The Hidden Fragility in the Heart of the Athletes: A Review of Genetic Biomarkers

 ,
,  , , and
, , and
Abstract
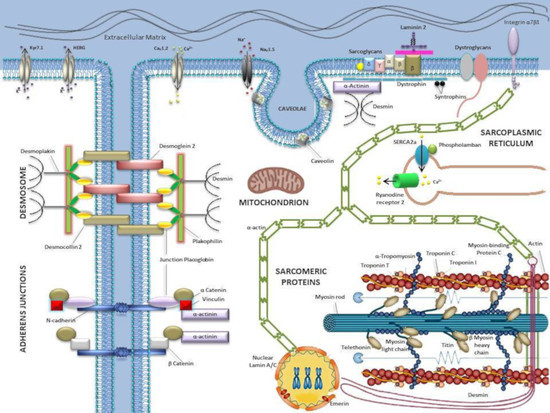
:
1. Introduction
2. Genetic Basis of Inherited Cardiac Channelopathies
2.1. Long QT Syndrome (LQTS)
2.2. Brugada Syndrome (BrS)
2.3. Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT)
3. Genetic Basis of Inherited Cardiomyopathies
3.1. Hypertrophic Cardiomyopathy (HCM)
3.2. Dilated Cardiomyopathy (DCM)
3.3. Restrictive Cardiomyopathy (RCM)
3.4. Arrhythmogenic Cardiomyopathy (ACM)
3.5. Left Ventricular Non-Compaction (LVNC)
4. Diagnostic Yield of Genetic Testing in Athletes
4.1. Evidence Acquisition: Database Searching, Study Eligibility and Inclusion/Exclusion Criteria
4.2. Evidence Synthesis
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACM | Arrhythmogenic cardiomyopathy |
| AHA/ACC | American Heart Association/American College of Cardiology |
| ARVC | Arrhythmogenic right ventricular cardiomyopathies |
| BRS | Brugada syndrome |
| CMR | Cardiac Magnetic Resonance |
| CPVT | Catecholaminergic polymorphic ventricular tachycardia |
| DCM | Dilated cardiomyopathy |
| ECG | Electrocardiogram |
| ESC | European Society of Cardiology |
| ESHG | European Society of Human Genetics |
| HCM | Hypertrophic cardiomyopathy |
| LQTS | Long QT syndrome |
| LVNC | Left ventricular non-compaction |
| MD | Mitochondrial disorders |
| MLPA | Multiplex Ligation-Dependent Probe Amplification |
| NGS | Next generation sequencing |
| NR | Not Reported |
| NSVT | Non-Sustained Ventricular Tachycardia |
| PRISMA | Preferred Reporting Items for Systematic Reviews and Meta-Analyses |
| QTc | corrected QT |
| RCM | Restrictive cardiomyopathy |
| SCD | Sudden cardiac death |
| SD | Standard Deviation |
| TWI | T-wave inversion |
| VF | Ventricular fibrillation |
| VA | Ventricular arrhythmias |
| Y | Year |
References
- Priori, S.G.; Blomstrom-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiene, G. Sudden cardiac death in the young: A genetic destiny? Clin. Med. 2018, 18, s17–s23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, J.C.; Lin, Y.J.; de Oliveira Figueiredo, M.J.; Sepehri Shamloo, A.; Alfie, A.; Boveda, S.; Dagres, N.; Di Toro, D.; Eckhardt, L.L.; Ellenbogen, K.; et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) expert consensus on risk assessment in cardiac arrhythmias: Use the right tool for the right outcome, in the right population. Europace 2020. [Google Scholar] [CrossRef]
- Friedlander, Y.; Siscovick, D.S.; Weinmann, S.; Austin, M.A.; Psaty, B.M.; Lemaitre, R.N.; Arbogast, P.; Raghunathan, T.E.; Cobb, L.A. Family history as a risk factor for primary cardiac arrest. Circulation 1998, 97, 155–160. [Google Scholar] [CrossRef]
- Dekker, L.R.; Bezzina, C.R.; Henriques, J.P.; Tanck, M.W.; Koch, K.T.; Alings, M.W.; Arnold, A.E.; de Boer, M.J.; Gorgels, A.P.; Michels, H.R.; et al. Familial sudden death is an important risk factor for primary ventricular fibrillation: A case-control study in acute myocardial infarction patients. Circulation 2006, 114, 1140–1145. [Google Scholar] [CrossRef] [Green Version]
- Bezzina, C.R.; Pazoki, R.; Bardai, A.; Marsman, R.F.; de Jong, J.; Blom, M.T.; Scicluna, B.P.; Jukema, J.W.; Bindraban, N.R.; Lichtner, P.; et al. Genome-wide association study identifies a susceptibility locus at 21q21 for ventricular fibrillation in acute myocardial infarction. Nat. Genet. 2010, 42, 688–691. [Google Scholar] [CrossRef] [Green Version]
- Arking, D.E.; Junttila, M.J.; Goyette, P.; Huertas-Vazquez, A.; Eijgelsheim, M.; Blom, M.T.; Newton-Cheh, C.; Reinier, K.; Teodorescu, C.; Uy-Evanado, A.; et al. Identification of a sudden cardiac death susceptibility locus at 2q24.2 through genome-wide association in European ancestry individuals. PLoS Genet. 2011, 7, e1002158. [Google Scholar] [CrossRef]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J. Am. Coll. Cardiol. 2018, 72, e91–e220. [Google Scholar] [CrossRef]
- Gehi, A.K.; Duong, T.D.; Metz, L.D.; Gomes, J.A.; Mehta, D. Risk stratification of individuals with the Brugada electrocardiogram: A meta-analysis. J. Cardiovasc. Electrophysiol. 2006, 17, 577–583. [Google Scholar] [CrossRef]
- Priori, S.G.; Napolitano, C.; Gasparini, M.; Pappone, C.; Della Bella, P.; Giordano, U.; Bloise, R.; Giustetto, C.; De Nardis, R.; Grillo, M.; et al. Natural history of Brugada syndrome: Insights for risk stratification and management. Circulation 2002, 105, 1342–1347. [Google Scholar] [CrossRef] [Green Version]
- El-Sherif, N.; Turitto, G. Electrolyte disorders and arrhythmogenesis. Cardiol. J. 2011, 18, 233–245. [Google Scholar] [PubMed]
- Laslett, D.B.; Cooper, J.M.; Greenberg, R.M.; Yesenosky, G.A.; Basil, A.; Gangireddy, C.; Whitman, I.R. Electrolyte Abnormalities in Patients Presenting with Ventricular Arrhythmia (from the LYTE-VT Study). Am. J. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Afzal, M.R.; Savona, S.; Mohamed, O.; Mohamed-Osman, A.; Kalbfleisch, S.J. Hypertension and Arrhythmias. Heart Fail. Clin. 2019, 15, 543–550. [Google Scholar] [CrossRef]
- Solomon, R.J. Ventricular arrhythmias in patients with myocardial infarction and ischaemia. Relationship to serum potassium and magnesium. Drugs 1984, 28 (Suppl. 1), 66–76. [Google Scholar] [CrossRef] [PubMed]
- Glinge, C.; Engstrom, T.; Midgley, S.E.; Tanck, M.W.T.; Madsen, J.E.H.; Pedersen, F.; Ravn Jacobsen, M.; Lodder, E.M.; Al-Hussainy, N.R.; Kjaer Stampe, N.; et al. Seasonality of ventricular fibrillation at first myocardial infarction and association with viral exposure. PLoS ONE 2020, 15, e0226936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enriquez, A.; Frankel, D.S. Arrhythmogenic effects of energy drinks. J. Cardiovasc. Electrophysiol. 2017, 28, 711–717. [Google Scholar] [CrossRef]
- Maurin, O.; Lemoine, S.; Jost, D.; Lanoe, V.; Renard, A.; Travers, S.; The Paris Fire Brigade Cardiac Arrest Work Group; Lapostolle, F.; Tourtier, J.P. Maternal out-of-hospital cardiac arrest: A retrospective observational study. Resuscitation 2019, 135, 205–211. [Google Scholar] [CrossRef]
- Alexandre, J.; Moslehi, J.J.; Bersell, K.R.; Funck-Brentano, C.; Roden, D.M.; Salem, J.E. Anticancer drug-induced cardiac rhythm disorders: Current knowledge and basic underlying mechanisms. Pharmacol. Ther. 2018, 189, 89–103. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Rizzoli, G.; Schiavon, M.; Thiene, G. Does sports activity enhance the risk of sudden death in adolescents and young adults? J. Am. Coll. Cardiol. 2003, 42, 1959–1963. [Google Scholar] [CrossRef]
- Schmied, C.; Borjesson, M. Sudden cardiac death in athletes. J. Intern. Med. 2014, 275, 93–103. [Google Scholar] [CrossRef]
- Bezzina, C.R.; Lahrouchi, N.; Priori, S.G. Genetics of sudden cardiac death. Circ. Res. 2015, 116, 1919–1936. [Google Scholar] [CrossRef] [PubMed]
- Girolami, F.; Frisso, G.; Benelli, M.; Crotti, L.; Iascone, M.; Mango, R.; Mazzaccara, C.; Pilichou, K.; Arbustini, E.; Tomberli, B.; et al. Contemporary genetic testing in inherited cardiac disease: Tools, ethical issues, and clinical applications. J Cardiovasc. Med. 2018, 19, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagu, B.; Charpentier, F.; Toumaniantz, G. Identifying potential functional impact of mutations and polymorphisms: Linking heart failure, increased risk of arrhythmias and sudden cardiac death. Front Physiol. 2013, 4, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detta, N.; Frisso, G.; Limongelli, G.; Marzullo, M.; Calabro, R.; Salvatore, F. Genetic analysis in a family affected by sick sinus syndrome may reduce the sudden death risk in a young aspiring competitive athlete. Int. J. Cardiol. 2014, 170, e63–e65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Peters, S.; Thompson, T.; Morgan, N.; Maccicoca, I.; Trainer, A.; Zentner, D.; Kalman, J.M.; Winship, I.; Vohra, J.K. Familial cardiological and targeted genetic evaluation: Low yield in sudden unexplained death and high yield in unexplained cardiac arrest syndromes. Heart Rhythm. 2013, 10, 1653–1660. [Google Scholar] [CrossRef]
- Monda, E.; Sarubbi, B.; Russo, M.G.; Caiazza, M.; Mazzaccara, C.; Magrelli, J.; Rubino, M.; Esposito, A.; Perna, A.; Passariello, A.; et al. Unexplained sudden cardiac arrest in children: Clinical and genetic characteristics of survivors. Eur. J. Prev. Cardiol. 2020. [Google Scholar] [CrossRef]
- Fellmann, F.; van El, C.G.; Charron, P.; Michaud, K.; Howard, H.C.; Boers, S.N.; Clarke, A.J.; Duguet, A.M.; Forzano, F.; Kauferstein, S.; et al. European recommendations integrating genetic testing into multidisciplinary management of sudden cardiac death. Eur. J. Hum. Genet. EJHG 2019, 27, 1763–1773. [Google Scholar] [CrossRef] [Green Version]
- Ackerman, M.J.; Tester, D.J.; Driscoll, D.J. Molecular autopsy of sudden unexplained death in the young. Am. J. Forensic Med. Pathol. 2001, 22, 105–111. [Google Scholar] [CrossRef]
- Tester, D.J.; Medeiros-Domingo, A.; Will, M.L.; Haglund, C.M.; Ackerman, M.J. Cardiac channel molecular autopsy: Insights from 173 consecutive cases of autopsy-negative sudden unexplained death referred for postmortem genetic testing. Mayo Clin. Proc. 2012, 87, 524–539. [Google Scholar] [CrossRef] [Green Version]
- Tester, D.J.; Spoon, D.B.; Valdivia, H.H.; Makielski, J.C.; Ackerman, M.J. Targeted mutational analysis of the RyR2-encoded cardiac ryanodine receptor in sudden unexplained death: A molecular autopsy of 49 medical examiner/coroner’s cases. Mayo Clin. Proc. 2004, 79, 1380–1384. [Google Scholar] [CrossRef]
- Hellenthal, N.; Gaertner-Rommel, A.; Klauke, B.; Paluszkiewicz, L.; Stuhr, M.; Kerner, T.; Farr, M.; Puschel, K.; Milting, H. Molecular autopsy of sudden unexplained deaths reveals genetic predispositions for cardiac diseases among young forensic cases. Europace 2017, 19, 1881–1890. [Google Scholar] [CrossRef] [PubMed]
- Hertz, C.L.; Christiansen, S.L.; Ferrero-Miliani, L.; Dahl, M.; Weeke, P.E.; LuCamp; Ottesen, G.L.; Frank-Hansen, R.; Bundgaard, H.; Morling, N. Next-generation sequencing of 100 candidate genes in young victims of suspected sudden cardiac death with structural abnormalities of the heart. Int. J. Leg. Med. 2016, 130, 91–102. [Google Scholar] [CrossRef]
- Christiansen, S.L.; Hertz, C.L.; Ferrero-Miliani, L.; Dahl, M.; Weeke, P.E.; LuCamp; Ottesen, G.L.; Frank-Hansen, R.; Bundgaard, H.; Morling, N. Genetic investigation of 100 heart genes in sudden unexplained death victims in a forensic setting. Eur. J. Hum. Genet. EJHG 2016, 24, 1797–1802. [Google Scholar] [CrossRef] [PubMed]
- Nunn, L.M.; Lopes, L.R.; Syrris, P.; Murphy, C.; Plagnol, V.; Firman, E.; Dalageorgou, C.; Zorio, E.; Domingo, D.; Murday, V.; et al. Diagnostic yield of molecular autopsy in patients with sudden arrhythmic death syndrome using targeted exome sequencing. Europace 2016, 18, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Semsarian, C.; Ingles, J.; Wilde, A.A. Sudden cardiac death in the young: The molecular autopsy and a practical approach to surviving relatives. Eur. Heart J. 2015, 36, 1290–1296. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Sanchez-Molero, O.; Fernandez, A.; Mademont-Soler, I.; Coll, M.; Perez-Serra, A.; Mates, J.; Del Olmo, B.; Pico, F.; Nogue-Navarro, L.; et al. Sudden Arrhythmic Death During Exercise: A Post-Mortem Genetic Analysis. Sports Med. 2017, 47, 2101–2115. [Google Scholar] [CrossRef]
- Miles, C.; Finocchiaro, G.; Papadakis, M.; Gray, B.; Westaby, J.; Ensam, B.; Basu, J.; Parry-Williams, G.; Papatheodorou, E.; Paterson, C.; et al. Sudden Death and Left Ventricular Involvement in Arrhythmogenic Cardiomyopathy. Circulation 2019, 139, 1786–1797. [Google Scholar] [CrossRef]
- Morentin, B.; Suarez-Mier, M.P.; Monzo, A.; Molina, P.; Lucena, J.S. Sports-related sudden cardiac death due to myocardial diseases on a population from 1-35 years: A multicentre forensic study in Spain. Forensic Sci. Res. 2019, 4, 257–266. [Google Scholar] [CrossRef]
- Martin, M.; Reguero, J.J.; Castro, M.G.; Coto, E.; Hernandez, E.; Carro, A.; Calvo, D.; de la Tassa, C.M. Hypertrophic cardiomyopathy and athlete’s heart: A tale of two entities. Eur. J. Echocardiogr. 2009, 10, 151–153. [Google Scholar] [CrossRef]
- Richard, P.; Denjoy, I.; Fressart, V.; Wilson, M.G.; Carre, F.; Charron, P. Advising a cardiac disease gene positive yet phenotype negative or borderline abnormal athlete: Is sporting disqualification really necessary? Br. J. Sports Med. 2012, 46 (Suppl. 1), i59–i68. [Google Scholar] [CrossRef]
- Kebed, K.Y.; Bos, J.M.; Anavekar, N.S.; Mulvagh, S.L.; Ackerman, M.J.; Ommen, S.R. Hypertrophic Cardiomyopathy, Athlete’s Heart, or Both: A Case of Hypertrophic Cardiomyopathy Regression. Circ. Cardiovasc. Imaging 2015, 8, e003312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Argenio, V.; Esposito, M.V.; Nunziato, M.; De Simone, A.; Buono, P.; Salvatore, F.; Frisso, G. Molecular diagnosis of Brugada syndrome via next-generation sequencing of a multigene panel in a young athlete. Med. Sport 2018, 71, 27–34. [Google Scholar] [CrossRef]
- Mazzaccara, C.; Redi, A.; Lemme, E.; Pelliccia, A.; Salvatore, F.; Frisso, G. Impact of molecular diagnostics in an asymptomatic amateur athlete found to be affected by hypertrophic cardiomyopathy. Med. Sport 2018, 71, 405–412. [Google Scholar] [CrossRef]
- Limongelli, G.; Nunziato, M.; Mazzaccara, C.; Intrieri, M.; D’Argenio, V.; Esposito, M.V.; Monda, E.; Maggio, F.D.; Frisso, G.; Salvatore, F. Genotype-Phenotype Correlation: A Triple DNA Mutational Event in a Boy Entering Sport Conveys an Additional Pathogenicity Risk. Genes 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Macarie, C.; Stoian, I.; Dermengiu, D.; Barbarii, L.; Piser, I.T.; Chioncel, O.; Carp, A. The electrocardiographic abnormalities in highly trained athletes compared to the genetic study related to causes of unexpected sudden cardiac death. J. Med. Life 2009, 2, 361–372. [Google Scholar]
- Kadota, C.; Arimura, T.; Hayashi, T.; Naruse, T.K.; Kawai, S.; Kimura, A. Screening of sarcomere gene mutations in young athletes with abnormal findings in electrocardiography: Identification of a MYH7 mutation and MYBPC3 mutations. J. Hum. Genet. 2015, 60, 641–645. [Google Scholar] [CrossRef]
- Sheikh, N.; Papadakis, M.; Wilson, M.; Malhotra, A.; Adamuz, C.; Homfray, T.; Monserrat, L.; Behr, E.R.; Sharma, S. Diagnostic Yield of Genetic Testing in Young Athletes With T-Wave Inversion. Circulation 2018, 138, 1184–1194. [Google Scholar] [CrossRef]
- Cronin, H.; Crinion, D.; Kerins, D.; Fahy, G.; Vaughan, C.J. Inferolateral T wave inversion in athletes: Phenotype-genotype correlation. Ir. J. Med. Sci. 2020. [Google Scholar] [CrossRef]
- Zorzi, A.; Vessella, T.; De Lazzari, M.; Cipriani, A.; Menegon, V.; Sarto, G.; Spagnol, R.; Merlo, L.; Pegoraro, C.; Marra, M.P.; et al. Screening young athletes for diseases at risk of sudden cardiac death: Role of stress testing for ventricular arrhythmias. Eur. J. Prev. Cardiol. 2020, 27, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Limongelli, G.; Nunziato, M.; D’Argenio, V.; Esposito, M.V.; Monda, E.; Mazzaccara, C.; Caiazza, M.; D’Aponte, A.; D’Andrea, A.; Bossone, E.; et al. Yield and clinical significance of genetic screening in elite and amateur athletes. Eur. J. Prev. Cardiol. 2020. [Google Scholar] [CrossRef]
- Skinner, J.R.; Winbo, A.; Abrams, D.; Vohra, J.; Wilde, A.A. Channelopathies That Lead to Sudden Cardiac Death: Clinical and Genetic Aspects. Heart Lung Circ. 2019, 28, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Giudicessi, J.R.; Ackerman, M.J. Genotype- and phenotype-guided management of congenital long QT syndrome. Curr. Probl. Cardiol. 2013, 38, 417–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieve, K.V.; Wilde, A.A. Inherited ion channel diseases: A brief review. Europace 2015, 17 (Suppl. 2), ii1–ii6. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Ackerman, M.J. The long QT syndrome: A transatlantic clinical approach to diagnosis and therapy. Eur. Heart J. 2013, 34, 3109–3116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, P.J.; Priori, S.G.; Spazzolini, C.; Moss, A.J.; Vincent, G.M.; Napolitano, C.; Denjoy, I.; Guicheney, P.; Breithardt, G.; Keating, M.T.; et al. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation 2001, 103, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, P.J.; Ackerman, M.J.; George, A.L., Jr.; Wilde, A.A.M. Impact of genetics on the clinical management of channelopathies. J. Am. Coll. Cardiol. 2013, 62, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Adler, A.; Novelli, V.; Amin, A.S.; Abiusi, E.; Care, M.; Nannenberg, E.A.; Feilotter, H.; Amenta, S.; Mazza, D.; Bikker, H.; et al. An International, Multicentered, Evidence-Based Reappraisal of Genes Reported to Cause Congenital Long QT Syndrome. Circulation 2020, 141, 418–428. [Google Scholar] [CrossRef]
- Altmann, H.M.; Tester, D.J.; Will, M.L.; Middha, S.; Evans, J.M.; Eckloff, B.W.; Ackerman, M.J. Homozygous/Compound Heterozygous Triadin Mutations Associated With Autosomal-Recessive Long-QT Syndrome and Pediatric Sudden Cardiac Arrest: Elucidation of the Triadin Knockout Syndrome. Circulation 2015, 131, 2051–2060. [Google Scholar] [CrossRef] [Green Version]
- Rooryck, C.; Kyndt, F.; Bozon, D.; Roux-Buisson, N.; Sacher, F.; Probst, V.; Thambo, J.B. New Family with Catecholaminergic Polymorphic Ventricular Tachycardia Linked to the Triadin Gene. J. Cardiovasc. Electrophysiol. 2015, 26, 1146–1150. [Google Scholar] [CrossRef]
- Takeda, I.; Takahashi, T.; Ueno, H.; Morino, H.; Ochi, K.; Nakamura, T.; Hosomi, N.; Kawakami, H.; Hashimoto, K.; Matsumoto, M. Autosomal recessive Andersen-Tawil syndrome with a novel mutation L94P in Kir2.1. Neurol. Clin. Neurosci. 2013, 1, 131–137. [Google Scholar] [CrossRef]
- Nakano, Y.; Shimizu, W. Genetics of long-QT syndrome. J. Hum. Genet. 2016, 61, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Velcea, A.E.; Siliste, C.; Vinereanu, D. Catecholaminergic Polymorphic Ventricular Tachycardia—Looking to the Future. Maedica 2017, 12, 306–310. [Google Scholar] [PubMed]
- Tabish, A.M.; Azzimato, V.; Alexiadis, A.; Buyandelger, B.; Knoll, R. Genetic epidemiology of titin-truncating variants in the etiology of dilated cardiomyopathy. Biophys. Rev. 2017, 9, 207–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershberger, R.E.; Morales, A.; Siegfried, J.D. Clinical and genetic issues in dilated cardiomyopathy: A review for genetics professionals. Genet. Med. 2010, 12, 655–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNally, E.M.; Mestroni, L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Genetics of hypertrophic cardiomyopathy after 20 years: Clinical perspectives. J. Am. Coll. Cardiol. 2012, 60, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Kostareva, A.; Kiselev, A.; Gudkova, A.; Frishman, G.; Ruepp, A.; Frishman, D.; Smolina, N.; Tarnovskaya, S.; Nilsson, D.; Zlotina, A.; et al. Genetic Spectrum of Idiopathic Restrictive Cardiomyopathy Uncovered by Next-Generation Sequencing. PLoS ONE 2016, 11, e0163362. [Google Scholar] [CrossRef] [Green Version]
- Towbin, J.A.; Lorts, A.; Jefferies, J.L. Left ventricular non-compaction cardiomyopathy. Lancet 2015, 386, 813–825. [Google Scholar] [CrossRef]
- Detta, N.; Frisso, G.; Salvatore, F. The multi-faceted aspects of the complex cardiac Nav1.5 protein in membrane function and pathophysiology. Biochim. Biophys. Acta 2015, 1854, 1502–1509. [Google Scholar] [CrossRef] [Green Version]
- Vutthikraivit, W.; Rattanawong, P.; Putthapiban, P.; Sukhumthammarat, W.; Vathesatogkit, P.; Ngarmukos, T.; Thakkinstian, A. Worldwide Prevalence of Brugada Syndrome: A Systematic Review and Meta-Analysis. Acta Cardiol. Sin. 2018, 34, 267–277. [Google Scholar] [CrossRef]
- Antzelevitch, C.; Yan, G.X.; Ackerman, M.J.; Borggrefe, M.; Corrado, D.; Guo, J.; Gussak, I.; Hasdemir, C.; Horie, M.; Huikuri, H.; et al. J-Wave syndromes expert consensus conference report: Emerging concepts and gaps in knowledge. J. Arrhythmia 2016, 32, 315–339. [Google Scholar] [CrossRef] [PubMed]
- Probst, V.; Veltmann, C.; Eckardt, L.; Meregalli, P.G.; Gaita, F.; Tan, H.L.; Babuty, D.; Sacher, F.; Giustetto, C.; Schulze-Bahr, E.; et al. Long-term prognosis of patients diagnosed with Brugada syndrome: Results from the FINGER Brugada Syndrome Registry. Circulation 2010, 121, 635–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brugada, J.; Campuzano, O.; Arbelo, E.; Sarquella-Brugada, G.; Brugada, R. Present Status of Brugada Syndrome: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 1046–1059. [Google Scholar] [CrossRef]
- Juang, J.J.; Horie, M. Genetics of Brugada syndrome. J. Arrhythmia 2016, 32, 418–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarquella-Brugada, G.; Campuzano, O.; Arbelo, E.; Brugada, J.; Brugada, R. Brugada syndrome: Clinical and genetic findings. Genet. Med. 2016, 18, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Coppola, G.; Corrado, E.; Curnis, A.; Maglia, G.; Oriente, D.; Mignano, A.; Brugada, P. Update on Brugada Syndrome 2019. Curr. Probl. Cardiol. 2019, 100454. [Google Scholar] [CrossRef]
- Perez-Riera, A.R.; Barbosa-Barros, R.; de Rezende Barbosa, M.P.C.; Daminello-Raimundo, R.; de Lucca, A.A., Jr.; de Abreu, L.C. Catecholaminergic polymorphic ventricular tachycardia, an update. Ann. Noninvasive Electrocardiol. 2018, 23, e12512. [Google Scholar] [CrossRef]
- Roston, T.M.; Yuchi, Z.; Kannankeril, P.J.; Hathaway, J.; Vinocur, J.M.; Etheridge, S.P.; Potts, J.E.; Maginot, K.R.; Salerno, J.C.; Cohen, M.I.; et al. The clinical and genetic spectrum of catecholaminergic polymorphic ventricular tachycardia: Findings from an international multicentre registry. Europace 2018, 20, 541–547. [Google Scholar] [CrossRef]
- Sumitomo, N. Current topics in catecholaminergic polymorphic ventricular tachycardia. J. Arrhythmia 2016, 32, 344–351. [Google Scholar] [CrossRef] [Green Version]
- Wleklinski, M.J.; Kannankeril, P.J.; Knollmann, B.C. Molecular and tissue mechanisms of catecholaminergic polymorphic ventricular tachycardia. J. Physiol. 2020. [Google Scholar] [CrossRef]
- Schaufelberger, M. Cardiomyopathy and pregnancy. Heart 2019, 105, 1543–1551. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Wu, W.; Liu, F.; Yang, K.; Li, J.; Liu, Y.; Wang, R.; Si, N.; Gao, P.; Zhang, S.; et al. Molecular analysis of inherited cardiomyopathy using next generation semiconductor sequencing technologies. J. Transl. Med. 2018, 16, 241. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef] [PubMed]
- Diez-Lopez, C.; Salazar-Mendiguchia, J. Clinical presentations of hypertrophic cardiomyopathy and implications for therapy. Glob. Cardiol. Sci. Pract. 2018, 2018, 19. [Google Scholar] [CrossRef] [Green Version]
- Limongelli, G.; Monda, E.; Tramonte, S.; Gragnano, F.; Masarone, D.; Frisso, G.; Esposito, A.; Gravino, R.; Ammendola, E.; Salerno, G.; et al. Prevalence and clinical significance of red flags in patients with hypertrophic cardiomyopathy. Int. J. Cardiol. 2020, 299, 186–191. [Google Scholar] [CrossRef]
- Yotti, R.; Seidman, C.E.; Seidman, J.G. Advances in the Genetic Basis and Pathogenesis of Sarcomere Cardiomyopathies. Annu. Rev. Genom. Hum. Genet. 2019, 20, 129–153. [Google Scholar] [CrossRef]
- Walsh, R.; Buchan, R.; Wilk, A.; John, S.; Felkin, L.E.; Thomson, K.L.; Chiaw, T.H.; Loong, C.C.W.; Pua, C.J.; Raphael, C.; et al. Defining the genetic architecture of hypertrophic cardiomyopathy: Re-evaluating the role of non-sarcomeric genes. Eur. Heart J. 2017, 38, 3461–3468. [Google Scholar] [CrossRef]
- Burke, M.A.; Cook, S.A.; Seidman, J.G.; Seidman, C.E. Clinical and Mechanistic Insights Into the Genetics of Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 68, 2871–2886. [Google Scholar] [CrossRef]
- Esposito, A.; Monda, E.; Gragnano, F.; Simone, F.; Cesaro, A.; Natale, F.; Concilio, C.; Moscarella, E.; Caiazza, M.; Pazzanese, V.; et al. Prevalence and clinical implications of hyperhomocysteinaemia in patients with hypertrophic cardiomyopathy and MTHFR C6777T polymorphism. Eur. J. Prev. Cardiol. 2019. [Google Scholar] [CrossRef]
- Ingles, J.; Doolan, A.; Chiu, C.; Seidman, J.; Seidman, C.; Semsarian, C. Compound and double mutations in patients with hypertrophic cardiomyopathy: Implications for genetic testing and counselling. J. Med. Genet. 2005, 42, e59. [Google Scholar] [CrossRef] [Green Version]
- Biagini, E.; Olivotto, I.; Iascone, M.; Parodi, M.I.; Girolami, F.; Frisso, G.; Autore, C.; Limongelli, G.; Cecconi, M.; Maron, B.J.; et al. Significance of sarcomere gene mutations analysis in the end-stage phase of hypertrophic cardiomyopathy. Am. J. Cardiol. 2014, 114, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Mazzaccara, C.; Limongelli, G.; Petretta, M.; Vastarella, R.; Pacileo, G.; Bonaduce, D.; Salvatore, F.; Frisso, G. A common polymorphism in the SCN5A gene is associated with dilated cardiomyopathy. J. Cardiovasc. Med. 2018, 19, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Bohm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzaccara, C.; Mirra, B.; Barretta, F.; Lombardo, B.; Scudiero, O.; Frisso, G. Sudden cardiac death in young athletes: Literature review of molecular basis. Cardiogenetics 2020, 10. [Google Scholar] [CrossRef] [Green Version]
- Gallego-Delgado, M.; Delgado, J.F.; Brossa-Loidi, V.; Palomo, J.; Marzoa-Rivas, R.; Perez-Villa, F.; Salazar-Mendiguchia, J.; Ruiz-Cano, M.J.; Gonzalez-Lopez, E.; Padron-Barthe, L.; et al. Idiopathic Restrictive Cardiomyopathy Is Primarily a Genetic Disease. J. Am. Coll. Cardiol. 2016, 67, 3021–3023. [Google Scholar] [CrossRef]
- Bates, M.G.; Bourke, J.P.; Giordano, C.; d’Amati, G.; Turnbull, D.M.; Taylor, R.W. Cardiac involvement in mitochondrial DNA disease: Clinical spectrum, diagnosis, and management. Eur. Heart J. 2012, 33, 3023–3033. [Google Scholar] [CrossRef] [Green Version]
- Pilichou, K.; Thiene, G.; Bauce, B.; Rigato, I.; Lazzarini, E.; Migliore, F.; Marra, M.P.; Rizzo, S.; Zorzi, A.; Daliento, L.; et al. Arrhythmogenic cardiomyopathy. Orphanet J. Rare Dis. 2016, 11. [Google Scholar] [CrossRef]
- Hoorntje, E.T.; Te Rijdt, W.P.; James, C.A.; Pilichou, K.; Basso, C.; Judge, D.P.; Bezzina, C.R.; van Tintelen, J.P. Arrhythmogenic cardiomyopathy: Pathology, genetics, and concepts in pathogenesis. Cardiovasc. Res. 2017, 113, 1521–1531. [Google Scholar] [CrossRef]
- Turkowski, K.L.; Tester, D.J.; Bos, J.M.; Haugaa, K.H.; Ackerman, M.J. Whole exome sequencing with genomic triangulation implicates CDH2-encoded N-cadherin as a novel pathogenic substrate for arrhythmogenic cardiomyopathy. Congenit. Heart Dis. 2017, 12, 226–235. [Google Scholar] [CrossRef]
- Poloni, G.; Calore, M.; Rigato, I.; Marras, E.; Minervini, G.; Mazzotti, E.; Lorenzon, A.; Li Mura, I.E.A.; Telatin, A.; Zara, I.; et al. A targeted next-generation gene panel reveals a novel heterozygous nonsense variant in the TP63 gene in patients with arrhythmogenic cardiomyopathy. Heart Rhythm. 2019, 16, 773–780. [Google Scholar] [CrossRef]
- Rigato, I.; Bauce, B.; Rampazzo, A.; Zorzi, A.; Pilichou, K.; Mazzotti, E.; Migliore, F.; Marra, M.P.; Lorenzon, A.; De Bortoli, M.; et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 533–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, U.; Minamisawa, M.; Koyama, J. Isolated left ventricular non-compaction cardiomyopathy in adults. J. Cardiol. 2015, 65, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbustini, E.; Weidemann, F.; Hall, J.L. Left ventricular noncompaction: A distinct cardiomyopathy or a trait shared by different cardiac diseases? J. Am. Coll. Cardiol. 2014, 64, 1840–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutton, B.; Salanti, G.; Caldwell, D.M.; Chaimani, A.; Schmid, C.H.; Cameron, C.; Ioannidis, J.P.; Straus, S.; Thorlund, K.; Jansen, J.P.; et al. The PRISMA extension statement for reporting of systematic reviews incorporating network meta-analyses of health care interventions: Checklist and explanations. Ann. Intern. Med. 2015, 162, 777–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Pelliccia, A.; Maron, B.J.; Culasso, F.; Di Paolo, F.M.; Spataro, A.; Biffi, A.; Caselli, G.; Piovano, P. Clinical significance of abnormal electrocardiographic patterns in trained athletes. Circulation 2000, 102, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Berdowski, J.; de Beus, M.F.; Blom, M.; Bardai, A.; Bots, M.L.; Doevendans, P.A.; Grobbee, D.E.; Tan, H.L.; Tijssen, J.G.; Koster, R.W.; et al. Exercise-related out-of-hospital cardiac arrest in the general population: Incidence and prognosis. Eur. Heart J. 2013, 34, 3616–3623. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.J.; Thompson, P.D.; Ackerman, M.J.; Balady, G.; Berger, S.; Cohen, D.; Dimeff, R.; Douglas, P.S.; Glover, D.W.; Hutter, A.M., Jr.; et al. Recommendations and considerations related to preparticipation screening for cardiovascular abnormalities in competitive athletes: 2007 update: A scientific statement from the American Heart Association Council on Nutrition, Physical Activity, and Metabolism: Endorsed by the American College of Cardiology Foundation. Circulation 2007, 115, 1643–1655. [Google Scholar] [CrossRef] [Green Version]
- Corrado, D.; Pelliccia, A.; Bjornstad, H.H.; Vanhees, L.; Biffi, A.; Borjesson, M.; Panhuyzen-Goedkoop, N.; Deligiannis, A.; Solberg, E.; Dugmore, D.; et al. Cardiovascular pre-participation screening of young competitive athletes for prevention of sudden death: Proposal for a common European protocol. Consensus Statement of the Study Group of Sport Cardiology of the Working Group of Cardiac Rehabilitation and Exercise Physiology and the Working Group of Myocardial and Pericardial Diseases of the European Society of Cardiology. Eur. Heart J. 2005, 26, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, B.; Izzo, V.; Terracciano, D.; Ranieri, A.; Mazzaccara, C.; Fimiani, F.; Cesaro, A.; Gentile, L.; Leggiero, E.; Pero, R.; et al. Laboratory medicine: Health evaluation in elite athletes. Clin. Chem. Lab. Med. 2019. [Google Scholar] [CrossRef]
- Frisso, G.; Detta, N.; Coppola, P.; Mazzaccara, C.; Pricolo, M.R.; D’Onofrio, A.; Limongelli, G.; Calabro, R.; Salvatore, F. Functional Studies and In Silico Analyses to Evaluate Non-Coding Variants in Inherited Cardiomyopathies. Int. J. Mol. Sci. 2016, 17, 1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidbuchel, H.; Prior, D.L.; La Gerche, A. Ventricular arrhythmias associated with long-term endurance sports: What is the evidence? Br. J. Sports Med. 2012, 46 (Suppl. 1), i44–i50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, N.; Papadakis, M.; Schnell, F.; Panoulas, V.; Malhotra, A.; Wilson, M.; Carre, F.; Sharma, S. Clinical Profile of Athletes With Hypertrophic Cardiomyopathy. Circ. Cardiovasc. Imaging 2015, 8, e003454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pricolo, M.R.; Herrero-Galan, E.; Mazzaccara, C.; Losi, M.A.; Alegre-Cebollada, J.; Frisso, G. Protein Thermodynamic Destabilization in the Assessment of Pathogenicity of a Variant of Uncertain Significance in Cardiac Myosin Binding Protein C. J. Cardiovasc. Transl. Res. 2020. [Google Scholar] [CrossRef]
- Levine, B.D.; Baggish, A.L.; Kovacs, R.J.; Link, M.S.; Maron, M.S.; Mitchell, J.H. Eligibility and Disqualification Recommendations for Competitive Athletes With Cardiovascular Abnormalities: Task Force 1: Classification of Sports: Dynamic, Static, and Impact: A Scientific Statement From the American Heart Association and American College of Cardiology. J. Am. Coll. Cardiol. 2015, 66, 2350–2355. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| DISEASE | Gene OMIM ID | GENE | TRANSCRIPT ID | Length bp | PROTEIN | Prevalence | REF. | Inheritance |
|---|---|---|---|---|---|---|---|---|
| Brugada Syndrome (BrS) | 601439 | ABCC9 | ENST00000261200.8 | 8293 | ATP-Binding Cassette, Subfamily C, Member 9 | 4–5% | [61] | AD |
| 600465 | ANK3 | ENST00000280772.7 | 17019 | Ankyrin 3 (G) | <5% | [62] | AD | |
| 611875 | CACNA1C | ENST00000399655.6 | 13744 | α subunit α1C of the Cav1.2 calcium channel | 6–7% | [61] | AD | |
| 114204 | CACNA2D1 | ENST00000356860.8 | 7542 | δ subunit Cavα2δ1 of calcium channel | <5% | [61] | AD | |
| 611876 | CACNB2 | ENST00000324631.13 | 6129 | β subunit Cavβ2b of calcium channel | 4–5% | [61] | AD | |
| 601513 | FGF12 | ENST00000445105.6 | 5406 | Fibroblast growth factor 12 | <5% | [62] | AD | |
| 611777 | GPD1L | ENST00000282541.10 | 3947 | Glycerol-3-phosphate dehydrogenase 1-like | <5% | [61] | AD | |
| 613123 | HCN4 | ENST00000261917.4 | 6922 | Hyperpolarization-activated cyclic nucleotide-gated channel 4 | <5% | [61] | AD | |
| 604674 | HEY2 | ENST00000368364.4 | 2626 | Hairy/Enhancer of Split-related with YRPW motif 2 | <5% | [61] | AD | |
| 605410 | KCND2 | ENST00000331113.9 | 5830 | α subunit of the KV4.2 potassium channel | <5% | [62] | AD | |
| 616399 | KCND3 | ENST00000369697.5 | 7396 | α subunit of the KV4.3 potassium channel | <5% | [61] | AD | |
| 613119 | KCNE3 | ENST00000310128.8 | 3143 | β subunit MiRP2 of potassium channel | <5% | [61] | AD | |
| 300328 | KCNE5 | ENST00000372101.3 | 1473 | Potassium channel, voltage-gated, ISK-related family, member 1-like | <5% | [61] | XLD | |
| 152427 | KCNH2 | ENST00000262186.10 | 4292 | α subunit of the HERG potassium channel | <5% | [61] [61] | AD | |
| 600935 | KCNJ8 | ENST00000240662.3 | 2274 | α subunit of the KIR6.1 potassium channel | <5% | [61] | AD | |
| 610846 | LRRC10 | ENST00000361484.5 | 2344 | Leucine-rich repeat-containing protein 10 | <5% | [62] | AD | |
| 602861 | PKP2 | ENST00000070846.10 | 4241 | Plakophilin 2 | <5% | [61] | AD | |
| 607954 | RANGRF | ENST00000226105.11 | 831 | RAN guanine nucleotide release factor | <5% | [61] | AD | |
| 612838 | SCN1B | ENST00000262631.11 | 1666 | β subunit Navβ1 of sodium channel | <5% | [61] | AD | |
| 601327 | SCN2B | ENST00000278947.6 | 4937 | β subunit Navβ2 of sodium channel | <5% | [61] | AD | |
| 613120 | SCN3B | ENST00000392770.6 | 6061 | β subunit Navβ3 of sodium channel | <5% | [61] | AD | |
| 601144 | SCN5A | ENST00000423572.7 | 8516 | α subunit of the Nav1.5 sodium channel | 20–25% | [61] | AD | |
| 604427 | SCN10A | ENST00000449082.3 | 6626 | α subunit of the Nav1.8 sodium channel | 2.5–5% | [62] | AD | |
| 603961 | SEMA3A | ENST00000265362.9 | 8113 | Semaphorin family protein | <5% | [62] | AD | |
| 602701 | SLMAP | ENST00000659705.1 | 6240 | Sarcolemma-associated protein | <5% | [61] | AD | |
| 606936 | TRPM4 | ENST00000252826.10 | 4058 | Calcium-activated non-selective ion channel | 8% | [61] | AD | |
| Long QT Syndrome (LQT) | 604001 | AKAP9 | ENST00000356239.8 | 12476 | A-kinase anchor protein 9 | <5% | [63] | AD |
| 106410 | ANK2 | ENST00000357077.9 | 14215 | Ankyrin 2 (B) | <5% | [63] | AD | |
| 114205 | CACNA1C | ENST00000399655.6 | 13744 | α subunit α1C of the Cav1.2 calcium channel | <5% | [63] | AD | |
| 114180 | CALM1 | ENST00000356978.9 | 4203 | Calmodulin 1 | <5% | [63] | AD | |
| 114182 | CALM2 | ENST00000272298.12 | 1210 | Calmodulin 2 | <5% | [63] | AD | |
| 114183 | CALM3 | ENST00000291295.14 | 2184 | Calmodulin 3 | <5% | [62] | AD | |
| 601253 | CAV3 | ENST00000343849.3 | 1422 | Caveolin 3 | <5% | [63] | AD | |
| 176261 | KCNE1 | ENST00000399286.3 | 3505 | Potassium voltage-gated channel subfamily E regulatory subunit 1 | <5% | [63] | AD/AR | |
| 603796 | KCNE2 | ENST00000290310. | 1051 | Potassium voltage-gated channel subfamily E regulatory subunit 2 | <5% | [63] | AD | |
| 152427 | KCNH2 | ENST00000262186.10 | 4292 | α subunit of the HERG potassium channel | 25–30% | [63] | AD | |
| 600681 | KCNJ2 | ENST00000243457.4 | 5391 | Potassium inwardly-rectifying channel, subfamily J, member 2 | <5% | [62] | AD/AR | |
| 600734 | KCNJ5 | ENST00000529694.6 | 6068 | Potassium inwardly-rectifying channel, subfamily J, member 5 | <5% | [63] | AD | |
| 607542 | KCNQ1 | ENST00000155840.12 | 3224 | Potassium voltage-gated channel subfamily KQT member 1 | 30–35% | [63] | AD/AR | |
| 602235 | KCNQ2 | ENST00000626839.2 | 9213 | Potassium voltage-gated channel subfamily Q member 2 | <5% | [62] | AD | |
| 180902 | RYR2 | ENST00000366574.7 | 16583 | Ryanodine Receptor 2 | <5% | [62] | AD | |
| 600235 | SCN1B | ENST00000262631.11 | 1666 | Sodium voltage-gated channel beta subunit 1 | <5% | [62] | AD | |
| 608256 | SCN4B | ENST00000324727.8 | 4484 | Sodium voltage-gated channel beta subunit 4 | <5% | [63] | AD | |
| 600163 | SCN5A | ENST00000423572.7 | 8516 | α subunit of the Nav1.5 sodium channel | 5–10% | [63] | AD | |
| 601017 | SNTA1 | ENST00000217381.3 | 2211 | α1-Syntrophin | <5% | [63] | AD | |
| 603283 | TRDN | ENST00000334268.9 | 4627 | Triadin | <5% | [62] | AR | |
| Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT) | 106410 | ANK2 | ENST00000357077.9 | 14215 | Ankyrin 2 (B) | <5% | [64] | AD |
| 114180 | CALM1 | ENST00000356978.9 | 4203 | Calmodulin 1 | <5% | [64] | AD | |
| 114182 | CALM2 | ENST00000272298.12 | 1210 | Calmodulin 2 | <5% | [64] | AD | |
| 114183 | CALM3 | ENST00000291295.14 | 2184 | Calmodulin 3 | <5% | [62] | AD | |
| 114251 | CASQ2 | ENST00000261448.5 | 2674 | Calsequestrin 2 | 5% | [64] | AR | |
| 600681 | KCNJ2 | ENST00000243457.4 | 5391 | Potassium inwardly-rectifying channel, subfamily J, member 2 | <5% | [64] | AR | |
| 180902 | RYR2 | ENST00000366574.7 | 16583 | Ryanodine Receptor 2 | 60% | [64] | AD | |
| 603283 | TRDN | ENST00000334268.9 | 4627 | Triadin | <5% | [64] | AR | |
| Arrhythmogenic Cardiomyopathy (ACM) | 114020 | CDH2 | ENST00000269141.8 | 4016 | Cadherin 2—N-Cadherin | <5% | [62] | AD |
| 607667 | CTNNA3 | ENST00000433211.7 | 10696 | Catenin, alpha 3 | <5% | [62] | AD | |
| 125660 | DES | ENST00000373960.4 | 2243 | Desmin | <5% | [62] | AD | |
| 125645 | DSC2 | ENST00000280904.11 | 12331 | Desmocollin 2 | 1–8% | [62] | AD/AR | |
| 125671 | DSG2 | ENST00000261590.13 | 5697 | Desmoglein 2 | 3–20% | [62] | AD | |
| 125647 | DSP | ENST00000379802.8 | 9697 | Desmoplakin | 3–15% | [62] | AD | |
| 102565 | FLNC | ENST00000325888.12 | 9188 | Filamin C | <5% | [62] | AD | |
| 173325 | JUP | ENST00000393931.8 | 3505 | Junction plakoglobin | <5% | [62] | AD | |
| 150330 | LMNA | ENST00000368300.9 | 3178 | Lamin A/C | <5% | [62] | AD | |
| 172405 | PLN | ENST00000357525.6 | 2989 | Phospholamban | <5% | [62] | AR | |
| 602861 | PKP2 | ENST00000070846.10 | 4241 | Plakophilin 2 | 30–40% | [62] | AD | |
| 180902 | RYR2 | ENST00000366574.7 | 16583 | Ryanodine receptor 2 | <5% | [62] | AD | |
| 601144 | SCN5A | ENST00000423572.7 | 8516 | α subunit of the Nav1.5 sodium channel | <5% | [62] | AD | |
| 190230 | TGFB3 | ENST00000238682.7 | 2522 | Transforming Growth Factor β 3 | <5% | [62] | AD | |
| 612048 | TMEM43 | ENST00000306077.5 | 3230 | Transmembrane protein 43 | <5% | [62] | AD | |
| 188840 | TTN | ENST00000589042.5 | 109224 | Titin | 0–10% | [62] | AD | |
| Dilated Cardiomyopathy (DCM) | 601439 | ABCC9 | ENST00000261200.8 | 8293 | ATP-Binding Cassette, Subfamily C, Member 9 | <5% | [62] | AD |
| 602330 | ABLIM1 | ENST00000392955.7 | 7408 | Limatin (actin-binding LIM domain protein) | <5% | [62] | AD | |
| 102540 | ACTC1 | ENST00000290378.6 | 1382 | Actin, alpha, cardiac muscle | <5% | [65,66,67] | AD | |
| 102573 | ACTN2 | ENST00000546208.5 | 5103 | Alpha-actinin 2 | <5% | [62] | AD | |
| 606844 | ALMS1 | ENST00000613296.5 | 12848 | ALMS1 centrosome and basal body associated protein | <5% | [62] | AR | |
| 609599 | ANKRD1 | ENST00000371697.3 | 1979 | Ankyrin repeat domain-containing protein 1 | <5% | [62] | AD | |
| 608662 | ANO5 | ENST00000324559.9 | 6742 | Anoctamin 5 | <5% | [62] | AR | |
| 603883 | BAG3 | ENST00000369085.8 | 2561 | BCL2-associated athanogene | <5% | [62] | AD | |
| 611414 | CALR3 | ENST00000269881.8 | 1263 | Calreticulin 3 | <5% | [62] | AD | |
| 114251 | CASQ2 | ENST00000261448.5 | 2674 | Calsequestrin 2 | <5% | [62] | AD | |
| 601253 | CAV3 | ENST00000343849.3 | 1422 | Caveolin 3 | <5% | [62] | AD | |
| 123590 | CRYAB | ENST00000533475.6 | 1117 | Alpha B crystallin | <5% | [62] | AD | |
| 600824 | CSRP3 | ENST00000533783.2 | 1457 | Cysteine- and glycine-rich protein 3 | <5% | [62] | AD | |
| 600435 | CTF1 | ENST00000279804.2 | 1664 | Cardiotrophin 1 | <5% | [62] | AD | |
| 128239 | DAG1 | ENST00000545947.5 | 5829 | Dystroglycan, alpha | <5% | [62] | AR | |
| 125660 | DES | ENST00000373960.4 | 2243 | Desmin | <5% | [62] | AD | |
| 300377 | DMD | ENST00000357033.8 | 13956 | Dystrophin | <5% | [62] | XLR | |
| 605377 | DMPK | ENST00000291270.9 | 2799 | Dystrophia myotonica protein kinase gene | <5% | [62] | AD | |
| 610746 | DOLK | ENST00000372586.4 | 2074 | Dolichol Kinase | <5% | [62] | AR | |
| 125645 | DSC2 | ENST00000251081.8 | 12377 | Desmocollin 2 | <5% | [62] | AD | |
| 125671 | DSG2 | ENST00000261590.13 | 5697 | Desmoglein 2 | <5% | [62] | AD | |
| 125647 | DSP | ENST00000379802.8 | 9697 | Desmoplakin | <5% | [62] | AD | |
| 601239 | DTNA | ENST00000283365.13 | 6522 | Dystrobrevin, alpha | <5% | [62] | AD/AR | |
| 300384 | EMD | ENST00000369842.9 | 1281 | Emerin | <5% | [62] | XLR | |
| 603550 | EYA4 | ENST00000355286.12 | 5701 | Eyes absent 4 | <5% | [62] | AD | |
| 300163 | FHL1 | ENST00000370683.6 | 2286 | Four-and—a-half LIM domains 1 | <5% | [62] | XLD/XLR | |
| 602633 | FHL2 | ENST00000409177.6 | 4881 | Four-and—a-half LIM domains 2 | <5% | [62] | AD | |
| 102565 | FLNC | ENST00000325888.12 | 9188 | Filamin C | <5% | [62] | AD | |
| 614518 | GATAD1 | ENST00000287957.5 | 4571 | GATA Zinc Finger Domain Containing Protein 1 | <5% | [62] | AD | |
| 300644 | GLA | ENST00000218516.4 | 1318 | Galactosidase, alpha | <5% | [62] | XLR | |
| 602366 | ILK | ENST00000299421.9 | 1759 | Integrin-linked kinase | <5% | [62] | AD | |
| 173325 | JUP | ENST00000393931.8 | 3505 | Junction plakoglobin | <5% | [62] | AD | |
| 156225 | LAMA2 | ENST00000421865.2 | 9640 | Laminin Alpha, 2 | <5% | [62] | AR | |
| 600133 | LAMA4 | ENST00000230538.12 | 7228 | Laminin Alpha, 4 | <5% | [62] | AD | |
| 309060 | LAMP2 | ENST00000200639.9 | 6535 | Lysosome-associated membrane protein 2 | <5% | [62] | XLR | |
| 605906 | LDB3 | ENST00000429277.6 | 5436 | LIM domain-binding 3 | <5% | [62] | AD | |
| 150330 | LMNA | ENST00000368300.9 | 3178 | Lamin A/C | 10% | [62] | AD | |
| 600958 | MYBPC3 | ENST00000545968.6 | 4217 | Myosin-binding protein C, cardiac | <5% | [65,66,67] | AD | |
| 160710 | MYH6 | ENST00000405093.8 | 5945 | Alpha-myosin heavy chain 6 | <5% | [62] | AD | |
| 160760 | MYH7 | ENST00000355349.4 | 6027 | Myosin, heavy chain 7, cardiac muscle, beta | <5% | [65,66,67] | AD | |
| 160781 | MYL2 | ENST00000228841.15 | 783 | Myosin light chain 2 | <5% | [65,66,67] | AD | |
| 160790 | MYL3 | ENST00000292327.6 | 885 | Myosin light chain 3 | <5% | [65,66,67] | AD/AR | |
| 605603 | MYOZ1 | ENST00000359322.5 | 1300 | Myozenin 1 | <5% | [62] | AD/AR | |
| 605602 | MYOZ2 | ENST00000307128.6 | 2549 | Myozenin 2 | <5% | [62] | AD | |
| 608517 | MYPN | ENST00000613327.4 | 6116 | Myopalladin | <5% | [62] | AD | |
| 605491 | NEBL | ENST00000377122.8 | 9216 | Nebulette | <5% | [62] | AD | |
| 613121 | NEXN | ENST00000330010.12 | 3195 | Nexilin | <5% | [62] | AD | |
| 600584 | NKX2-5 | ENST00000329198.5 | 1558 | NK2 homeobox 5; cardiac specific homeobox 1 | <5% | [62] | AD | |
| 605900 | PDLIM1 | ENST00000329399.7 | 1431 | C-terminal LIM domain protein 1 | <5% | [62] | AD | |
| 605889 | PDLIM3 | ENST00000284767.12 | 2802 | PDZ and LIM domain protein 3 | <5% | [62] | AD | |
| 603422 | PDLIM4 | ENST00000253754.8 | 2257 | PDZ and LIM domain protein 4 | <5% | [62] | AD | |
| 602861 | PKP2 | ENST00000070846.10 | 4241 | Plakophilin 2 | <5% | [62] | AD | |
| 172405 | PLN | ENST00000357525.6 | 2989 | Phospholamban | <5% | [62] | AR | |
| 605557 | PRDM16 | ENST00000270722.10 | 8698 | PR domain containing 16 | <5% | [62] | AD | |
| 602743 | PRKAG2 | ENST00000287878.9 | 3279 | Protein Kinase, AMP-Activated, Non-Catalytic, Gamma 2 | <5% | [62] | AD | |
| 104311 | PSEN1 | ENST00000324501.10 | 6018 | Presenilin 1 | <5% | [62] | AD | |
| 600759 | PSEN2 | ENST00000366783.8 | 2249 | Presenilin 2 | <5% | [62] | AD | |
| 176876 | PTPN11 | ENST00000351677.7 | 6073 | Protein-Tyrosine Phosphatase, Non-Receptor Type, 11 | <5% | [62] | AD | |
| 613171 | RBM20 | ENST00000369519.4 | 7293 | RNA-binding motif protein 20 | 1–5% | [62] | AD | |
| 609591 | RIT1 | ENST00000368323.8 | 3390 | RIC-like protein without CAAX motif 1 | <5% | [62] | AD | |
| 180902 | RYR2 | ENST00000366574.7 | 16583 | Ryanodine Receptor 2 | <5% | [62] | AD | |
| 601144 | SCN5A | ENST00000423572.7 | 8516 | α subunit of the Nav1.5 sodium channel | <5% | [62] | AD | |
| 601411 | SGCD | ENST00000435422.7 | 9755 | Delta-sarcoglycan | <5% | [62] | AD | |
| 603377 | SLC22A5 | ENST00000245407.8 | 3277 | Solute Carrier Family 22 (Organic Cation Transporter), Member 5 | <5% | [62] | AR | |
| 601017 | SNTA1 | ENST00000217381.3 | 2211 | α1-Syntrophin | <5% | [62] | AD | |
| 182530 | SOS1 | ENST00000402219.7 | 8318 | SOS Ras/Rac guanine nucleotide exchange factor 1 | <5% | [62] | AD | |
| 607723 | SUN1 | ENST00000401592.6 | 4010 | SAD1 and UNC84 domain-containing protein 1 | <5% | [62] | AD | |
| 613569 | SUN2 | ENST00000405510.5 | 4055 | SAD1 and UNC84 domain-containing protein 2 | <5% | [62] | AD | |
| 608441 | SYNE1 | ENST00000367255.10 | 27708 | Nesprin 1, Synaptic nuclear envelop protein 1 | <5% | [62] | AD | |
| 608442 | SYNE2 | ENST00000358025.7 | 21842 | Nesprin 2, Synaptic nuclear envelop protein 2 | <5% | [62] | AD | |
| 300394 | TAZ | ENST00000601016.6 | 1906 | Tafazzin | <5% | [62] | XLR | |
| 601620 | TBX5 | ENST00000405440.7 | 3733 | T-box 5 | <5% | [62] | AD | |
| 604488 | TCAP | ENST00000309889.3 | 960 | Titin-cap; telethonin | <5% | [62] | AD/AR | |
| 603306 | TCF21 | ENST00000367882.5 | 1280 | Transcription factor 21, epicardin | <5% | [62] | AD | |
| 190230 | TGFB3 | ENST00000238682.7 | 2522 | Transforming Growth Factor β 3 | <5% | [62] | AD | |
| 612048 | TMEM43 | ENST00000306077.5 | 3230 | Transmembrane protein 43 | <5% | [62] | AD | |
| 188380 | TMPO | ENST00000556029.5 | 4242 | Thymopoietin | <5% | [62] | AD | |
| 191040 | TNNC1 | ENST00000232975.8 | 687 | Cardiac troponin C | <5% | [62] | AD | |
| 191044 | TNNI3 | ENST00000344887.10 | 843 | Cardiac troponin I3 | <5% | [65,66,67] | AD | |
| 191045 | TNNT2 | ENST00000656932.1 | 1165 | Cardiac troponin T2 | <5% | [65,66,67] | AD | |
| 191010 | TPM1 | ENST00000403994.9 | 1217 | Tropomyosin 1 | <5% | [65,66,67] | AD | |
| 188840 | TTN | ENST00000589042.5 | 109224 | Titin | 12–25% | [65,66,67] | AD | |
| 176300 | TTR | ENST00000237014.8 | 616 | Transthyretin | <5% | [62] | AD | |
| 193065 | VCL | ENST00000211998.10 | 5497 | Vinculin | <5% | [62] | AD | |
| Hypertrophic Cardiomyopathy (HCM) | 102540 | ACTC1 | ENST00000290378.6 | 1382 | Actin, alpha, cardiac muscle | <5% | [68] | AD |
| 102573 | ACTN2 | ENST00000546208.5 | 5103 | Alpha-actinin 2 | <5% | [62] | AD | |
| 603883 | BAG3 | ENST00000369085.8 | 2561 | BCL2-associated athanogene | <5% | [62] | AD | |
| 611414 | CALR3 | ENST00000269881.8 | 1263 | Calreticulin 3 | <5% | [62] | AD | |
| 601253 | CAV3 | ENST00000343849.3 | 1422 | Caveolin 3 | <5% | [62] | AD | |
| 123590 | CRYAB | ENST00000650687.2 | 774 | Alpha B crystallin | <5% | [62] | AD | |
| 600824 | CSRP3 | ENST00000265968.9 | 1283 | Cysteine- and glycine-rich protein 3 | <5% | [62] | AD | |
| 125660 | DES | ENST00000373960.4 | 2243 | Desmin | <5% | [62] | AD | |
| 300163 | FHL1 | ENST00000370683.6 | 2286 | Four-and—a-half LIM domains 1 | <5% | [62] | XLD/XLR | |
| 602633 | FHL2 | ENST00000409177.6 | 4881 | Four-and—a-half LIM domains 2 | <5% | [62] | AD | |
| 102565 | FLNC | ENST00000325888.12 | 9188 | Filamin C | <5% | [62] | AD | |
| 300644 | GLA | ENST00000218516.4 | 1318 | Galactosidase, alpha | <5% | [62] | XLR | |
| 602366 | ILK | ENST00000299421.9 | 1759 | Integrin-linked kinase | <5% | [62] | AD | |
| 605267 | JPH2 | ENST00000372980.4 | 9502 | Junctophilin 2 | <5% | [62] | AD | |
| 309060 | LAMP2 | ENST00000200639.9 | 6535 | Lysosome-associated membrane protein 2 | <5% | [62] | XLR | |
| 605906 | LDB3 | ENST00000429277.6 | 5436 | LIM domain-binding 3 | <5% | [62] | AD | |
| 600958 | MYBPC3 | ENST00000545968.6 | 4217 | Myosin-binding protein C, cardiac | 30–40% | [62] | AD | |
| 160710 | MYH6 | ENST00000405093.8 | 5945 | Alpha-myosin heavy chain 6 | <5% | [62] | AD | |
| 160760 | MYH7 | ENST00000355349.4 | 6027 | Myosin, heavy chain 7, cardiac muscle, beta | 20–30% | [62] | AD | |
| 609928 | MYH7B | ENST00000262873.12 | 6293 | Myosin Heavy Chain 7B | <5% | [62] | AD | |
| 160781 | MYL2 | ENST00000228841.15 | 783 | Myosin light chain 2 | <5% | [62] | AD | |
| 160790 | MYL3 | ENST00000292327.6 | 885 | Myosin light chain 3 | <5% | [62] | AD/AR | |
| 606566 | MYLK2 | ENST00000375985.5 | 2799 | Myosin light chain kinase 2 | <5% | [62] | AD | |
| 605603 | MYOZ1 | ENST00000359322.5 | 1300 | Myozenin 1 | <5% | [62] | AD/AR | |
| 605602 | MYOZ2 | ENST00000307128.6 | 2549 | Myozenin 2 | <5% | [62] | AD | |
| 608517 | MYPN | ENST00000613327.4 | 6116 | Myopalladin | <5% | [62] | AD | |
| 613121 | NEXN | ENST00000330010.12 | 3195 | Nexilin | <5% | [62] | AD | |
| 605900 | PDLIM1 | ENST00000329399.7 | 1431 | C-terminal LIM domain protein 1 | <5% | [62] | AD | |
| 605889 | PDLIM3 | ENST00000284767.12 | 2802 | PDZ and LIM domain protein 3 | <5% | [62] | AD | |
| 603422 | PDLIM4 | ENST00000253754.8 | 2257 | PDZ and LIM domain protein 4 | <5% | [62] | AD | |
| 172405 | PLN | ENST00000357525.6 | 2989 | Phospholamban | <5% | [62] | AD | |
| 602743 | PRKAG2 | ENST00000287878.9 | 3279 | Protein Kinase, AMP-Activated, Non-Catalytic, Gamma 2 | <5% | [62] | AD | |
| 176876 | PTPN11 | ENST00000351677.7 | 6073 | Protein-Tyrosine Phosphatase, Non-Receptor Type, 11 | <5% | [62] | AD | |
| 604488 | TCAP | ENST00000309889.3 | 960 | Titin-cap; telethonin | <5% | [62] | AD/AR | |
| 612418 | TMEM70 | ENST00000312184.6 | 2032 | Mitochondrial complex V (ATP synthase) deficiency, nuclear type 2 | <5% | [62] | AR | |
| 191040 | TNNC1 | ENST00000232975.8 | 687 | Cardiac troponin C | <5% | [68] | AD | |
| 191044 | TNNI3 | ENST00000344887.10 | 843 | Cardiac troponin I3 | 2–5% | [68] | AD | |
| 191045 | TNNT2 | ENST00000656932.1 | 1165 | Cardiac troponin T2 | 10–20% | [68] | AD | |
| 191010 | TPM1 | ENST00000403994.9 | 1217 | Tropomyosin 1 | 2–5% | [68] | AD | |
| 188840 | TTN | ENST00000589042.5 | 109224 | Titin | <5% | [62] | AD | |
| 176300 | TTR | ENST00000237014.8 | 616 | Transthyretin | <5% | [62] | AD | |
| 193065 | VCL | ENST00000211998.10 | 5497 | Vinculin | <5% | [62] | AD | |
| Restrictive cardiomyopathy (RCM) | 125660 | DES | ENST00000373960.4 | 2243 | Desmin | <5% | [69] | AD |
| 102565 | FLNC | ENST00000325888.12 | 9188 | Filamin C | <5% | [69] | AD | |
| 309060 | LAMP2 | ENST00000200639.9 | 6535 | Lysosome-associated membrane protein 2 | <5% | [69] | XLR | |
| 150330 | LMNA | ENST00000368300.9 | 3178 | Lamin A/C | <5% | [69] | AD | |
| 600958 | MYBPC3 | ENST00000545968.6 | 4217 | Myosin-binding protein C, cardiac | <5% | [69] | AD | |
| 160760 | MYH7 | ENST00000355349.4 | 6027 | Myosin, heavy chain 7, cardiac muscle, beta | <5% | [69] | AD | |
| 604488 | TCAP | ENST00000309889.3 | 960 | Titin-cap; telethonin | <5% | [69] | AD/AR | |
| 191044 | TNNI3 | ENST00000344887.10 | 843 | Cardiac troponin I3 | <5% | [69] | AD | |
| 191045 | TNNT2 | ENST00000656932.1 | 1165 | Cardiac troponin T2 | <5% | [69] | AD | |
| 191010 | TPM1 | ENST00000403994.9 | 1217 | Tropomyosin 1 | <5% | [69] | AD | |
| Left ventricular non-compaction (LVNC) | 102540 | ACTC1 | ENST00000290378.6 | 1382 | Actin, alpha, cardiac muscle | <5% | [70] | AD |
| 601239 | DTNA | ENST00000283365.13 | 6522 | Dystrobrevin, alpha | <5% | [70] | AD/AR | |
| 605906 | LDB3 | ENST00000429277.6 | 5436 | LIM domain-binding 3 | <5% | [70] | AD | |
| 150330 | LMNA | ENST00000368300.9 | 3178 | Lamin A/C | <5% | [70] | AD | |
| 600958 | MYBPC3 | ENST00000545968.6 | 4217 | Myosin-binding protein C, cardiac | 8% | [70] | AD | |
| 160760 | MYH7 | ENST00000355349.4 | 6027 | Myosin, heavy chain 7, cardiac muscle, beta | 13% | [70] | AD | |
| 191045 | TNNT2 | ENST00000656932.1 | 1165 | Cardiac troponin T2 | <5% | [70] | AD |
| Authors | Athletes (N) | Sex | Ethnicity | Age Range (y) | Age (y) or Mean Age (y + SD) | Reason for Enrolment after Clinical Evaluation | SCD Familiarity | Technology of Genetic Test | Positive Genetic Test (%) | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| N. Detta et al., 2014 | 1 | 1 M | White | NA | 12 | ECG: HR 50 bpm, with phases of sinus atrial blocks | No | Sanger (SCN5A, LMNA A/C,EMD, GJ5A, HCN4) | 100 | [24] |
| V. D’Argenio et al., 2018 | 1 | 1 M | White | NA | 8 | ECG: BrS pattern | No | NGS Panel (75 genes) | 100 | [42] |
| C. Mazzaccara et al., 2018 | 1 | 1 M | White | NA | 47 | HCM | No | Sanger (8 sarcomeric genes) | 100 | [43] |
| G. Limongelli et al., 2020 | 1 | 1 M | White | NA | 12 | Putative ACM | Yes | Sanger (PKP2, DSP, DSG2, DSC2, RYR2, JUP). NGS panel (138 genes) | 100 | [44] |
| C. Kadota et al., 2015 | 102 | NR | NR | 18–28 | NR | ECG abnormalities; ECO abnormalities (N = 7) | No | Sanger: MYBPC3, TNNT2, TNNI3, MYH7 genes | 4.9 | [46] |
| N. Sheikh et al., 2018 | 100 | 94 M 6 F | 50 Black 50 White | 14–35 | 22.7 + 7.3 (Black) 25.1 + 7.1 (White) | ECG: T-wave inversion. | NR | NGS panel (311 genes) | Black: 6 White: 14 | [47] |
| H. Cronin et al., 2020 | 10 | 10 M | White | 18–54 | 39 | ECG: deep T-wave inversion inferolaterally. All athletes were phenotypically normal | No | NGS panel (133 genes) | 0 | [48] |
| G. Limongelli et al., 2020 | 61 | 56 M 5 F | White | NR | 26 + 12.8 | Clinical suspicion for inherited cardiac disease | Yes | NGS panel (138 genes) | 13 | [50] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barretta, F.; Mirra, B.; Monda, E.; Caiazza, M.; Lombardo, B.; Tinto, N.; Scudiero, O.; Frisso, G.; Mazzaccara, C. The Hidden Fragility in the Heart of the Athletes: A Review of Genetic Biomarkers. Int. J. Mol. Sci. 2020, 21, 6682. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186682
Barretta F, Mirra B, Monda E, Caiazza M, Lombardo B, Tinto N, Scudiero O, Frisso G, Mazzaccara C. The Hidden Fragility in the Heart of the Athletes: A Review of Genetic Biomarkers. International Journal of Molecular Sciences. 2020; 21(18):6682. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186682
Chicago/Turabian StyleBarretta, Ferdinando, Bruno Mirra, Emanuele Monda, Martina Caiazza, Barbara Lombardo, Nadia Tinto, Olga Scudiero, Giulia Frisso, and Cristina Mazzaccara. 2020. "The Hidden Fragility in the Heart of the Athletes: A Review of Genetic Biomarkers" International Journal of Molecular Sciences 21, no. 18: 6682. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186682