In Vitro-Transcribed mRNA Chimeric Antigen Receptor T Cell (IVT mRNA CAR T) Therapy in Hematologic and Solid Tumor Management: A Preclinical Update


Abstract
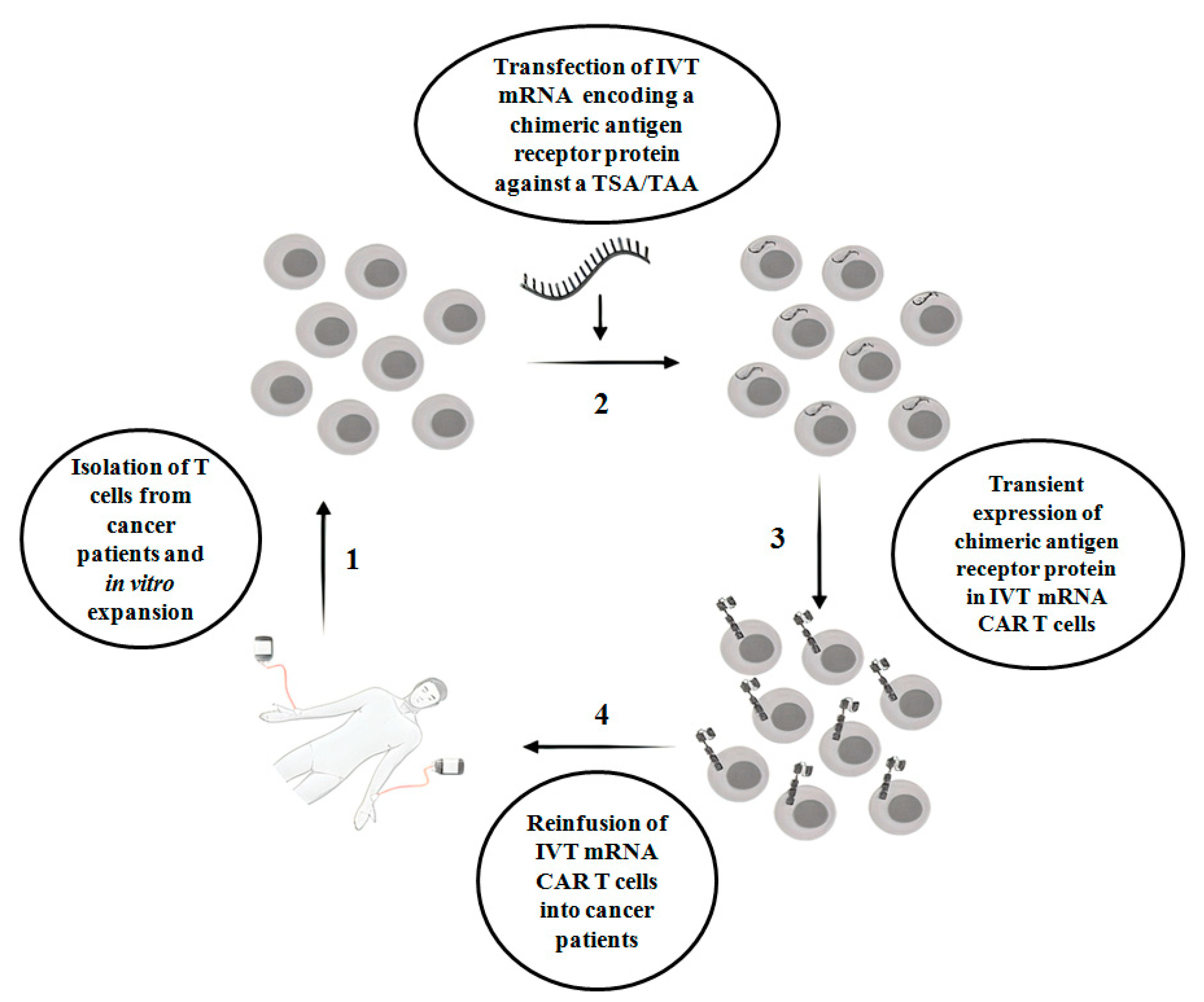
:1. Introduction
2. Effect of IVT mRNA CAR T Cells in Preclinical Studies Related to Hematologic Malignancies
2.1. Chronic Lymphocytic Leukemia (CLL)
2.2. Acute Lymphoblastic Leukemia (ALL)
2.3. Acute Myeloid Leukemia (AML)
3. Effect of IVT mRNA CAR T Cells in Preclinical Studies Related to Solid Malignancies
3.1. Mesothelioma and Colon Cancers
3.2. Ovarian and Breast Cancers
3.3. Neuroblastoma and Glioblastoma Multiforme
3.4. Melanoma
4. IVT mRNA-Based Clinical Trials in Hematologic and Solid Tumors
5. Future Directions
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CAR T | Chimeric antigen receptor T cell |
| IVT mRNA CAR T | in vitro transcribed anti-mRNA-based CAR T cells |
| TCR | T cell receptor |
| CAR | Chimeric antigen receptor |
| scFv | Single-chain variable fragment |
| TAA | Tumor-associated antigen |
| EGFR | Epidermal growth factor receptor |
| CEA | Carcinoembryonic antigen |
| ERBB2 | Epidermal growth factor receptor 2 |
| PSMA | Prostate-specific membrane antigen |
| B-CLL | B cell chronic lymphocytic leukemia |
| B-NHL | B cell non-Hodgkin’s lymphoma |
| FcγR | Fragment c gamma receptor |
| BiTEs | Bispecific T cell engager |
| AML | Acute myeloid leukemia |
| NKG2D | Natural killer group 2D |
| ESFT | Ewing’s sarcoma family of tumors |
| TGFβRIImut | Transforming growth factor β receptor II frameshift mutation |
| GM-CSF | Granulocyte–macrophage colony Stimulating factor |
| FRα | Folate receptor alpha |
| EpCAM | Epithelial cell adhesion molecule |
| GBM | Glioblastoma multiforme |
| gp100 | Glycoprotein 100 |
| TRP-1 | Tyrosinase-related protein 1 |
| TRP-2 | Tyrosinase-related protein 2 |
| GD2 | Disialoganglioside 2 |
| L1-CAM | L1 cell adhesion molecule |
| MCSP | Melanoma-associated chondroitin sulfate proteoglycan |
| TETARs | T cells expressing two additional receptors |
| CSPG4 | Chondroitin sulfate proteoglycan 4 |
| NKT | Natural killer T |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
| CARTmeso cells | CAR T cells redirected for mesothelin |
| IgG | Immunoglobulin G |
| PD1 | Programmed cell death protein 1 |
| PDL1 | Programmed death ligand 1 |
References
- Cheadle, E.J.; Gornall, H.; Baldan, V.; Hanson, V.; Hawkins, R.E.; Gilham, D.E. CAR T cells: Driving the road from the laboratory to the clinic. Immunol. Rev. 2014, 257, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, M.; Mount, N. Genetically modified T cells in cancer therapy: Opportunities and challenges. Dis. Models Mech. 2015, 8, 337–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. Driving T-cell immunotherapy to solid tumors. Nat. Biotechnol. 2018, 36, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Kenderian, S.S.; Porter, D.L.; Gill, S. Chimeric Antigen Receptor T Cells and Hematopoietic Cell Transplantation: How Not to Put the CART Before the Horse. Biol. Blood Marrow Transplant. 2017, 23, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Ping, J.; Huang, Z.; Zhang, X.; Zhou, J.; Wang, G.; Liu, S.; Ma, J. CAR-T Cell Therapy in Cancer: Tribulations and Road Ahead. J. Immunol. Res. 2020, 2020, 1924379. [Google Scholar] [CrossRef] [Green Version]
- Miliotou, A.N.; Papadopoulou, L.C. In Vitro-Transcribed (IVT)-mRNA CAR Therapy Development. Methods Mol. Biol. 2020, 2086, 87–117. [Google Scholar] [CrossRef]
- Braun, D.A.; Wu, C.J. Antigen Discovery and Therapeutic Targeting in Hematologic Malignancies. Cancer J. 2017, 23, 115–124. [Google Scholar] [CrossRef]
- Rabinovich, P.M.; Komarovskaya, M.E.; Ye, Z.J.; Imai, C.; Campana, D.; Bahceci, E.; Weissman, S.M. Synthetic messenger RNA as a tool for gene therapy. Hum. Gene Ther. 2006, 17, 1027–1035. [Google Scholar] [CrossRef]
- Rabinovich, P.M.; Komarovskaya, M.E.; Wrzesinski, S.H.; Alderman, J.L.; Budak-Alpdogan, T.; Karpikov, A.; Guo, H.F.; Flavell, R.A.; Cheung, N.K.; Weissman, S.M.; et al. Chimeric Receptor mRNA Transfection as a Tool to Generate Antineoplastic Lymphocytes. Hum. Gene Ther. 2009, 20, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Almasbak, H.; Rian, E.; Hoel, H.J.; Pule, M.; Walchli, S.; Kvalheim, G.; Gaudernack, G.; Rasmussen, A.M. Transiently redirected T cells for adoptive transfer. Cytotherapy 2011, 13, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Koksal, H.; Dillard, P.; Josefsson, S.E.; Maggadottir, S.M.; Pollmann, S.; Fane, A.; Blaker, Y.N.; Beiske, K.; Huse, K.; Kolstad, A.; et al. Preclinical development of CD37CAR T-cell therapy for treatment of B-cell lymphoma. Blood Adv. 2019, 3, 1230–1243. [Google Scholar] [CrossRef] [PubMed]
- Panjwani, M.K.; Smith, J.B.; Schutsky, K.; Gnanandarajah, J.; O’Connor, C.M.; Powell, D.J., Jr.; Mason, N.J. Feasibility and Safety of RNA-transfected CD20-specific Chimeric Antigen Receptor T Cells in Dogs with Spontaneous B Cell Lymphoma. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 1602–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, D.M.; Zhao, Y.; Liu, X.; Jiang, S.; Carpenito, C.; Kalos, M.; Carroll, R.G.; June, C.H.; Grupp, S.A. Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum. Gene Ther. 2011, 22, 1575–1586. [Google Scholar] [CrossRef] [Green Version]
- Barrett, D.M.; Liu, X.; Jiang, S.; June, C.H.; Grupp, S.A.; Zhao, Y. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum. Gene Ther. 2013, 24, 717–727. [Google Scholar] [CrossRef] [Green Version]
- Almasbak, H.; Walseng, E.; Kristian, A.; Myhre, M.R.; Suso, E.M.; Munthe, L.A.; Andersen, J.T.; Wang, M.Y.; Kvalheim, G.; Gaudernack, G.; et al. Inclusion of an IgG1-Fc spacer abrogates efficacy of CD19 CAR T cells in a xenograft mouse model. Gene Ther. 2015, 22, 391–403. [Google Scholar] [CrossRef]
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol. Res. 2015, 3, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Till, B.G.; Jensen, M.C.; Wang, J.; Qian, X.; Gopal, A.K.; Maloney, D.G.; Lindgren, C.G.; Lin, Y.; Pagel, J.M.; Budde, L.E.; et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: Pilot clinical trial results. Blood 2012, 119, 3940–3950. [Google Scholar] [CrossRef] [Green Version]
- Al-Hussaini, M.; Rettig, M.P.; Ritchey, J.K.; Karpova, D.; Uy, G.L.; Eissenberg, L.G.; Gao, F.; Eades, W.C.; Bonvini, E.; Chichili, G.R.; et al. Targeting CD123 in acute myeloid leukemia using a T-cell-directed dual-affinity retargeting platform. Blood 2016, 127, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Kenderian, S.S.; Ruella, M.; Shestova, O.; Klichinsky, M.; Aikawa, V.; Morrissette, J.J.D.; Scholler, J.; Song, D.; Porter, D.L.; Carroll, M.; et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia 2015, 29, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Tasian, S.K.; Kenderian, S.S.; Shen, F.; Ruella, M.; Shestova, O.; Kozlowski, M.; Li, Y.; Schrank-Hacker, A.; Morrissette, J.J.D.; Carroll, M.; et al. Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood 2017, 129, 2395–2407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, J.B.; Choudhari, N.; Perazzelli, J.; Storm, J.; Hofmann, T.J.; Jain, P.; Storm, P.B.; Pardi, N.; Weissman, D.; Waanders, A.J.; et al. Purification of mRNA Encoding Chimeric Antigen Receptor Is Critical for Generation of a Robust T-Cell Response. Hum. Gene Ther. 2019, 30, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Li, A.; Liu, Q.; Li, T.; Yuan, X.; Han, X.; Wu, K. Chimeric antigen receptor T cells: A novel therapy for solid tumors. J. Hematol. Oncol. 2017, 10, 78. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.; Mullins, C.S.; Linnebacher, M. Colorectal cancer vaccines: Tumor-associated antigens vs. neoantigens. World J. Gastroenterol. 2018, 24, 5418–5432. [Google Scholar] [CrossRef]
- Zhao, Y.B.; Moon, E.; Carpenito, C.; Paulos, C.M.; Liu, X.J.; Brennan, A.L.; Chew, A.; Carroll, R.G.; Scholler, J.; Levine, B.L.; et al. Multiple Injections of Electroporated Autologous T Cells Expressing a Chimeric Antigen Receptor Mediate Regression of Human Disseminated Tumor. Cancer Res. 2010, 70, 9053–9061. [Google Scholar] [CrossRef] [Green Version]
- Lehner, M.; Gotz, G.; Proff, J.; Schaft, N.; Dorrie, J.; Full, F.; Ensser, A.; Muller, Y.A.; Cerwenka, A.; Abken, H.; et al. Redirecting T cells to Ewing’s sarcoma family of tumors by a chimeric NKG2D receptor expressed by lentiviral transduction or mRNA transfection. PLoS ONE 2012, 7, e31210. [Google Scholar] [CrossRef]
- Mirzaei, H.R.; Rodriguez, A.; Shepphird, J.; Brown, C.E.; Badie, B. Chimeric Antigen Receptors T Cell Therapy in Solid Tumor: Challenges and Clinical Applications. Front. Immunol. 2017, 8, 1850. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.H.; Lee, J.M.; Cho, H.I.; Kim, E.K.; Kim, H.S.; Park, M.Y.; Kim, T.G. Adoptive immunotherapy using human peripheral blood lymphocytes transferred with RNA encoding Her-2/neu-specific chimeric immune receptor in ovarian cancer xenograft model. Cancer Gene Ther. 2009, 16, 489–497. [Google Scholar] [CrossRef] [Green Version]
- Schutsky, K.; Song, D.G.; Lynn, R.; Smith, J.B.; Poussin, M.; Figini, M.; Zhao, Y.B.; Powell, D.J. Rigorous optimization and validation of potent RNA CAR T cell therapy for the treatment of common epithelial cancers expressing folate receptor. Oncotarget 2015, 6, 28911–28928. [Google Scholar] [CrossRef] [Green Version]
- Hung, C.F.; Xu, X.Q.; Li, L.H.; Ma, Y.; Jin, Q.; Viley, A.; Allen, C.; Natarajan, P.; Shivakumar, R.; Peshwa, M.V.; et al. Development of Anti-Human Mesothelin-Targeted Chimeric Antigen Receptor Messenger RNA-transfected Peripheral Blood Lymphocytes for Ovarian Cancer Therapy. Hum. Gene Ther. 2018, 29, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Ang, W.X.; Li, Z.D.; Chi, Z.X.; Du, S.H.; Chen, C.; Tay, J.C.K.; Toh, H.C.; Connolly, J.E.; Xu, X.H.; Wang, S. Intraperitoneal immunotherapy with T cells stably and transiently expressing anti-EpCAM CAR in xenograft models of peritoneal carcinomatosis. Oncotarget 2017, 8, 13545–13559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchou, J.; Zhao, Y.B.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.J.; Lacey, S.F.; et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Zhao, S.; Zhang, Y.; Wang, Y.; Zhang, Z.; Yang, M.; Zhu, Y.; Zhang, G.; Guo, G.; Tong, A.; et al. B7-H3 as a Novel CAR-T Therapeutic Target for Glioblastoma. Mol. Ther. Oncolytics 2019, 14, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Liu, X.; Hulitt, J.; Jiang, S.; June, C.H.; Grupp, S.A.; Barrett, D.M.; Zhao, Y. Nature of tumor control by permanently and transiently modified GD2 chimeric antigen receptor T cells in xenograft models of neuroblastoma. Cancer Immunol. Res. 2014, 2, 1059–1070. [Google Scholar] [CrossRef] [Green Version]
- Caruso, H.G.; Torikai, H.; Zhang, L.; Matti, S.; Dai, J.L.; Do, K.A.; Singh, H.; Huls, H.; Lee, D.A.; Champlin, R.E.; et al. Redirecting T-Cell Specificity to EGFR Using mRNA to Self-limit Expression of Chimeric Antigen Receptor. J.Immunother. 2016, 39, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Ilieva, K.M.; Cheung, A.; Mele, S.; Chiaruttini, G.; Crescioli, S.; Griffin, M.; Nakamura, M.; Spicer, J.F.; Tsoka, S.; Lacy, K.E.; et al. Chondroitin Sulfate Proteoglycan 4 and Its Potential As an Antibody Immunotherapy Target across Different Tumor Types. Front. Immunol. 2017, 8, 1911. [Google Scholar] [CrossRef] [Green Version]
- Krug, C.; Birkholz, K.; Paulus, A.; Schwenkert, M.; Schmidt, P.; Hoffmann, N.; Hombach, A.; Fey, G.; Abken, H.; Schuler, G.; et al. Stability and activity of MCSP-specific chimeric antigen receptors (CARs) depend on the scFv antigen-binding domain and the protein backbone. Cancer Immunol. Immun. 2015, 64, 1623–1635. [Google Scholar] [CrossRef]
- Harrer, D.C.; Simon, B.; Fujii, S.I.; Shimizu, K.; Uslu, U.; Schuler, G.; Gerer, K.F.; Hoyer, S.; Dorrie, J.; Schaft, N. RNA-transfection of gamma/delta T cells with a chimeric antigen receptor or an alpha/beta T-cell receptor: A safer alternative to genetically engineered alpha/beta T cells for the immunotherapy of melanoma. BMC Cancer 2017, 17, 551. [Google Scholar] [CrossRef]
- Uslu, U.; Schuler, G.; Dorrie, J.; Schaft, N. Combining a chimeric antigen receptor and a conventional T-cell receptor to generate T cells expressing two additional receptors (TETARs) for a multi-hit immunotherapy of melanoma. Exp. Dermatol. 2016, 25, 872–879. [Google Scholar] [CrossRef]
- Simon, B.; Harrer, D.C.; Schuler-Thurner, B.; Schuler, G.; Uslu, U. Arming T Cells with a gp100-Specific TCR and a CSPG4-Specific CAR Using Combined DNA- and RNA-Based Receptor Transfer. Cancers 2019, 11, 696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesinger, M.; Marz, J.; Kummer, M.; Schuler, G.; Dorrie, J.; Schuler-Thurner, B.; Schaft, N. Clinical-Scale Production of CAR-T Cells for the Treatment of Melanoma Patients by mRNA Transfection of a CSPG4-Specific CAR under Full GMP Compliance. Cancers 2019, 11, 1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, B.; Wiesinger, M.; Marz, J.; Wistuba-Hamprecht, K.; Weide, B.; Schuler-Thurner, B.; Schuler, G.; Dorrie, J.; Uslu, U. The Generation of CAR-Transfected Natural Killer T Cells for the Immunotherapy of Melanoma. Int. J. Mol. Sci. 2018, 19, 2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walseng, E.; Koksal, H.; Sektioglu, I.M.; Fane, A.; Skorstad, G.; Kvalheim, G.; Gaudernack, G.; Inderberg, E.M.; Walchli, S. A TCR-based Chimeric Antigen Receptor. Sci. Rep. 2017, 7, 10713. [Google Scholar] [CrossRef] [PubMed]
- Inoo, K.; Inagaki, R.; Fujiwara, K.; Sasawatari, S.; Kamigaki, T.; Nakagawa, S.; Okada, N. Immunological quality and performance of tumor vessel-targeting CAR-T cells prepared by mRNA-EP for clinical research. Mol. Ther. Oncolytics 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Cummins, K.D.; Frey, N.; Nelson, A.M.; Schmidt, A.; Luger, S.; Isaacs, R.E.; Lacey, S.F.; Hexner, E.; Melenhorst, J.J.; June, C.H.; et al. Treating Relapsed/Refractory (RR) AML with Biodegradable Anti-CD123 CAR Modified T Cells. Blood 2017, 130, 1359. [Google Scholar]
- Svoboda, J.; Rheingold, S.R.; Gill, S.I.; Grupp, S.A.; Lacey, S.F.; Kulikovskaya, I.; Suhoski, M.M.; Melenhorst, J.J.; Loudon, B.; Mato, A.R.; et al. Nonviral RNA chimeric antigen receptor-modified T cells in patients with Hodgkin lymphoma. Blood 2018, 132, 1022–1026. [Google Scholar] [CrossRef]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.J.; Zhao, Y.B.; Kalos, M.; June, C.H. T Cells Expressing Chimeric Antigen Receptors Can Cause Anaphylaxis in Humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Caruso, H.G.; Heimberger, A.B.; Cooper, L.J.N. Steering CAR T cells to distinguish friend from foe. Oncoimmunology 2019, 8, e1271857. [Google Scholar] [CrossRef]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Type of Study | Target | Dose and Administration | Tumor Type | Results | Reference |
|---|---|---|---|---|---|
| in vitro | CD19 | - | Leukemia, lymphoma | Cytotoxicity | [10] |
| in vivo | CD19 | Multiple doses 5 × 106; i.p. | lymphoma | Tumor growth inhibition | [11] |
| in vitro | CD19 | - | Leukemia, lymphoma | Degranulation and IFN-γ secretion | [12] |
| In vitro and in vivo | CD37 | Multiple doses 107; intratumorally | Lymphoma | Cytotoxicity; tumor growth reduction | [13] |
| in vivo | Canine CD20-ζ | 2.4 ×108, 5.4 × 107, 1.1 × 108 cells i.v.; 1.16 × 108 cells i.n. | Lymphoma | Antitumor activity | [14] |
| In vitro and in vivo | CD19 | 5 × 106, 1 × 107, or 2.5 × 107 cells; i.v. | Leukemia | Cytotoxicity | [15] |
| in vivo | CD19 | Multiple doses 1 × 107 or 2 × 107, 5 × 106 and 5 × 106; i.v. | Acute lymphoblastic leukemia | Increased cytotoxicity with split doses | [16] |
| in vivo | CD19 | 20 × 106 or 10 × 106; i.v. | Acute lymphoblastic leukemia | Abrogation of CD19-based cytotoxicity due to the presence of IgG1-CH2CH3 spacer in CAR construct | [17] |
| in vivo | CD19 | 2 × 107; i.v. | Acute lymphoblastic leukemia | Cytotoxicity | [23] |
| In vitro and in vivo | CD33 | 5 × 106; i.v. | Acute myeloid leukemia | Cytotoxicity; tumor growth reduction | [21] |
| in vivo | CD123 | 1 × 107; i.v. | Acute myeloid leukemia | Cytotoxicity | [22] |
| Type of Study | Target | Dose and Administration | Tumor Type | Results | Reference |
|---|---|---|---|---|---|
| in vivo | Mesothelin | 10–15 × 106; intratumorally | Mesothelioma | Tumor growth reduction | [26] |
| in vitro | NKG2D | - | Ewing’s sarcoma family of tumors | Short-lived expression of mRNA | [27] |
| in vitro and in vivo | Her2 | 5 × 106; intratumorally or i.p. | Ovarian cancer | Cytotoxicity; tumor growth reduction | [29] |
| in vitro and in vivo | FRα | Multiple doses 107; i.p. | Ovarian cancer | Tumor growth inhibition | [30] |
| in vitro and in vivo | Mesothelin | 1 × 107 or 1 × 108; i.p. | Ovarian cancer | Tumor growth inhibition | [31] |
| in vivo | Epithelial cell adhesion molecule | 1 × 107; i.p. | Ovarian and colorectal cancer | Tumor growth inhibition | [32] |
| in vitro and in vivo | c-Met | 2 × 107; i.p. | Breast and ovarian cancer | Cytotoxicity; tumor growth inhibition | [33] |
| in vivo | Disialoganglioside GD2 | 5 × 106; intratumorally | Neuroblastoma | Cytotoxicity | [35] |
| in vitro | EGFR | - | Glioblastoma | Cytotoxicity | [36] |
| in vitro and in vivo | MCSP | Melanoma | Cytotoxicity | [38] | |
| in vitro | gp100/HLA-A2 or MCSP | - | Melanoma | Cytotoxicity | [39] |
| in vitro | gp100 and MCSP | - | Melanoma | Cytotoxicity | [40] |
| in vitro | gp100 and CSPG4 | - | Melanoma | Cytotoxicity | [41] |
| in vitro | CSPG4 | - | Melanoma | Cytotoxicity | [43] |
| in vivo | VEGFR2 | 5 × 106; i.v. | Melanoma | Tumor growth reduction | [45] |
| Phase | National Clinical Trial (NCT) No. | Target | Dose and Administration | Tumor Type | Results | Reference |
|---|---|---|---|---|---|---|
| Early Phase 1 | NCT02623582 | CD123 | 3 or 6 doses 4 × 106cells/kg; i.v. | Relapsed/refractory acute myeloid leukemia | Safe method; no antitumor effects | [46] |
| Early Phase 1 | NCT02277522 (adult); NCT02624258 (pediatric) | CD19 | 6 doses in the range 7.46 × 105–2.11 × 106; i.v. | Hodgkin’s lymphoma | No severe toxicity | [47] |
| Phase 1 | NCT01355965 | Mesothelin | Cohort 1: 1 × 108 and 1 × 109; Extended cohort: 3 doses of 1 × 108 cells followed by 3 doses of 1 × 109; i.v. | Malignant pleural mesothelioma | Severe anaphylaxis in one patient; partial antitumor response | [48,50] |
| Phase 1 | NCT01897415 | Mesothelin | 3 doses weekly 1–3 × 108/m2; i.v. | Metastatic pancreatic ductal adenocarcinoma | Increased expression of antitumor antibodies | [51] |
| Phase 0 | NCT01837602 | c-Met | 3 × 107 or 3 × 108; intratumoral | Metastatic breast cancer | Anti-cancer effects | [33] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soundara Rajan, T.; Gugliandolo, A.; Bramanti, P.; Mazzon, E. In Vitro-Transcribed mRNA Chimeric Antigen Receptor T Cell (IVT mRNA CAR T) Therapy in Hematologic and Solid Tumor Management: A Preclinical Update. Int. J. Mol. Sci. 2020, 21, 6514. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186514
Soundara Rajan T, Gugliandolo A, Bramanti P, Mazzon E. In Vitro-Transcribed mRNA Chimeric Antigen Receptor T Cell (IVT mRNA CAR T) Therapy in Hematologic and Solid Tumor Management: A Preclinical Update. International Journal of Molecular Sciences. 2020; 21(18):6514. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186514
Chicago/Turabian StyleSoundara Rajan, Thangavelu, Agnese Gugliandolo, Placido Bramanti, and Emanuela Mazzon. 2020. "In Vitro-Transcribed mRNA Chimeric Antigen Receptor T Cell (IVT mRNA CAR T) Therapy in Hematologic and Solid Tumor Management: A Preclinical Update" International Journal of Molecular Sciences 21, no. 18: 6514. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186514