Consensus-Based Pharmacophore Mapping for New Set of N-(disubstituted-phenyl)-3-hydroxyl-naphthalene-2-carboxamides

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
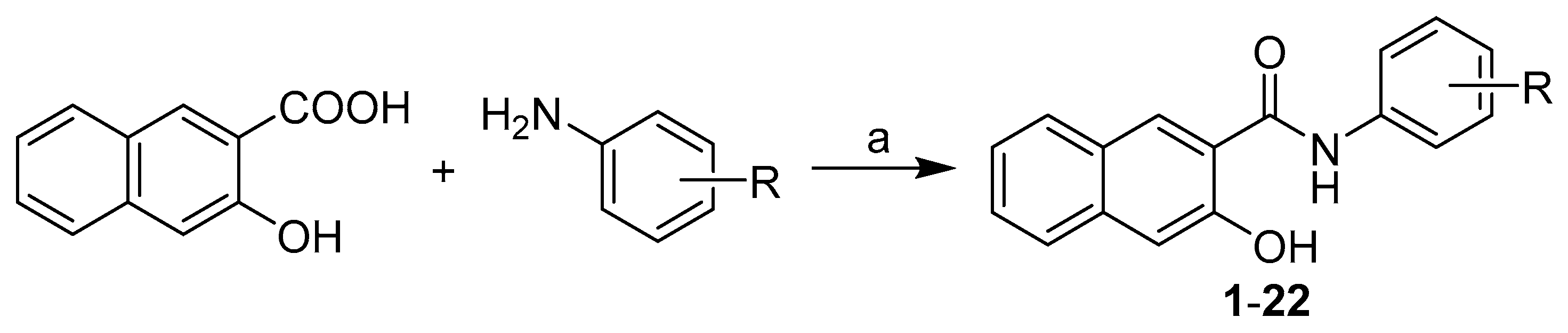
2.1. Synthesis and Physicochemical Properties
2.2. In Vitro Antimicrobial Activity
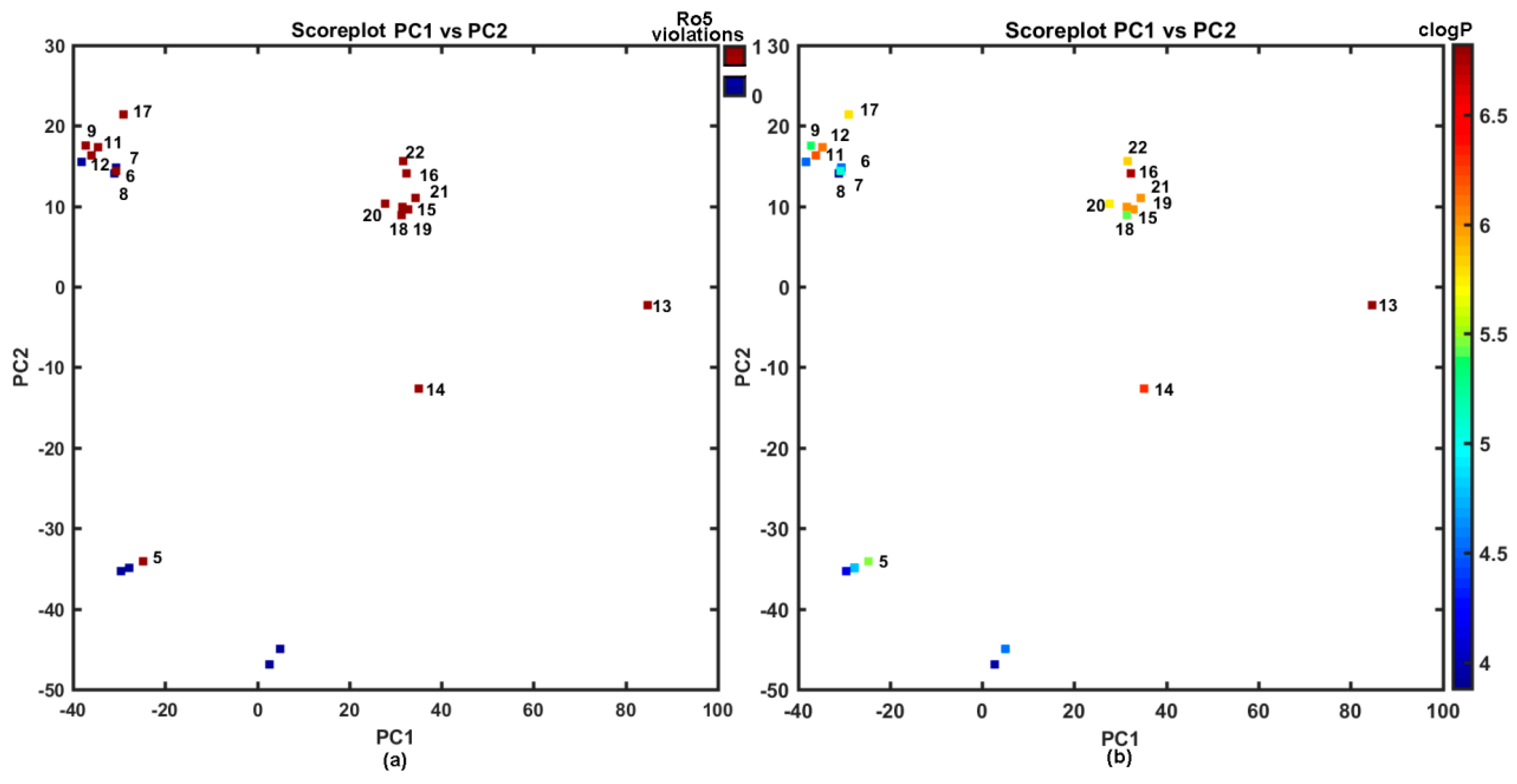
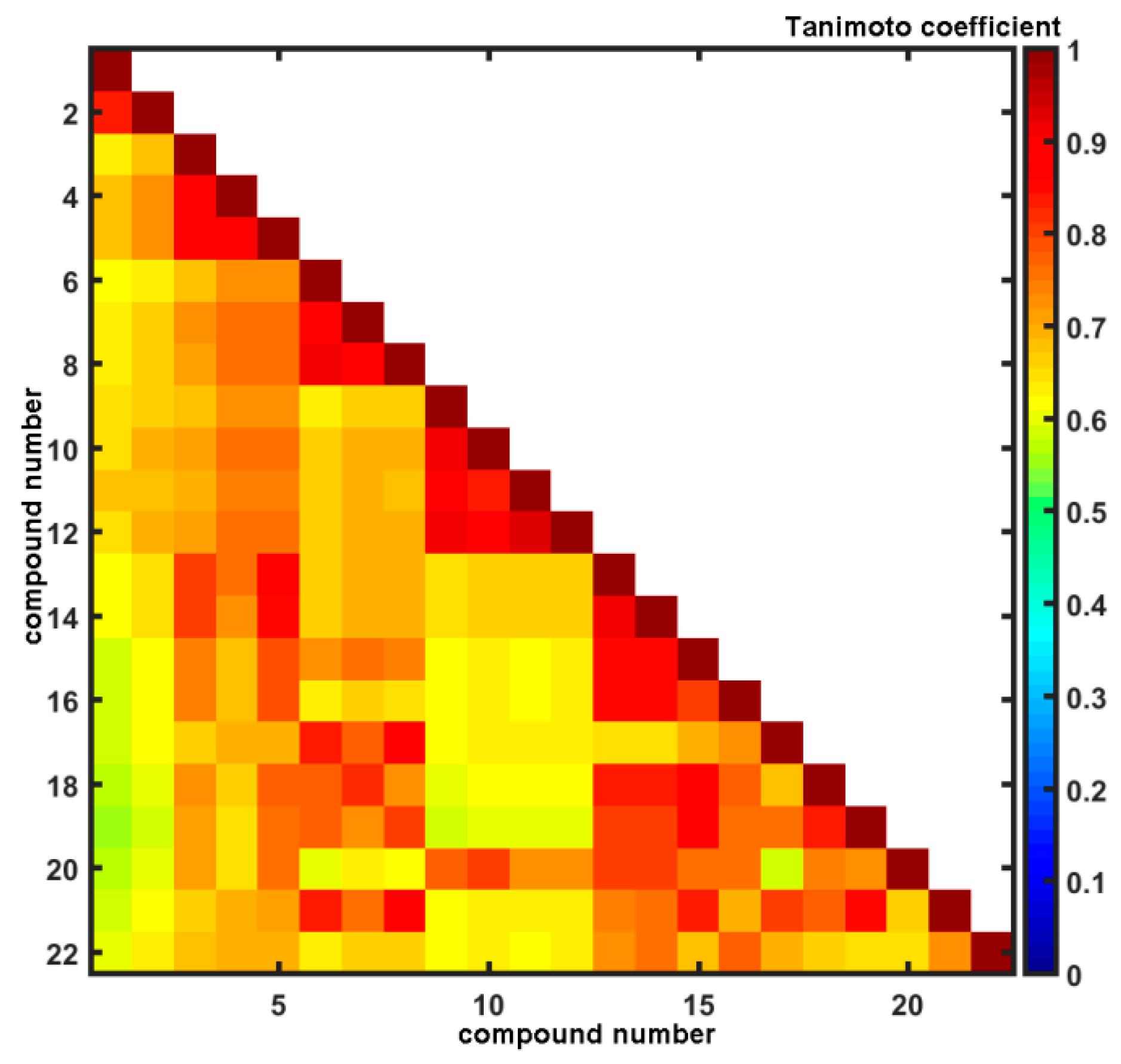
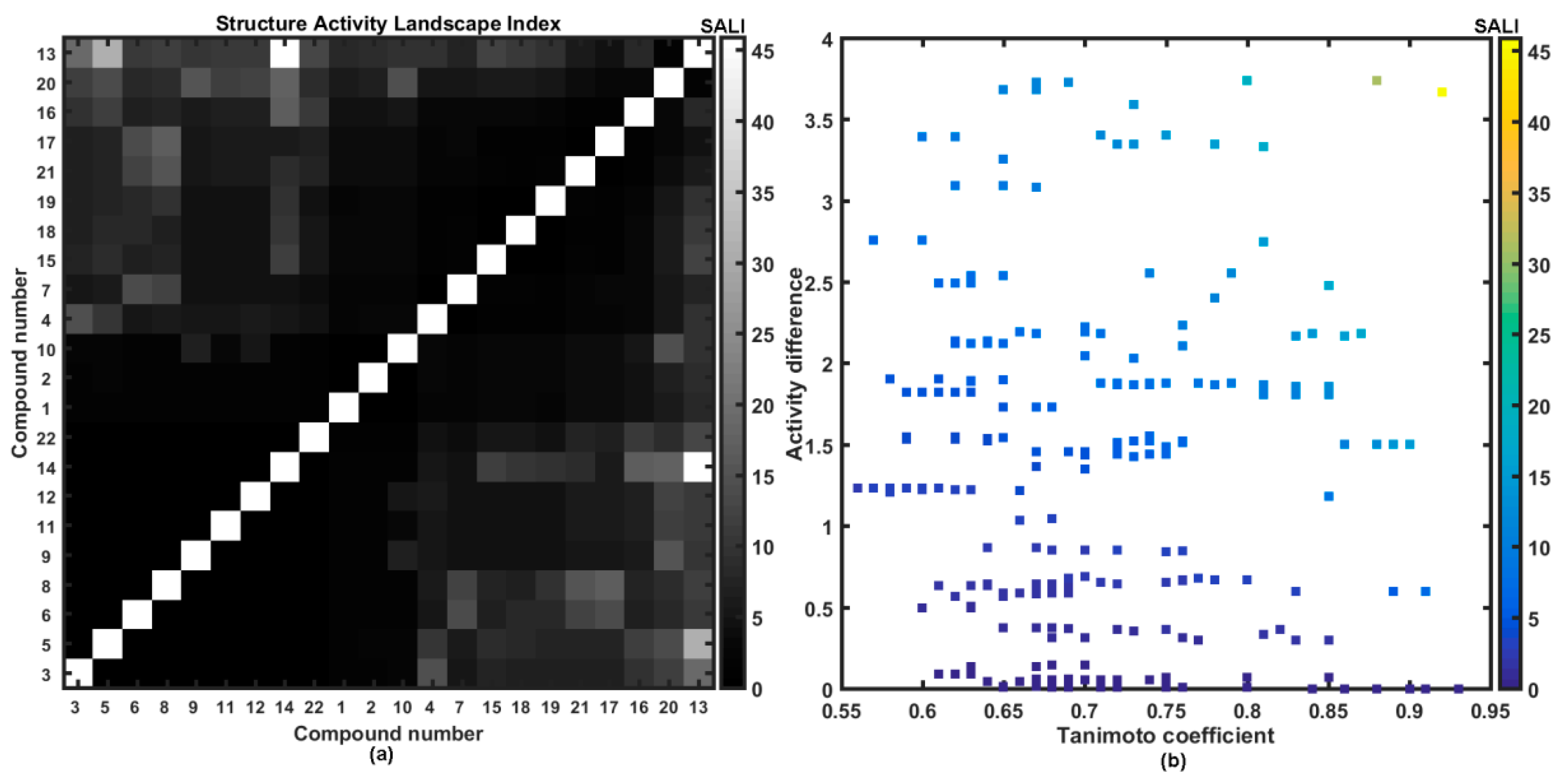
2.3. Molecular Similarity Assessment
2.4. Probability-Driven Pharmacophore Mapping
2.5. Advanced Antimicrobial Evaluation
2.5.1. Combined Effect
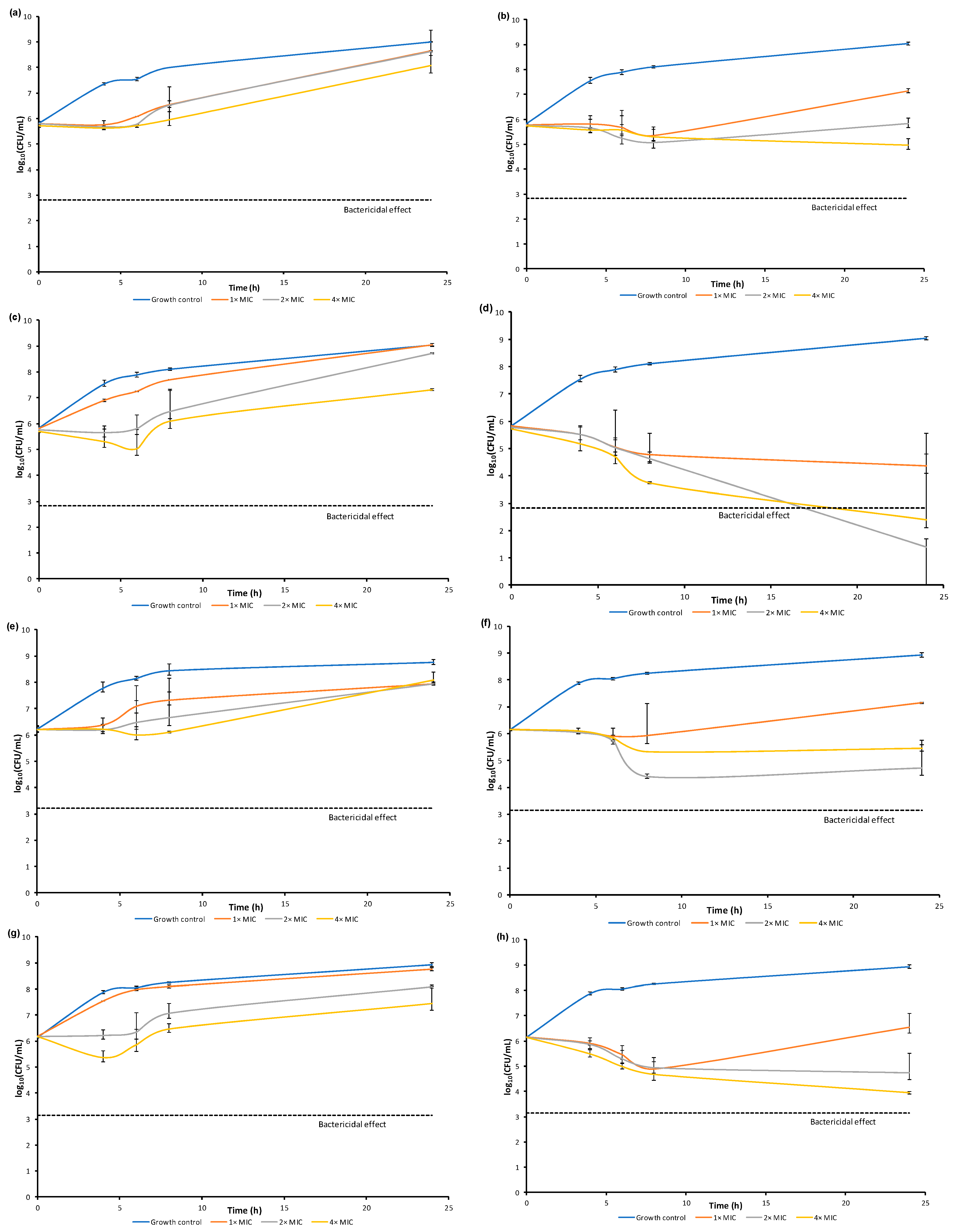
2.5.2. Time-Kill Assay
3. Materials and Methods
3.1. General Methods
3.2. Chemistry
General Procedure for Synthesis of Carboxamide Derivatives 1–22
3.3. Biological Testing
3.3.1. In Vitro Antibacterial Evaluation
3.3.2. In Vitro Antimycobacterial Assessment
3.3.3. MTT Assay
3.3.4. Combined Effect with Clinically Used Drugs
3.3.5. Dynamic of Antibacterial Activity
3.4. Theoretical Calculations
3.4.1. Model Building and Molecular Modeling
3.4.2. Similarity-Driven Activity Landscape
3.4.3. Selection-Driven Surface Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Valent, P.; Groner, B.; Schumacher, U.; Superti-Furga, G.; Busslinger, M.; Kralovics, R.; Zielinski, C.; Penninger, J.M.; Kerjaschki, D.; Stingl, G.; et al. Paul Ehrlich (1854–1915) and his contributions to the foundation and birth of translational medicine. J. Innate Immun. 2016, 8, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Devillers, J. Methods for building QSARs. Methods Mol. Biol. 2013, 930, 3–27. [Google Scholar] [PubMed]
- Bak, A.; Pizova, H.; Kozik, V.; Vorcakova, K.; Kos, J.; Treml, J.; Odehnalova, K.; Oravec, M.; Imramovsky, A.; Bobal, P.; et al. SAR-mediated similarity assessment of the property profile for new, silicon-based AChE/BChE inhibitors. Int. J. Mol. Sci. 2019, 20, 5385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colquhoun, D. The quantitative analysis of drug–receptor interactions: A short history. Trends Pharmacol. Sci. 2006, 27, 149–157. [Google Scholar] [CrossRef]
- Bak, A.; Kozik, V.; Malik, I.; Jampilek, J.; Smolinski, A. Probability-driven 3D pharmacophore mapping of antimycobacterial potential of hybrid molecules combining phenylcarbamoyloxy and N-arylpiperazine fragments. SAR QSAR Environ. Res. 2018, 29, 801–821. [Google Scholar] [CrossRef]
- Hann, M.; Oprea, T. Pursuing the leadlikeness concept in pharmaceutical research. Curr. Opin. Chem. Biol. 2004, 8, 255–263. [Google Scholar] [CrossRef]
- Grammatica, P. Principles of QSAR models validation: Internal and external. Qsar Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Mod. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Merlot, C.; Domine, D.; Cleva, C.; Church, D.J. Chemical substructures in drug discovery. Drug Discov. Today 2003, 8, 594–602. [Google Scholar] [CrossRef]
- Reymond, J.L.; van Deursen, R.; Blum, L.C.; Ruddigkeit, L. Chemical space as a source for new drugs. MedChemComm 2010, 1, 30–38. [Google Scholar] [CrossRef]
- Peltason, L.; Bajorath, J. Systematic computational analysis of structure-activity relationships: Concepts, challenges and recent advances. Future Med. Chem. 2009, 1, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Polanski, J.; Bak, A.; Gieleciak, R.; Magdziarz, T. Modeling robust QSAR. J. Chem. Inf. Model. 2003, 46, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Bak, A.; Kozik, V.; Smolinski, A.; Jampilek, J. Multidimensional (3D/4D-QSAR) probability-guided pharmacophore mapping: Investigation of activity profile for a series of drug absorption promoters. RSC Adv. 2016, 6, 76183–76205. [Google Scholar] [CrossRef]
- Kubinyi, H. Hansch Analysis and Related Approaches; Wiley-VCH Verlag GmbH: Weinheim, Germany, 1993. [Google Scholar]
- Maggiora, G.M.; Shanmugasundaram, V. Molecular similarity measures. Methods Mol. Biol. 2011, 672, 39–100. [Google Scholar]
- Van de Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Lopez-Lopez, E.; Prieto-Martínez, F.D.; Medina-Franco, J.L. Activity landscape and molecular modeling to explore the SAR of dual epigenetic inhibitors: A focus on G9a and DNMT1. Molecules 2018, 23, 3282. [Google Scholar] [CrossRef] [Green Version]
- Guha, R.; Van Drie, J.H. Assessing how well a modeling protocol captures a structure—Activity landscape. J. Chem. Inf. Model. 2008, 48, 1716–1728. [Google Scholar] [CrossRef]
- Bajorath, J.; Peltason, L.; Wawer, M.; Guha, R.; Lajiness, M.S.; Van Drie, J.H. Navigating structure—Activity landscapes. Drug Discov. Today 2009, 14, 698–705. [Google Scholar] [CrossRef]
- Hu, H.; Stumpfe, D.; Bajorath, J. Systematic identification of target set-dependent activity cliffs. Future Sci. OA 2019, 5, 363. [Google Scholar] [CrossRef] [Green Version]
- Gonec, T.; Kos, J.; Zadrazilova, I.; Pesko, M.; Keltosova, S.; Tengler, J.; Bobal, P.; Kollar, P.; Cizek, A.; Kralova, K.; et al. Antimycobacterial and herbicidal activity of ring-substituted 1-hydroxynaphthalene-2-carboxanilides. Bioorg. Med. Chem. 2013, 21, 6531–6541. [Google Scholar] [CrossRef]
- Gonec, T.; Kos, J.; Zadrazilova, I.; Pesko, M.; Govender, R.; Keltosova, S.; Chambel, B.; Pereira, D.; Kollar, P.; Imramovsky, A.; et al. Antibacterial and herbicidal activity of ring-substituted 2-hydroxynaphthalene-1-carboxanilides. Molecules 2013, 18, 9397–9419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonec, T.; Zadrazilova, I.; Nevin, E.; Kauerova, T.; Pesko, M.; Kos, J.; Oravec, M.; Kollar, P.; Coffey, A.; O’Mahony, J.; et al. Synthesis and biological evaluation of N-alkoxyphenyl-3-hydroxynaphthalene- 2-carboxanilides. Molecules 2015, 20, 9767–9787. [Google Scholar] [CrossRef] [PubMed]
- Kos, J.; Nevin, E.; Soral, M.; Kushkevych, I.; Gonec, T.; Bobal, P.; Kollar, P.; Coffey, A.; O’Mahony, J.; Liptaj, T.; et al. Synthesis and antimycobacterial properties of ring-substituted 6-hydroxynaphthalene- 2-carboxanilides. Bioorg. Med. Chem. 2015, 23, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Gonec, T.; Pospisilova, S.; Kauerova, T.; Kos, J.; Dohanosova, J.; Oravec, M.; Kollar, P.; Coffey, A.; Liptaj, T.; Cizek, A.; et al. N-Alkoxyphenylhydroxynaphthalenecarboxamides and their antimycobacterial activity. Molecules 2016, 21, 1068. [Google Scholar] [CrossRef]
- Michnova, H.; Pospisilova, S.; Gonec, T.; Kapustikova, I.; Kollar, P.; Kozik, V.; Musiol, R.; Jendrzejewska, I.; Vanco, J.; Travnicek, Z.; et al. Bioactivity of methoxylated and methylated 1-hydroxynaphthalene-2-carboxanilides: Comparative molecular surface analysis. Molecules 2019, 24, 2991. [Google Scholar] [CrossRef] [Green Version]
- Kauerova, T.; Kos, J.; Gonec, T.; Jampilek, J.; Kollar, P. Antiproliferative and pro-apoptotic effect of novel nitro-substituted hydroxynaphthanilides on human cancer cell lines. Int. J. Mol. Sci. 2016, 17, 1219. [Google Scholar] [CrossRef]
- Kauerova, T.; Gonec, T.; Jampilek, J.; Hafner, S.; Gaiser, A.K.; Syrovets, T.; Fedr, R.; Soucek, K.; Kollar, P. Ring-substituted 1-hydroxynaphthalene-2-carboxanilides inhibit proliferation and trigger mitochondria-mediated apoptosis. Int. J. Mol. Sci. 2020, 21, 3416. [Google Scholar] [CrossRef]
- Imramovsky, A.; Pesko, M.; Kralova, K.; Vejsova, M.; Stolarikova, J.; Vinsova, J.; Jampilek, J. Investigating spectrum of biological activity of 4- and 5-chloro-2-hydroxy-N-[2-(arylamino)-1-alkyl-2-oxoethyl]- benzamides. Molecules 2011, 16, 2414–2430. [Google Scholar] [CrossRef] [Green Version]
- Pauk, K.; Zadrazilova, I.; Imramovsky, A.; Vinsova, J.; Pokorna, M.; Masarikova, M.; Cizek, A.; Jampilek, J. New derivatives of salicylamides: Preparation and antimicrobial activity against various bacterial species. Bioorg. Med. Chem. 2013, 21, 6574–6581. [Google Scholar] [CrossRef]
- Kos, J.; Zadrazilova, I.; Pesko, M.; Keltosova, S.; Tengler, J.; Gonec, T.; Bobal, P.; Kauerova, T.; Oravec, M.; Kollar, P.; et al. Antibacterial and herbicidal activity of ring-substituted 3-hydroxynaphthalene- 2-carboxanilides. Molecules 2013, 18, 7977–7997. [Google Scholar] [CrossRef] [Green Version]
- Kos, J.; Kapustikova, I.; Clements, C.; Gray, A.I.; Jampilek, J. 3-Hydroxynaphthalene-2-carboxanilides and their antitrypanosomal activity. Monatsh. Chem. 2018, 149, 887–892. [Google Scholar] [CrossRef]
- DrugBank—Niclosamide. Available online: https://www.drugbank.ca/drugs/DB06803 (accessed on 27 August 2020).
- Gajdar, J.; Tsami, K.; Michnova, H.; Gonec, T.; Brazdova, M.; Soldanova, Z.; Fojta, M.; Jampilek, J.; Barek, J.; Fischer, J. Electrochemistry of ring-substituted 1-hydroxynaphthalene-2-carboxanilides: Relation to structure and biological activity. Electrochim. Acta 2020, 332, 135485. [Google Scholar] [CrossRef]
- Pospisilova, S.; Kos, J.; Michnova, H.; Kapustikova, I.; Strharsky, T.; Oravec, M.; Moricz, A.M.; Bakonyi, J.; Kauerova, T.; Kollar, P.; et al. Synthesis and spectrum of biological activities of novel N-arylcinnamamides. Int. J. Mol. Sci. 2018, 19, 2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spaczynska, E.; Mrozek-Wilczkiewicz, A.; Malarz, K.; Kos, J.; Gonec, T.; Oravec, M.; Gawecki, R.; Bak, A.; Dohanosova, J.; Kapustikova, I.; et al. Design and synthesis of anticancer 1-hydroxynaphthalene-2-carboxanilides with p53 independent mechanism of action. Sci. Rep. 2019, 9, 6387. [Google Scholar] [CrossRef] [Green Version]
- Gonec, T.; Kos, J.; Pesko, M.; Dohanosova, J.; Oravec, M.; Liptaj, T.; Kralova, K.; Jampilek, J. Halogenated 1-hydroxynaphthalene-2-carboxanilides affecting photosynthetic electron transport in photosystem II. Molecules 2017, 22, 1709. [Google Scholar] [CrossRef] [Green Version]
- Likus-Cieslik, J.; Smolinski, A.; Pietrzykowski, M.; Bak, A. Sulphur contamination impact on seasonal and surface water chemistry on a reforested area of a former sulphur mine. Land Degrad. Dev. 2019, 30, 212–225. [Google Scholar] [CrossRef]
- Bak, A.; Kozik, V.; Smolinski, A.; Jampilek, J. In silico estimation of basic activity-relevant parameters for a set of drug absorption promoters. SAR QSAR Environ. Res. 2017, 28, 427–449. [Google Scholar] [CrossRef]
- Hann, M.M.; Keserü, G.M. Finding the sweet spot: The role of nature and nurture in medicinal chemistry. Nat. Rev. Drug Discov. 2012, 11, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Zadrazilova, I.; Pospisilova, S.; Masarikova, M.; Imramovsky, A.; Ferriz, J.M.; Vinsova, J.; Cizek, A.; Jampilek, J. Salicylanilide carbamates: Promising antibacterial agents with high in vitro activity against methicillin-resistant Staphylococcus aureus (MRSA). Eur. J. Pharm. Sci. 2015, 77, 197–207. [Google Scholar] [CrossRef]
- National Committee for Clinical Laboratory Standards. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 11th ed.; M07; NCCLS: Wayne, PA, USA, 2018.
- National Committee for Clinical Laboratory Standards. Susceptibility Testing of Mycobacteria, Nocardiae, and Other Aerobic Actinomycetes, 3rd ed.; Approved Standard, M24Approved Standard, M24; NCCLS: Wayne, PA, USA, 2020.
- Nubel, U.; Dordel, J.; Kurt, K.; Strommenger, B.; Westh, H.; Shukla, S.K.; Zemlickova, H.; Leblois, R.; Wirth, T.; Jombart, T.; et al. A timescale for evolution, population expansion, and spatial spread of an emerging clone of methicillin-resistant Staphylococcus aureus. PLoS Pathog. 2010, 6, e1000855. [Google Scholar] [CrossRef] [Green Version]
- Measuring Cell Viability/Cytotoxicity. Dojindo EU GmbH, Munich, Germany. Available online: https://www.dojindo.eu.com/Protocol/Dojindo-Cell-Proliferation-Protocol.pdf (accessed on 27 August 2020).
- Bueno, J. Antitubercular in vitro drug discovery: Tools for begin the search. In Understanding Tuberculosis—New Approaches to Fighting Against Drug Resistance; IntechOpen: Rijeka, Croatia, 2012; pp. 147–168. [Google Scholar]
- International Organization for Standardization. ISO 10993-5:2009 Biological Evaluation of Medical Devices Part 5: Tests for in Vitro Cytotoxicity; International Organization for Standardization: Geneva, Switzerland, 2009; last revision 2017. [Google Scholar]
- Jampilek, J. Design and discovery of new antibacterial agents: Advances, perspectives, challenges. Curr. Med. Chem. 2018, 25, 4972–5006. [Google Scholar] [CrossRef]
- Imramovsky, A.; Pesko, M.; Ferriz, J.M.; Kralova, K.; Vinsova, J.; Jampilek, J. Photosynthesis—Inhibiting efficiency of 4-chloro-2-(chlorophenylcarbamoyl)phenyl alkylcarbamates. Bioorg. Med. Chem. Lett. 2011, 21, 4564–4567. [Google Scholar] [CrossRef] [PubMed]
- Kralova, K.; Perina, M.; Waisser, K.; Jampilek, J. Structure-activity relationships of n-benzylsalicylamides for inhibition of photosynthetic electron transport. Med. Chem. 2015, 11, 156–164. [Google Scholar] [CrossRef]
- Gonec, T.; Kralova, K.; Pesko, M.; Jampilek, J. Antimycobacterial N-alkoxyphenylhydroxy- naphthalenecarboxamides affecting photosystem II. Bioorg. Med. Chem. Lett. 2017, 27, 1881–1885. [Google Scholar] [CrossRef] [PubMed]
- Kos, J.; Zadrazilova, I.; Nevin, E.; Soral, M.; Gonec, T.; Kollar, P.; Oravec, M.; Coffey, A.; O’Mahony, J.; Liptaj, T.; et al. Ring-substituted 8-hydroxyquinoline-2-carboxanilides as potential antimycobacterial agents. Bioorg. Med. Chem. 2015, 23, 4188–4196. [Google Scholar] [CrossRef] [PubMed]
- Jampilek, J.; Kralova, K.; Pesko, M.; Kos, J. Ring-substituted 8-hydroxyquinoline-2-carboxanilides as photosystem II inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 3862–3865. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2010. [Google Scholar]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Clark, D.E.; Pickett, S.E. Computational methods for the prediction of ‘drug-likeness’. Drug Discov. Today 2000, 5, 49–58. [Google Scholar] [CrossRef]
- Pizova, H.; Havelkova, M.; Stepankova, S.; Bak, A.; Kauerova, T.; Kozik, V.; Oravec, M.; Imramovsky, A.; Kollar, P.; Bobal, P.; et al. Proline-based carbamates as cholinesterase inhibitors. Molecules 2017, 22, 1969. [Google Scholar] [CrossRef] [Green Version]
- Holliday, J.D.; Salim, N.; Whittle, M.; Willett, P. Analysis and display of the size dependence of chemical similarity coefficients. J. Chem. Inf. Comput. Sci. 2003, 43, 819–828. [Google Scholar] [CrossRef]
- Gieleciak, R.; Magdziarz, T.; Bak, A.; Polanski, J. Modeling robust QSAR. 1. Coding molecules in 3D-QSAR—From a point to surface sectors and molecular volumes. J. Chem. Inf. Model. 2005, 45, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Stouch, T.R.; Kenyon, J.R.; Johnson, S.R.; Chen, X.Q.; Doweyko, A.; Li, Y. In silico ADME/Tox: Why models fail. J. Comput. Aided Mol. Des. 2003, 17, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Doweyko, A.M. QSAR: Dead or alive? J. Comput. Aided Mol. Des. 2008, 22, 81–89. [Google Scholar] [CrossRef]
- Bak, A.; Polanski, J. Modeling robust QSAR 3: SOM-4D-QSAR with iterative variable elimination IVE-PLS: Application to steroid, azo dye, and benzoic acid series. J. Chem. Inf. Model. 2007, 47, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Schwalbe, R.; Steele-Moore, L.; Goodwin, A.C. Antimicrobial Susceptibility Testing Protocols, 1st ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Bonapace, C.R.; Bosso, J.A.; Friedrich, L.V.; White, R.L. Comparison of methods of interpretation of checkerboard synergy testing. Diagn. Microbiol. Infect. Dis. 2002, 44, 363–366. [Google Scholar] [CrossRef]
- Abate, G.; Mshana, R.N.; Miorner, H. Evaluation of a colorimetric assay based on 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) for rapid detection of rifampicin resistance in Mycobacterium tuberculosis. Int. J. Tuberc. Lung Dis. 1998, 2, 1011–1016. [Google Scholar]
- Stanton, D.T. QSAR and QSPR model interpretation using partial least squares (PLS) analysis. Curr. Comput. Aided Drug Des. 2012, 8, 107–127. [Google Scholar] [CrossRef]
- Xie, X.Q.; Chen, J.Z. Data mining a small molecule drug screening representative subset from NIH PubChem. J. Chem. Inf. Model. 2008, 48, 465–475. [Google Scholar] [CrossRef]
- Polanski, J.; Gieleciak, R.; Magdziarz, T.; Bak, A. GRID formalism for the comparative molecular surface analysis: Application to the CoMFA benchmark steroids, azo dyes, and HEPT derivatives. J. Chem. Inf. Comput Sci. 2004, 44, 1423–1435. [Google Scholar] [CrossRef]
- Zupan, J.; Gasteiger, J. Neural Networks and Drug Design for Chemists, 2nd ed.; Wiley-VCH: Weinheim, Germany, 1999. [Google Scholar]
- Centner, V.; Massart, D.L.; de Noord, O.E.; de Jong, S.; Vandeginste, B.M.V.; Sterna, C. Elimination of uninformative variables for multivariate calibration. Anal. Chem. 1996, 68, 3851–3858. [Google Scholar] [CrossRef]
- Dearden, J.C.; Cronin, M.T.; Kaiser, K.L. How not to develop a quantitative structure-activity or structure-property relationship (QSAR/QSPR)? Sar Qsar Environ. Res. 2009, 20, 241–266. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| No. | R | MIC [μM] | ||||
| SA | MRSA 63718 | MRSA SA 630 | MRSA SA 3202 | MT | ||
| 1. | 2,5-OCH3 | 198 | 792 | 792 | 792 | 792 |
| 2. | 3,5-OCH3 | 198 | 792 | 792 | 792 | 792 |
| 3. | 2,5-CH3 | 879 | 879 | 879 | 879 | 256 |
| 4. | 2,6-CH3 | 27.4 | 54.9 | 27.4 | 27.4 | 54.9 |
| 5. | 3,5-CH3 | 879 | 879 | 879 | 879 | 110 |
| 6. | 2,5-F | 856 | 856 | 856 | 856 | 879 |
| 7. | 2,6-F | 26.7 | 856 | 53.4 | 107 | 107 |
| 8. | 3,5-F | 856 | 856 | 214 | 856 | 53.4 |
| 9. | 2,5-Cl | 770 | 770 | 770 | 770 | 770 |
| 10. | 2,6-Cl | 193 | 385 | 385 | 385 | 385 |
| 11. | 3,4-Cl | 770 | 770 | 770 | 770 | 770 |
| 12. | 3,5-Cl | 770 | 770 | 770 | 770 | 12.0 |
| 13. | 3,5-CF3 | 0.158 | 0.626 | 0.158 | 0.626 | 10.0 |
| 14. | 3-CF3-4-CH3 | 741 | 371 | 741 | 371 | 741 |
| 15. | 3-CF3-4-F | 11.4 | 11.4 | 11.4 | 11.4 | 22.9 |
| 16. | 3-CF3-4-Br | 2.43 | 4.78 | 4.78 | 4.78 | 9.75 |
| 17. | 3-F-4-Br | 5.54 | 5.54 | 5.54 | 5.54 | 11.1 |
| 18. | 5-CF3-2-F | 11.4 | 11.4 | 11.4 | 11.4 | 11.4 |
| 19. | 5-CF3-3-F | 11.4 | 11.4 | 11.4 | 11.4 | 11.4 |
| 20. | 5-CF3-2-Cl | 0.342 | 0.684 | 0.342 | 0.342 | 10.9 |
| 21. | 4-CF3-3-F | 5.72 | 5.72 | 5.72 | 5.72 | 11.4 |
| 22. | 4-CF3-2-Br | 624 | 624 | 624 | 624 | 624 |
| AMP | – | 5.72 | 45.8 | 45.8 | 45.8 | – |
| CPX | – | 0.751 | 48.3 | 48.3 | 48.3 | – |
| INH | – | – | – | – | – | 36.5 |
| RIF | – | – | – | – | – | 9.72 |
| Combination (MIC [μg/mL]) | FIC Index | Comb. Effect (MICs [μg/mL]) |
|---|---|---|
| Comp. 13 + CPX (0.125/128) | 1.000 | IND |
| Comp. 16 + CPX (8/128) | 0.375 | SYN 1/32 |
| Comp. 17 + CPX (1/128) | 0.500 | SYN 0.25/32 |
| Comp. 20 + CPX (0,5/128) | 1.004 | IND |
| Comp. 21 + CPX (0.125/128) | 0.75 | ADD 0.0625/32; 0.03125/64 |
| Comp. 13 + OXA (0.125/64) | 1.004 | IND |
| Comp. 16 + OXA (8/64) | 1.000 | IND |
| Comp. 17 + OXA (1/64) | 1.125 | IND |
| Comp. 20 + OXA (0,5/64) | 1.002 | IND |
| Comp. 21 + OXA (0,125/64) | 1.125 | IND |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bak, A.; Kos, J.; Michnova, H.; Gonec, T.; Pospisilova, S.; Kozik, V.; Cizek, A.; Smolinski, A.; Jampilek, J. Consensus-Based Pharmacophore Mapping for New Set of N-(disubstituted-phenyl)-3-hydroxyl-naphthalene-2-carboxamides. Int. J. Mol. Sci. 2020, 21, 6583. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186583
Bak A, Kos J, Michnova H, Gonec T, Pospisilova S, Kozik V, Cizek A, Smolinski A, Jampilek J. Consensus-Based Pharmacophore Mapping for New Set of N-(disubstituted-phenyl)-3-hydroxyl-naphthalene-2-carboxamides. International Journal of Molecular Sciences. 2020; 21(18):6583. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186583
Chicago/Turabian StyleBak, Andrzej, Jiri Kos, Hana Michnova, Tomas Gonec, Sarka Pospisilova, Violetta Kozik, Alois Cizek, Adam Smolinski, and Josef Jampilek. 2020. "Consensus-Based Pharmacophore Mapping for New Set of N-(disubstituted-phenyl)-3-hydroxyl-naphthalene-2-carboxamides" International Journal of Molecular Sciences 21, no. 18: 6583. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186583