleus to trigger RAS activation in myeloid leukemia. Blood 2014, 124, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Kimmelman, A.C. Metabolic Dependencies in RAS-Driven Cancers. Clin. Cancer Res. 2015, 21, 1828–1834. [Google Scholar] [CrossRef] [Green Version]
- Pajak, B.; Siwiak, E.; Sołtyka, M.; Priebe, A.; Zieliński, R.; Fokt, I.; Ziemniak, M.; Jaśkiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int. J. Mol. Sci. 2019, 21, 234. [Google Scholar] [CrossRef] [Green Version]
- Chan, L.N.; Chen, Z.; Braas, D.; Lee, J.-W.; Xiao, G.; Geng, H.; Cosgun, K.N.; Hurtz, C.; Shojaee, S.; Cazzaniga, V.; et al. Metabolic gatekeeper function of B-lymphoid transcription factors. Nature 2017, 542, 479–483. [Google Scholar] [CrossRef]
- Gibson, T.M.; Ehrhardt, M.J.; Ness, K.K. Obesity and Metabolic Syndrome Among Adult Survivors of Childhood Leukemia. Curr. Treat. Options Oncol. 2016, 17, 17. [Google Scholar] [CrossRef] [Green Version]
- Dyczynski, M.; Vesterlund, M.; Björklund, A.-C.; Zachariadis, V.; Janssen, J.; Gallart-Ayala, H.; Daskalaki, E.; Wheelock, C.E.; Lehtiö, J.; Grandér, D.; et al. Metabolic reprogramming of acute lymphoblastic leukemia cells in response to glucocorticoid treatment. Cell Death Dis. 2018, 9, 846. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, K.; Firth, M.J.; Beesley, A.H.; Freitas, J.R.; Ford, J.; Senanayake, S.; De Klerk, N.; Baker, D.L.; Kees, U.R. Prediction of relapse in paediatric pre-B acute lymphoblastic leukaemia using a three-gene risk index. Br. J. Haematol. 2008, 140, 656–664. [Google Scholar] [CrossRef]
- Leni, Z.; Ćwiek, P.; Dimitrova, V.; Dulcey, A.S.; Zamboni, N.; Simillion, C.; Rossi, G.; Leibundgut, K.; Arcaro, A. 2-Deoxy-d-glucose Restore Glucocorticoid Sensitivity in Acute Lymphoblastic Leukemia via Modification of N-Linked Glycosylation in an Oxygen Tension-Independent Manner. Oxidative Med. Cell. Longev. 2017, 2017, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosilio, C.; Lounnas, N.; Nebout, M.; Imbert, V.; Hagenbeek, T.; Spits, H.; Asnafi, V.; Pontier-Bres, R.; Reverso, J.; Michiels, J.-F.; et al. The metabolic perturbators metformin, phenformin and AICAR interfere with the growth and survival of murine PTEN-deficient T cell lymphomas and human T-ALL/T-LL cancer cells. Cancer Lett. 2013, 336, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Biondani, G.; Peyron, J.F. Metformin, an Anti-diabetic Drug to Target Leukemia. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Chen, C.; Jin, Y.; Fuentes-Mattei, E.; Velazquez-Tores, G.; Benito, J.M.; Konopleva, M.; Andreeff, M.; Lee, M.-H.; Yeung, S.-C.J. Differential impact of structurally different anti-diabetic drugs on proliferation and chemosensitivity of acute lymphoblastic leukemia cells. Cell Cycle 2012, 11, 2314–2326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trucco, M.; Barredo, J.C.; Goldberg, J.; Leclerc, G.M.; Hale, G.A.; Gill, J.; Setty, B.A.; Smith, T.; Lush, R.; Lee, J.K.; et al. A phase I window, dose escalating and safety trial of metformin in combination with induction chemotherapy in relapsed refractory acute lymphoblastic leukemia: Metformin with induction chemotherapy of vincristine, dexamethasone, PEG-asparaginase, and doxo. Pediatr. Blood Cancer 2018, 65, e27224. [Google Scholar] [CrossRef] [PubMed]
- Serravalle, S.; Bertuccio, S.N.; Astolfi, A.; Melchionda, F.; Pession, A. Synergistic Cytotoxic Effect of L-Asparaginase Combined with Decitabine as a Demethylating Agent in Pediatric T-ALL, with Specific Epigenetic Signature. BioMed Res. Int. 2016, 2016, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.; Taurino, G.; Bianchi, M.G.; Kilberg, M.S.; Bussolati, O. Asparagine Synthetase in Cancer: Beyond Acute Lymphoblastic Leukemia. Front. Oncol. 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Hermanova, I.; Zaliova, M.; Trka, J.; Starkova, J. Low expression of asparagine synthetase in lymphoid blasts precludes its role in sensitivity to L-asparaginase. Exp. Hematol. 2012, 40, 657–665. [Google Scholar] [CrossRef]
- Heřmanová, I.; Arruabarrena-Aristorena, A.; Valis, K.; Nůsková, H.; Alberich-Jorda, M.; Fiser, K.; Fernández-Ruiz, S.; Kavan, D.; Pecinova, A.; Niso-Santano, M.; et al. Pharmacological inhibition of fatty-acid oxidation synergistically enhances the effect of l-asparaginase in childhood ALL cells. Leukemia 2015, 30, 209–218. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Chen, S.-C.; Andersson, A.; Phillips, L.A.; Li, Y.; Sotzen, J.; Kundu, M.; Downing, J.R.; Melnick, A.; Mullighan, C.G. Integrated genetic and epigenetic analysis of childhood acute lymphoblastic leukemia. J. Clin. Investig. 2013, 123, 3099–3111. [Google Scholar] [CrossRef] [Green Version]
- Bhatla, T.; Wang, J.; Morrison, D.J.; Raetz, E.A.; Burke, M.J.; Brown, P.; Carroll, W.L. Epigenetic reprogramming reverses the relapse-specific gene expression signature and restores chemosensitivity in childhood B-lymphoblastic leukemia. Blood 2012, 119, 5201–5210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt Stimulates Aerobic Glycolysis in Cancer Cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frolova, O.; Buglio, D.; Samudio, I.; McQueen, T.; Kantarjian, H.; Weiser, M.A.; Andreeff, M.; Konopleva, M. Relationship between mTOR-Mediated Upregulation of Glycolysis and Chemosensitivity of ALL Blasts. Blood 2006, 108, 1833. [Google Scholar] [CrossRef]
- Mian, A.A.; Zafar, U.; Ottmann, O.; Ruthardt, M.A.; Lalani, E.-N.M. Activation of AKT/mTOR Pathway in Ph+ Acute Lymphoblastic Leukemia (ALL) Leads to Non-Mutational Resistance. Blood 2019, 134, 2570. [Google Scholar] [CrossRef]
- Maude, S.L.; Tasian, S.K.; Vincent, T.; Hall, J.W.; Sheen, C.; Roberts, K.G.; Seif, A.E.; Barrett, D.M.; Chen, I.-M.; Collins, J.R.; et al. Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood 2012, 120, 3510–3518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrue, C.; Saland, E.; Vergez, F.; Serhan, N.; Delabesse, E.; De Mas, V.M.-; Hospital, M.-A.; Tamburini, J.; Manenti, S.; Sarry, J.E.; et al. Anti-leukemic activity of 2-deoxy-d-glucose through inhibition of N-linked glycosylation in acute myeloid leukemia with FLT3-ITD or cKIT mutations. Mol. Cancer Ther. 2015, 14, 2364–2373. [Google Scholar] [CrossRef] [Green Version]
- Bertuccio, S.N.; Serravalle, S.; Astolfi, A.; Lonetti, A.; Indio, V.; Leszl, A.; Pession, A.; Melchionda, F. Identification of a cytogenetic and molecular subgroup of acute myeloid leukemias showing sensitivity to L-Asparaginase. Oncotarget 2017, 8, 109915–109923. [Google Scholar] [CrossRef] [Green Version]
- Herranz, D.; Ambesi-Impiombato, A.; Sudderth, J.; Sanchez-Martin, M.; Belver, L.; Tosello, V.; Xu, L.; Wendorff, A.A.; Castillo, M.; Haydu, J.E.; et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat. Med. 2015, 21, 1182–1189. [Google Scholar] [CrossRef]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.J.; Agip, A.-N.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [Green Version]
- Tan, P.; Tiong, I.S.; Fleming, S.; Pomilio, G.; Cummings, N.; Droogleever, M.; McManus, J.; Schwarer, A.; Catalano, J.; Patil, S.; et al. The mTOR inhibitor everolimus in combination with azacitidine in patients with relapsed/refractory acute myeloid leukemia: A phase Ib/II study. Oncotarget 2016, 8, 52269–52280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Chapuis, N.; Bardet, V.; Willems, L.; Tamburini, J.; Knight, Z.A.; Shokat, K.M.; Azar, N.; Ifrah, N.; Dreyfus, F.; et al. PI-103, a Dual Inhibitor of Class I Phosphatidylinositide 3-Kinase and mTOR, Has Anti-Leukemic Activity in Acute Myeloid Leukemia. Blood 2007, 110, 876. [Google Scholar] [CrossRef]
- Kampa-Schittenhelm, K.M.; Heinrich, M.C.; Akmut, F.; Rasp, K.H.; Illing, B.; Döhner, H.; Döhner, K.; Schittenhelm, M.M. Cell cycle-dependent activity of the novel dual PI3K-MTORC1/2 inhibitor NVP-BGT226 in acute leukemia. Mol. Cancer 2013, 1476–4598, 12–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Jiang, L.; Lin, X.-H.; Tseng, K.-F.; Liu, Y.; Zhang, X.; Dong, R.-H.; Lu, Z.-G.; Wang, X.-J. The PI3K/mTOR dual inhibitor BEZ235 suppresses proliferation and migration and reverses multidrug resistance in acute myeloid leukemia. Acta Pharmacol. Sin. 2017, 38, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, A.; Sanda, T.; Grebliunaite, R.; Carracedo, A.; Salmena, L.; Ahn, Y.; Dahlberg, S.E.; Neuberg, N.; Moreau, L.A.; Winter, S.S.; et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood 2009, 114, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Masetti, R.; Castelli, I.; Astolfi, A.; Bertuccio, S.N.; Indio, V.; Togni, M.; Belotti, T.; Serravalle, S.; Tarantino, G.; Zecca, M.; et al. Genomic complexity and dynamics of clonal evolution in childhood acute myeloid leukemia studied with whole-exome sequencing. Oncotarget 2016, 7, 56746–56757. [Google Scholar] [CrossRef] [PubMed]
- Stockard, B.; Garrett, T.; Guingab-Cagmat, J.; Meshinchi, S.; Lamba, J.K. Distinct Metabolic features differentiating FLT3-ITD AML from FLT3-WT childhood Acute Myeloid Leukemia. Sci. Rep. 2018, 8, 5534. [Google Scholar] [CrossRef] [PubMed]
- Lou, S.; Balluff, B.; Cleven, A.H.G.; Bovée, J.V.M.G.; McDonnell, L.A. Prognostic Metabolite Biomarkers for Soft Tissue Sarcomas Discovered by Mass Spectrometry Imaging. J. Am. Soc. Mass Spectrom. 2016, 28, 376–383. [Google Scholar] [CrossRef] [Green Version]
- Altman, J.K.; Sassano, A.; Platanias, L.C. Targeting mTOR for the treatment of AML. New agents and new directions. Oncotarget 2011, 2, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Michelozzi, I.M.; Granata, V.; De Ponti, G.; Alberti, G.; Tomasoni, C.; Antolini, L.; Gambacorti-Passerini, C.; Gentner, B.; Dazzi, F.; Biondi, A.; et al. Acute myeloid leukaemia niche regulates response to L-asparaginase. Br. J. Haematol. 2019, 186, 420–430. [Google Scholar] [CrossRef]
- Mirabilii, S.; Ricciardi, M.R.; Tafuri, A. mTOR Regulation of Metabolism in Hematologic Malignancies. Cells 2020, 9, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasian, S.K.; Teachey, D.T.; Rheingold, S. Targeting the PI3K/mTOR Pathway in Pediatric Hematologic Malignancies. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, T.; Takemoto, N.; Sato, T.; Takimoto, R.; Tanaka, I.; Fujimi, A.; Akiyama, T.; Kuroda, H.; Kawano, Y.; Kobune, M.; et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat. Med. 2003, 9, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Lonetti, A.; Pession, A.; Masetti, R. Targeted Therapies for Pediatric AML: Gaps and Perspective. Front. Pediatr. 2019, 7. [Google Scholar] [CrossRef]
- Kuhlen, M.; Klusmann, J.-H.; Hoell, J.I. Molecular Approaches to Treating Pediatric Leukemias. Front. Pediatr. 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Barrett, D.; Brown, V.I.; Grupp, S.A.; Teachey, D.T. Targeting the PI3KAKTmTOR signaling axis in children with hematologic malignancies. Pediatric Drugs 2012, 14, 299–316. [Google Scholar]
- Khandia, R.; Dadar, M.; Munjhal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef] [Green Version]
- Altman, J.K.; Szilard, A.; Goussetis, D.J.; Sassano, A.; Colamonici, M.; Gounaris, E.; Frankfurt, O.; Giles, F.J.; Eklund, E.A.; Beauchamp, E.; et al. Autophagy is a survival mechanism of acute myelogenous leukemia precursors during dual mTORC2/mTORC1 targeting. Clin. Cancer Res. 2014, 20, 2400–2409. [Google Scholar] [CrossRef] [Green Version]
- Herschbein, L.; Liesveld, J.L. Dueling for dual inhibition: Means to enhance effectiveness of PI3K/Akt/mTOR inhibitors in AML. Blood Rev. 2018, 32, 235–248. [Google Scholar] [CrossRef]


{kind=link}
| Drug | Target | Condition | Reference |
|---|---|---|---|
| 2-deoxy-d-glucose | Hexokinase II | ALL AML | [41] (Leni et al., 2017) [57] (Larrue et al., 2015) |
| L-Asparaginase | Asparagine availability | ALL AML | [46] (Serravalle et al., 2016) [58] (Bertuccio et al., 2017) |
| BPTES | Glutaminase | ALL | [59] (Herranz et al., 2015) |
| Etomoxir | Fatty acid oxidation | ALL | [49] (Hermanova et al., 2016) |
| IACS 010759 | OXPHOS | AML | [60] (Molina et al., 2018) |
| Metformin | Mitochondrial Complex I | ALL | [45] (Trucco et al., 2018) |
| Sirolimus Temsirolimus Everolimus | mTOR | ALL AML | [56] (Maude et al., 2012) [61] (Tan et al., 2017) |
| NVP-BEZ235 BGT226 PI-103 PF-04691502 | PI3K/mTOR | AML | [62] (Park et al., 2007) [63] (Kampa-Schittenhelm et al., 2013) [64] (Deng et al., 2017) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anselmi, L.; Bertuccio, S.N.; Lonetti, A.; Prete, A.; Masetti, R.; Pession, A. Insights on the Interplay between Cells Metabolism and Signaling: A Therapeutic Perspective in Pediatric Acute Leukemias. Int. J. Mol. Sci. 2020, 21, 6251. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176251
Anselmi L, Bertuccio SN, Lonetti A, Prete A, Masetti R, Pession A. Insights on the Interplay between Cells Metabolism and Signaling: A Therapeutic Perspective in Pediatric Acute Leukemias. International Journal of Molecular Sciences. 2020; 21(17):6251. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176251
Chicago/Turabian StyleAnselmi, Laura, Salvatore Nicola Bertuccio, Annalisa Lonetti, Arcangelo Prete, Riccardo Masetti, and Andrea Pession. 2020. "Insights on the Interplay between Cells Metabolism and Signaling: A Therapeutic Perspective in Pediatric Acute Leukemias" International Journal of Molecular Sciences 21, no. 17: 6251. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176251