Epitranscriptomics in Normal and Malignant Hematopoiesis

 ,
, 
Abstract
:1. Introduction
2. Methods for Detection and Mapping of RNA Modifications
3. Epitranscriptomics and Hematopoiesis
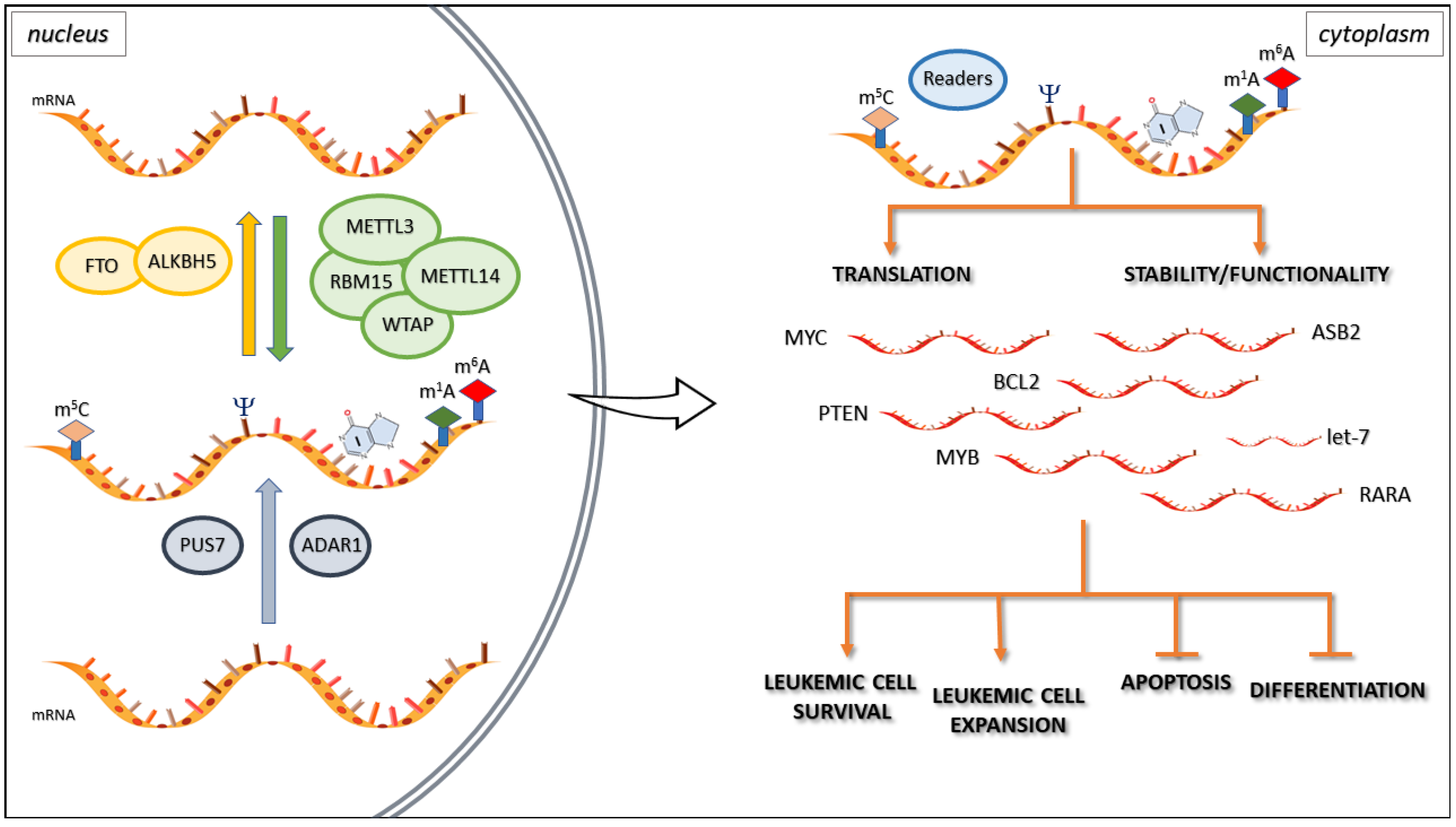
4. Epitranscriptomics in Hematological Ma Lignancies
5. A Therapeutic Opportunity
6. An Evolutionary Vision
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| tRNA | Transfer RNA |
| rRNA | Ribosomal RNA |
| snRNA | Spliceosomal RNA |
| sno RNA | Small nucleolar RNA |
| mRNA | Messenger RNA |
| lncRNA | long non-coding RNA |
| m6A | N6-methyladenosine |
| m5C | 5-methylcytidine |
| m1A | N1-methyladenosine |
| Ψ | Pseudouridylation |
| NGS | Next-generation sequencing |
| HSCs | Hematopoietic stem cells |
| METTL3 | Methyltransferase-like 3 |
| BM | Bone marrow |
| METTL14 | Methyltransferase-like 14 |
| LSC | Leukemia stem cell |
| LIC | Leukemia initiating cell |
| AML | Acute myeloid leukemia |
| ANG | Angiogenin |
| MyePro | Myeloid-restricted progenitor |
| LKS | Lin− Sca-1+ c-Kit+ cells |
| tRFs | tRNA-derived fragments |
| mTOGs | Mini 5′ terminal oligoguanines |
| EV | Extracellular vesicles |
| ADAR1 | Adenosine deaminase acting on RNA-1 |
| FTO | Fat mass and obesity-associated |
| ATRA | All-trans-retinoic acid |
| WTAP | Wilms tumor 1-associated protein |
| SNP | Single nucleotide polymorphism |
| UTRs | Untranslated regions |
| MCL | Mantle cell lymphoma |
| EBV | Epstein-Barr virus |
| BL | Burkitt’s lymphoma |
| scaRNAs | Small Cajal body-specific RNAs |
| CLL | Chronic lymphocytic leukemia |
| PTCL | Peripheral T-cell lymphoma |
| PTCL-NOS | PTCL-not otherwise specified |
| AITL | Angioimmunoblastic T-cell lymphoma |
| CML | Chronic myeloid leukemia |
| TKIs | Tyrosine kinase inhibitors |
| BC | Blast crisis |
| IFN | Interferon |
| MDA5 | Melanoma differentiation-associated protein 5 |
| PKR | Protein kinase R |
| TLRs | Toll-like receptors |
| DCs | Dendritic cells |
| IGF2BPs | Insulin-like growth factor 2 binding proteins |
| R-2HG | (R)-2-hydroxyglutarate |
References
- Boccaletto, P.; MacHnicka, M.A.; Purta, E.; Pitkowski, P.; Baginski, B.; Wirecki, T.K.; De Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef] [PubMed]
- Globisch, D.; Pearson, D.; Hienzsch, A.; Brückl, T.; Wagner, M.; Thoma, I.; Thumbs, P.; Reiter, V.; Kneuttinger, A.C.; Müller, M.; et al. Systems-Based Analysis of Modified tRNA Bases. Angew. Chemie Int. Ed. 2011, 50, 9739–9742. [Google Scholar] [CrossRef] [PubMed]
- Kamminga, L.M.; Luteijn, M.J.; Den Broeder, M.J.; Redl, S.; Kaaij, L.J.T.; Roovers, E.F.; Ladurner, P.; Berezikov, E.; Ketting, R.F. Hen1 is required for oocyte development and piRNA stability in zebrafish. EMBO J. 2010, 29, 3688–3700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, T.A.; Rim, Y.S.; Zhang, C.; Dowen, R.H.; Phillips, C.M.; Fischer, S.E.J.; Ruvkun, G. PIWI associated siRNAs and piRNAs specifically require the Caenorhabditis elegans HEN1 ortholog henn-1. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Barros-Silva, D.; Henrique, R.; Jerónimo, C. The Emerging Role of Epitranscriptomics in Cancer: Focus on Urological Tumors. Genes 2018, 9, 552. [Google Scholar] [CrossRef] [Green Version]
- Helm, M.; Motorin, Y. Detecting RNA modifications in the epitranscriptome: Predict and validate. Nat. Rev. Genet. 2017, 18, 275–291. [Google Scholar] [CrossRef]
- Nachtergaele, S.; He, C. Chemical Modifications in the Life of an mRNA Transcript. Annu. Rev. Genet. 2018, 52, 349–372. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Pan, T. N6-methyladenosine-encoded epitranscriptomics. Nat. Struct. Mol. Biol. 2016, 23, 98–102. [Google Scholar] [CrossRef]
- Pan, T. N6-methyl-adenosine modification in messenger and long non-coding RNA. Trends Biochem. Sci. 2013, 38, 204–209. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.M.; Bowman, M.; Madden, S.L.; Rauscher, F.J.; Sukumar, S. RNA editing in the Wilms’ tumor susceptibility gene, WT1. Genes Dev. 1994, 8, 720–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajaddod, M.; Jantsch, M.F.; Licht, K. The dynamic epitranscriptome: A to I editing modulates genetic information. Chromosoma 2016, 125, 51–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohn, W.E.; Volkin, E. Nucleoside-5′-phosphates from ribonucleic acid. Nature 1951, 167, 483–484. [Google Scholar] [CrossRef]
- Zhao, Y.; Dunker, W.; Yu, Y.T.; Karijolich, J. The role of noncoding RNA pseudouridylation in nuclear gene expression events. Front. Bioeng. Biotechnol. 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karijolich, J.; Yi, C.; Yu, Y.T. Transcriptome-wide dynamics of RNA pseudouridylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 581–585. [Google Scholar] [CrossRef] [Green Version]
- Decatur, W.A.; Fournier, M.J. rRNA modifications and ribosome function. Trends Biochem. Sci. 2002, 27, 344–351. [Google Scholar] [CrossRef]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Hsu, P.J.; Chen, Y.S.; Yang, Y.G. Dynamic transcriptomic m6A decoration: Writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018, 28, 616–624. [Google Scholar] [CrossRef] [Green Version]
- Jonkhout, N.; Tran, J.; Smith, M.A.; Schonrock, N.; Mattick, J.S.; Novoa, E.M. The RNA modification landscape in human disease. RNA 2017, 23, 1754–1769. [Google Scholar]
- DAVIS, F.F.; ALLEN, F.W. Ribonucleic acids from yeast which contain a fifth nucleotide. J. Biol. Chem. 1957, 227, 907–915. [Google Scholar]
- Thüring, K.; Schmid, K.; Keller, P.; Helm, M. Analysis of RNA modifications by liquid chromatography–tandem mass spectrometry. Methods 2016, 107, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xiong, X.; Yi, C. Epitranscriptome sequencing technologies: Decoding RNA modifications. Nat. Methods 2016, 14, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Minervini, C.F.; Cumbo, C.; Orsini, P.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. Nanopore Sequencing in Blood Diseases: A Wide Range of Opportunities. Front. Genet. 2020, 11, 76. [Google Scholar] [CrossRef] [PubMed]
- Minervini, C.F.; Cumbo, C.; Orsini, P.; Brunetti, C.; Anelli, L.; Zagaria, A.; Minervini, A.; Casieri, P.; Coccaro, N.; Tota, G.; et al. TP53 gene mutation analysis in chronic lymphocytic leukemia by nanopore MinION sequencing. Diagn. Pathol. 2016, 11, 96. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Begik, O.; Lucas, M.C.; Ramirez, J.M.; Mason, C.E.; Wiener, D.; Schwartz, S.; Mattick, J.S.; Smith, M.A.; Novoa, E.M. Accurate detection of m6A RNA modifications in native RNA sequences. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.M.; Jain, M.; Mulroney, L.; Garalde, D.R.; Akeson, M. Reading canonical and modified nucleobases in 16S ribosomal RNA using nanopore native RNA sequencing. PLoS ONE 2019, 14, e0216709. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Vu, L.P.; Pickering, B.F.; Cheng, Y.; Zaccara, S.; Nguyen, D.; Minuesa, G.; Chou, T.; Chow, A.; Saletore, Y.; Mackay, M.; et al. The N 6 -methyladenosine (m 6 A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017, 23, 1369–1376. [Google Scholar] [CrossRef]
- Weng, H.; Huang, H.; Wu, H.; Qin, X.; Zhao, B.S.; Dong, L.; Shi, H.; Skibbe, J.; Shen, C.; Hu, C.; et al. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m6A Modification. Cell Stem Cell 2018, 22, 191–205. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Renda, M.J.; Wang, L.; Cheng, E.-C.; Niu, C.; Morris, S.W.; Chi, A.S.; Krause, D.S. Rbm15 modulates Notch-induced transcriptional activation and affects myeloid differentiation. Mol. Cell. Biol. 2007, 27, 3056–3064. [Google Scholar] [CrossRef] [Green Version]
- Tuorto, F.; Herbst, F.; Alerasool, N.; Bender, S.; Popp, O.; Federico, G.; Reitter, S.; Liebers, R.; Stoecklin, G.; Gröne, H.; et al. The tRNA methyltransferase Dnmt2 is required for accurate polypeptide synthesis during haematopoiesis. EMBO J. 2015, 34, 2350–2362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncalves, K.A.; Silberstein, L.; Li, S.; Severe, N.; Hu, M.G.; Yang, H.; Scadden, D.T.; Hu, G. fu Angiogenin Promotes Hematopoietic Regeneration by Dichotomously Regulating Quiescence of Stem and Progenitor Cells. Cell 2016, 166, 894–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzzi, N.; Cieśla, M.; Ngoc, P.C.T.; Lang, S.; Arora, S.; Dimitriou, M.; Pimková, K.; Sommarin, M.N.E.; Munita, R.; Lubas, M.; et al. Pseudouridylation of tRNA-Derived Fragments Steers Translational Control in Stem Cells. Cell 2018, 173, 1204–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiou, N.T.; Kageyama, R.; Ansel, K.M. Selective Export into Extracellular Vesicles and Function of tRNA Fragments during T Cell Activation. Cell Rep. 2018, 25, 3356–3370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcu-Malina, V.; Goldberg, S.; Vax, E.; Amariglio, N.; Goldstein, I.; Rechavi, G. ADAR1 is vital for B cell lineage development in the mouse bone marrow. Oncotarget 2016, 7, 54370–54379. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Khillan, J.; Gadue, P.; Nishikura, K. Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science 2000, 290, 1765–1768. [Google Scholar] [CrossRef]
- Hartner, J.C.; Walkley, C.R.; Lu, J.; Orkin, S.H. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat. Immunol. 2009, 10, 109–115. [Google Scholar] [CrossRef]
- Essers, M.A.G.; Offner, S.; Blanco-Bose, W.E.; Waibler, Z.; Kalinke, U.; Duchosal, M.A.; Trumpp, A. IFNα activates dormant haematopoietic stem cells in vivo. Nature 2009, 458, 904–908. [Google Scholar] [CrossRef]
- Weng, H.; Huang, H.; Chen, J. RNA N 6-Methyladenosine Modification in Normal and Malignant Hematopoiesis. Adv. Exp. Med. Biol. 2019, 1143, 75–93. [Google Scholar]
- Ianniello, Z.; Paiardini, A.; Fatica, A. N6-Methyladenosine (M6A): A promising new molecular target in acute myeloid leukemia. Front. Oncol. 2019, 9, 251. [Google Scholar] [CrossRef] [Green Version]
- Martin, G.H.; Park, C.Y. Meddling with METTLs in Normal and Leukemia Stem Cells. Cell Stem Cell 2018, 139–141. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, I.; Tzelepis, K.; Pandolfini, L.; Shi, J.; Millán-Zambrano, G.; Robson, S.C.; Aspris, D.; Migliori, V.; Bannister, A.J.; Han, N.; et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature 2017, 552, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Bansal, H.; Yihua, Q.; Iyer, S.P.; Ganapathy, S.; Proia, D.; Penalva, L.O.; Uren, P.J.; Suresh, U.; Carew, J.S.; Karnad, A.B.; et al. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia 2014, 28, 1171–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorci, M.; Ianniello, Z.; Cruciani, S.; Larivera, S.; Ginistrelli, L.C.; Capuano, E.; Marchioni, M.; Fazi, F.; Fatica, A. METTL3 regulates WTAP protein homeostasis. Cell Death Dis. 2018, 9, 796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N6-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef] [Green Version]
- Church, C.; Moir, L.; McMurray, F.; Girard, C.; Banks, G.T.; Teboul, L.; Wells, S.; Brüning, J.C.; Nolan, P.M.; Ashcroft, F.M.; et al. Overexpression of Fto leads to increased food intake and results in obesity. Nat. Genet. 2010, 42, 1086–1092. [Google Scholar] [CrossRef]
- Söderberg, K.C.; Kaprio, J.; Verkasalo, P.K.; Pukkala, E.; Koskenvuo, M.; Lundqvist, E.; Feychting, M. Overweight, obesity and risk of haematological malignancies: A cohort study of Swedish and Finnish twins. Eur. J. Cancer 2009, 45, 1232–1238. [Google Scholar] [CrossRef]
- Castillo, J.J.; Mull, N.; Reagan, J.L.; Nemr, S.; Mitri, J. Increased incidence of non-Hodgkin lymphoma, leukemia, and myeloma in patients with diabetes mellitus type 2: A meta-analysis of observational studies. Blood 2012, 119, 4845–4850. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Chen, Q.; Wang, L.; Ke, D.; Yuan, Z. Association between FTO gene polymorphism and cancer risk: Evidence from 16,277 cases and 31,153 controls. Tumour Biol. 2012, 33, 1237–1243. [Google Scholar] [CrossRef]
- Paris, J.; Morgan, M.; Campos, J.; Spencer, G.J.; Shmakova, A.; Ivanova, I.; Mapperley, C.; Lawson, H.; Wotherspoon, D.A.; Sepulveda, C.; et al. Targeting the RNA m6A Reader YTHDF2 Selectively Compromises Cancer Stem Cells in Acute Myeloid Leukemia. Cell Stem Cell 2019, 25, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; He, X.; Hu, J.; Yang, P.; Liu, C.; Wang, J.; An, R.; Zhen, J.; Pang, M.; Hu, K.; et al. Dysregulation of N6-methyladenosine regulators predicts poor patient survival in mantle cell lymphoma. Oncol. Lett. 2019, 18, 3682–3690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, F.; Singh, R.K.; Pei, Y.; Zhang, S.; Sun, K.; Robertson, E.S. EBV epitranscriptome reprogramming by METTL14 is critical for viral-associated tumorigenesis. PLoS Pathog. 2019, 15, e1007796. [Google Scholar] [CrossRef] [PubMed]
- Bazot, Q.; Paschos, K.; Allday, M.J. Epstein-Barr Virus (EBV) Latent Protein EBNA3A Directly Targets and Silences the STK39 Gene in B Cells Infected by EBV. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowling, V.H.; Turner, S.A.; Cole, M.D. Burkitt’s lymphoma-associated c-Myc mutations converge on a dramatically altered target gene response and implicate Nol5a/Nop56 in oncogenesis. Oncogene 2014, 33, 3519–3527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronchetti, D.; Todoerti, K.; Tuana, G.; Agnelli, L.; Mosca, L.; Lionetti, M.; Fabris, S.; Colapietro, P.; Miozzo, M.; Ferrarini, M.; et al. The expression pattern of small nucleolar and small Cajal body-specific RNAs characterizes distinct molecular subtypes of multiple myeloma. Blood Cancer J. 2012, 2, e96. [Google Scholar] [CrossRef]
- Ronchetti, D.; Mosca, L.; Cutrona, G.; Tuana, G.; Gentile, M.; Fabris, S.; Agnelli, L.; Ciceri, G.; Matis, S.; Massucco, C.; et al. Small nucleolar RNAs as new biomarkers in chronic lymphocytic leukemia. BMC Med. Genom. 2013, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Valleron, W.; Ysebaert, L.; Berquet, L.; Fataccioli, V.; Quelen, C.; Martin, A.; Parrens, M.; Lamant, L.; De Leval, L.; Gisselbrecht, C.; et al. Small nucleolar RNA expression profiling identifies potential prognostic markers in peripheral T-cell lymphoma. Blood 2012, 120, 3997–4005. [Google Scholar] [CrossRef]
- Steinman, R.A.; Yang, Q.; Gasparetto, M.; Robinson, L.J.; Liu, X.; Lenzner, D.E.; Hou, J.; Smith, C.; Wang, Q. Deletion of the RNA-editing enzyme ADAR1 causes regression of established chronic myelogenous leukemia in mice. Int. J. Cancer 2013, 132, 1741–1750. [Google Scholar] [CrossRef]
- Zipeto, M.A.; Court, A.C.; Sadarangani, A.; Delos Santos, N.P.; Balaian, L.; Chun, H.J.; Pineda, G.; Morris, S.R.; Mason, C.N.; Geron, I.; et al. ADAR1 Activation Drives Leukemia Stem Cell Self-Renewal by Impairing Let-7 Biogenesis. Cell Stem Cell 2016, 19, 177–191. [Google Scholar] [CrossRef]
- Cully, M. Chemical inhibitors make their RNA epigenetic mark. Nat. Rev. Drug Discov. 2019, 18, 892–894. [Google Scholar] [CrossRef]
- Huang, Y.; Su, R.; Sheng, Y.; Dong, L.; Dong, Z.; Xu, H.; Ni, T.; Zhang, Z.S.; Zhang, T.; Li, C.; et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 677–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Werf, I.; Jamieson, C. The Yin and Yang of RNA Methylation: An Imbalance of Erasers Enhances Sensitivity to FTO Demethylase Small-Molecule Targeting in Leukemia Stem Cells. Cancer Cell 2019, 35, 540–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobzhansky, T. Nothing in Biology Makes Sense except in the Light of Evolution. Am. Biol. Teach. 1973, 35, 125–129. [Google Scholar] [CrossRef]
- Matzke, M.A.; Mette, M.F.; Aufsatz, W.; Jakowitsch, J.; Matzke, A.J.M. Host defenses to parasitic sequences and the evolution of epigenetic control mechanisms. Genetica 1999, 107, 271–287. [Google Scholar] [CrossRef]
- Polson, A.G.; Bass, B.L. Preferential selection of adenosines for modification by double-stranded RNA adenosine deaminase. EMBO J. 1994, 13, 5701–5711. [Google Scholar] [CrossRef]
- O’Connell, M.A.; Mannion, N.M.; Keegan, L.P. The Epitranscriptome and Innate Immunity. PLoS Genet. 2015, 11, e1005687. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.D.; Öhman, M. ADAR1 editing and its role in cancer. Genes 2019, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- Ishizuka, J.J.; Manguso, R.T.; Cheruiyot, C.K.; Bi, K.; Panda, A.; Iracheta-Vellve, A.; Miller, B.C.; Du, P.P.; Yates, K.B.; Dubrot, J.; et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 2019, 565, 43–48. [Google Scholar] [CrossRef]
- Vitali, P.; Scadden, A.D.J. Double-stranded RNAs containing multiple IU pairs are sufficient to suppress interferon induction and apoptosis. Nat. Struct. Mol. Biol. 2010, 17, 1043–1050. [Google Scholar] [CrossRef]
- Darnell, R.R.; Shengdong, K.E.; Darnell, J.E. Pre-mRNA processing includes N6 methylation of adenosine residues that are retained in mRNA exons and the fallacy of “RNA epigenetics. ” RNA 2018, 24, 262–267. [Google Scholar] [CrossRef] [Green Version]
- Shulman, Z.; Stern-Ginossar, N. The RNA modification N6-methyladenosine as a novel regulator of the immune system. Nat. Immunol. 2020, 21, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Zhang, Z.; Xue, M.; Zhao, B.S.; Harder, O.; Li, A.; Liang, X.; Gao, T.Z.; Xu, Y.; Zhou, J.; et al. N 6-methyladenosine modification enables viral RNA to escape recognition by RNA sensor RIG-I. Nat. Microbiol. 2020, 5, 584–598. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Modification | Writers | Readers | Eraser |
|---|---|---|---|
| m6A | METTL3 | ||
| METTL14 | |||
| WTAP | YTH domain proteins | FTO | |
| RBM15 | IGF2BP family | ALKBH5 | |
| RBM15B | HNRNPA2B1 | ||
| KIAA1429 | |||
| m5C | NSUN1/2/3/4/5 | ALYREF | Still not identified |
| DNMT2 | YBX1 | ||
| m1A | TRMT6/10C/61A | YTHDF1/3 | ALKBH1/H3 |
| YTHDC1 | |||
| PseudoUridine | PUS genes | Not identified | Not identified possible irreversible modification |
| DKC1 | |||
| A-to-I Editing | ADAR genes | Not identified | Not identified possible irreversible modification |
| ADAT genes | |||
| C-to-U Editing | AID/APOBEC gene family | Not identified | Not identified possible irreversible modification |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minervini, C.F.; Parciante, E.; Impera, L.; Anelli, L.; Zagaria, A.; Specchia, G.; Musto, P.; Albano, F. Epitranscriptomics in Normal and Malignant Hematopoiesis. Int. J. Mol. Sci. 2020, 21, 6578. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186578
Minervini CF, Parciante E, Impera L, Anelli L, Zagaria A, Specchia G, Musto P, Albano F. Epitranscriptomics in Normal and Malignant Hematopoiesis. International Journal of Molecular Sciences. 2020; 21(18):6578. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186578
Chicago/Turabian StyleMinervini, Crescenzio Francesco, Elisa Parciante, Luciana Impera, Luisa Anelli, Antonella Zagaria, Giorgina Specchia, Pellegrino Musto, and Francesco Albano. 2020. "Epitranscriptomics in Normal and Malignant Hematopoiesis" International Journal of Molecular Sciences 21, no. 18: 6578. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186578