Mechanisms Underlying the Regulation of Mitochondrial Respiratory Chain Complexes by Nuclear Steroid Receptors



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
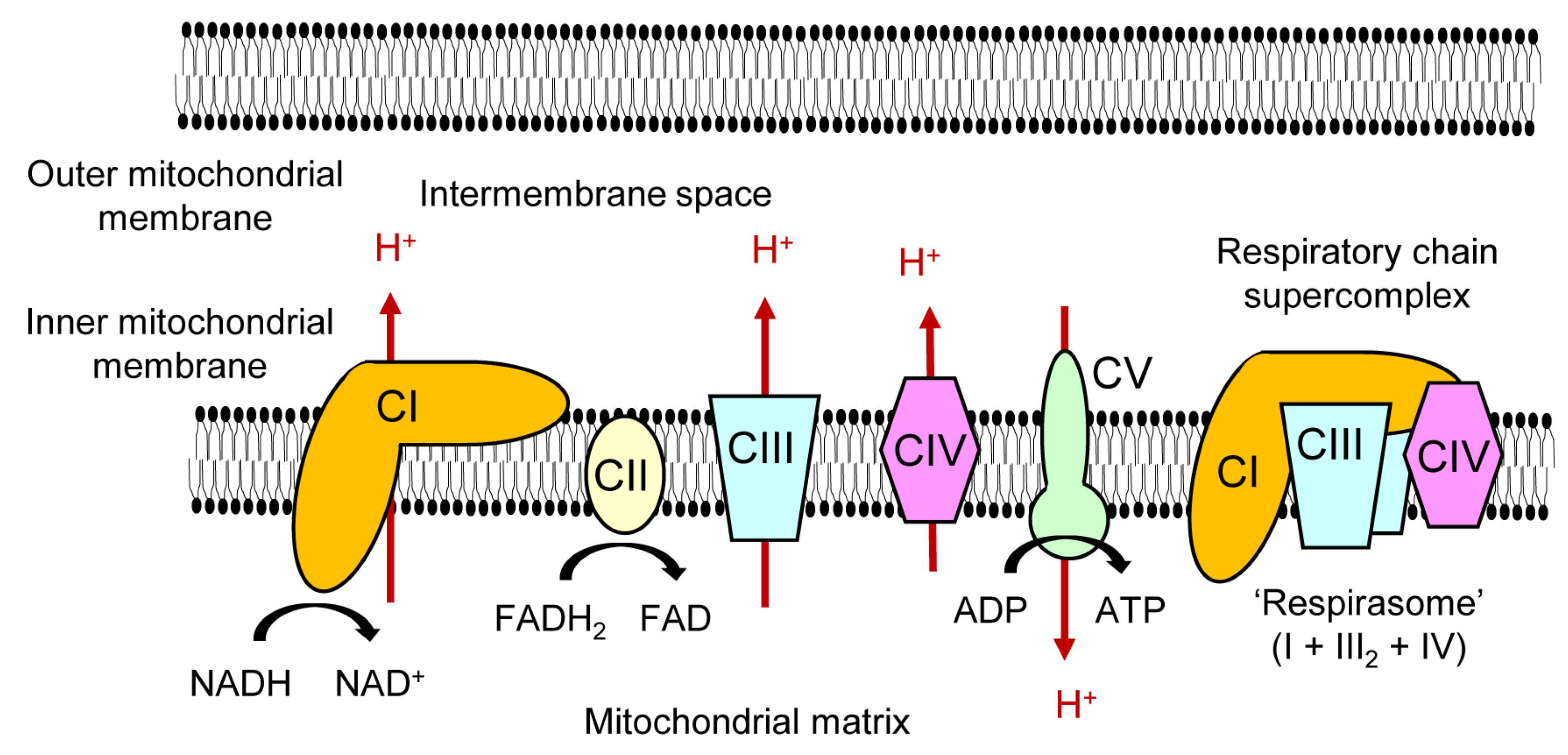
2. Regulatory Mechanisms of Nuclear Receptors Affecting Mitochondrial Respiratory Chain Complexes
3. ERs in the Regulation of Mitochondrial OXPHOS Complexes
4. ERRs in the Regulation of Mitochondrial OXPHOS Complexes
5. GRs in the Regulation of Mitochondrial OXPHOS Complexes
6. MR in the Regulation of Mitochondrial OXPHOS Complexes
7. PRs in the Regulation of Mitochondrial OXPHOS Complexes
8. AR in the Regulation of Mitochondrial OXPHOS Complexes
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Boczonadi, V.; Ricci, G.; Horvath, R. Mitochondrial DNA transcription and translation: Clinical syndromes. Essays Biochem. 2018, 62, 321–340. [Google Scholar] [CrossRef]
- Mayr, J.A.; Haack, T.B.; Freisinger, P.; Karall, D.; Makowski, C.; Koch, J.; Feichtinger, R.G.; Zimmermann, F.A.; Rolinski, B.; Ahting, U.; et al. Spectrum of combined respiratory chain defects. J. Inherit. Metab. Dis. 2015, 38, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Protasoni, M.; Pérez-Pérez, R.; Lobo-Jarne, T.; Harbour, M.E.; Ding, S.; Peñas, A.; Diaz, F.; Moraes, C.T.; Fearnley, I.M.; Zeviani, M.; et al. Respiratory supercomplexes act as a platform for complex III -mediated maturation of human mitochondrial complexes I and IV. EMBO J. 2020, 39, e102817. [Google Scholar] [CrossRef]
- Schägger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, K.; Shiba, S.; Horie-Inoue, K.; Shimokata, K.; Inoue, S. A stabilizing factor for mitochondrial respiratory supercomplex assembly regulates energy metabolism in muscle. Nat. Commun. 2013, 4, 2147. [Google Scholar] [CrossRef] [Green Version]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acín-Pérez, R.; Latorre-Pellicer, A.; Colás, C.; Balsa, E.; Perales-Clemente, E.; Quirós, P.M.; Calvo, E.; Rodríguez-Hernández, M.A.; et al. Supercomplex Assembly Determines Electron Flux in the Mitochondrial Electron Transport Chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef]
- Genova, M.L.; Lenaz, G. Functional role of mitochondrial respiratory supercomplexes. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 427–443. [Google Scholar] [CrossRef] [Green Version]
- Maranzana, E.; Barbero, G.; Falasca, A.I.; Lenaz, G.; Genova, M.L. Mitochondrial Respiratory Supercomplex Association Limits Production of Reactive Oxygen Species from Complex I. Antioxid. Redox Signal 2013, 19, 1469–1480. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Fabuel, I.; Le Douce, J.; Logan, A.; James, A.M.; Bonvento, G.; Murphy, M.P.; Almeida, A.; Bolaños, J.P. Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 13063–13068. [Google Scholar] [CrossRef] [Green Version]
- Schägger, H.; De Coo, R.; Bauer, M.F.; Hofmann, S.; Godinot, C.; Brandt, U. Significance of Respirasomes for the Assembly/Stability of Human Respiratory Chain Complex I. J. Boil. Chem. 2004, 279, 36349–36353. [Google Scholar] [CrossRef] [Green Version]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The Nuclear Receptor Superfamily: The Second Decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef] [Green Version]
- Mazaira, G.I.; Zgajnar, N.R.; Lotufo, C.M.; Daneri-Becerra, C.; Sivils, J.C.; Soto, O.B.; Cox, M.B.; Galigniana, M.D. The Nuclear Receptor Field: A Historical Overview and Future Challenges. Nucl. Recept. Res. 2018, 5, 101320. [Google Scholar] [CrossRef] [PubMed]
- Sandelin, A.; Wasserman, W.W. Prediction of Nuclear Hormone Receptor Response Elements. Mol. Endocrinol. 2005, 19, 595–606. [Google Scholar] [CrossRef]
- Azuma, K.; Inoue, S. Genomic and non-genomic actions of estrogen: Recent developments. Biomol. Concepts 2012, 3, 365–370. [Google Scholar] [CrossRef]
- Xiong, J.; Kuang, X.; Lu, T.; Liu, X.; Cheng, B.; Wang, W.; Wei, D.; Li, X.; Zhang, Z.; Fang, Q.; et al. Fenretinide-induced Apoptosis of Acute Myeloid Leukemia Cells via NR4A1 Translocation into Mitochondria and Bcl-2 Transformation. J. Cancer 2019, 10, 6767–6778. [Google Scholar] [CrossRef] [Green Version]
- Mattingly, K.A.; Ivanova, M.M.; Riggs, K.A.; Wickramasinghe, N.S.; Barch, M.J.; Klinge, C.M. Estradiol Stimulates Transcription of Nuclear Respiratory Factor-1 and Increases Mitochondrial Biogenesis. Mol. Endocrinol. 2008, 22, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Dufour, C.R.; Wilson, B.J.; Huss, J.M.; Kelly, D.P.; Alaynick, W.A.; Downes, M.R.; Evans, R.M.; Blanchette, M.; Giguère, V. Genome-wide Orchestration of Cardiac Functions by the Orphan Nuclear Receptors ERRα and γ. Cell Metab. 2007, 5, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Koh, J.-H.; Hancock, C.R.; Terada, S.; Higashida, K.; Holloszy, J.O.; Han, D.-H. PPARβ Is Essential for Maintaining Normal Levels of PGC-1α and Mitochondria and for the Increase in Muscle Mitochondria Induced by Exercise. Cell Metab. 2017, 25, 1176–1185.e5. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Transcriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cells. Gene 2002, 286, 81–89. [Google Scholar] [CrossRef]
- Kang, D.; Kim, S.H.; Hamasaki, N. Mitochondrial transcription factor A (TFAM): Roles in maintenance of mtDNA and cellular functions. Mitochondrion 2007, 7, 39–44. [Google Scholar] [CrossRef]
- Ikeuchi, M.; Matsusaka, H.; Kang, D.; Matsushima, S.; Ide, T.; Kubota, T.; Fujiwara, T.; Hamasaki, N.; Takeshita, A.; Sunagawa, K.; et al. Overexpression of Mitochondrial Transcription Factor A Ameliorates Mitochondrial Deficiencies and Cardiac Failure After Myocardial Infarction. Circulation 2005, 112, 683–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hondares, E.; Mora, O.; Yubero, P.; De La Concepción, M.R.; Iglesias, R.; Giralt, M.; Villarroya, F.; De La Concepción, M.L.R. Thiazolidinediones and Rexinoids Induce Peroxisome Proliferator-Activated Receptor-Coactivator (PGC)-1α Gene Transcription: An Autoregulatory Loop Controls PGC-1α Expression in Adipocytes via Peroxisome Proliferator-Activated Receptor-γ Coactivation. Endocrinology 2006, 147, 2829–2838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Liu, J.; Saha, P.; Huang, J.; Chan, L.; Spiegelman, B.; Moore, D.D. The orphan nuclear receptor SHP regulates PGC-1α expression and energy production in brown adipocytes. Cell Metab. 2005, 2, 227–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bost, F.; Kaminski, L. The metabolic modulator PGC-1α in cancer. Am. J. Cancer Res. 2019, 9, 198–211. [Google Scholar]
- Shao, D.; Liu, Y.; Liu, X.; Zhu, L.; Cui, Y.; Cui, A.; Qiao, A.; Kong, X.; Liu, Y.; Chen, Q.; et al. PGC-1β-Regulated mitochondrial biogenesis and function in myotubes is mediated by NRF-1 and ERRα. Mitochondrion 2010, 10, 516–527. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogenic control of mitochondrial function. Redox Boil. 2020, 31, 101435. [Google Scholar] [CrossRef]
- Kemper, M.F.; Stirone, C.; Krause, D.N.; Duckles, S.P.; Procaccio, V. Genomic and non-genomic regulation of PGC1 isoforms by estrogen to increase cerebral vascular mitochondrial biogenesis and reactive oxygen species protection. Eur. J. Pharmacol. 2013, 723, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Galmés-Pascual, B.M.; Nadal-Casellas, A.; Bauza-Thorbrügge, M.; Sbert-Roig, M.; García-Palmer, F.J.; Proenza, A.M.; Gianotti, M.; Llado, I. 17β-estradiol improves hepatic mitochondrial biogenesis and function through PGC1B. J. Endocrinol. 2017, 232, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Inoue, S.; Hiroi, H.; Orimo, A.; Kawashima, H.; Muramatsu, M. Isolation of Estrogen-Responsive Genes with a CpG Island Library. Mol. Cell. Boil. 1998, 18, 442–449. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, K.; Horie-Inoue, K.; Suzuki, T.; Hobo, R.; Nakasato, N.; Takeda, S.; Inoue, S. Mitochondrial supercomplex assembly promotes breast and endometrial tumorigenesis by metabolic alterations and enhanced hypoxia tolerance. Nat. Commun. 2019, 10, 4108–4115. [Google Scholar] [CrossRef]
- Nagai, S.; Ikeda, K.; Horie-Inoue, K.; Shiba, S.; Nagasawa, S.; Takeda, S.; Inoue, S. Estrogen modulates exercise endurance along with mitochondrial uncoupling protein 3 downregulation in skeletal muscle of female mice. Biochem. Biophys. Res. Commun. 2016, 480, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Nagai, S.; Ikeda, K.; Horie-Inoue, K.; Takeda, S.; Inoue, S. Estrogen signaling increases nuclear receptor subfamily 4 group A member 1 expression and energy production in skeletal muscle cells. Endocr. J. 2018, 65, 1209–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, M.S.; Hancock, C.R.; Ray, J.D.; Kener, K.B.; Draney, C.; Garland, K.; Hardman, J.; Bikman, B.; Tessem, J.S. β-Cell deletion of Nr4a1 and Nr4a3 nuclear receptors impedes mitochondrial respiration and insulin secretion. Am. J. Physiol. Metab. 2016, 311, E186–E201. [Google Scholar] [CrossRef] [Green Version]
- Klinge, C.M. Estrogens regulate life and death in mitochondria. J. Bioenerg. Biomembr. 2017, 49, 307–324. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Liu, R.; Perez, E.J.; Wen, Y.; Stevens, S.M.; Valencia, T.; Brun-Zinkernagel, A.M.; Prokai, L.; Will, Y.; Dykens, J.; et al. Mitochondrial localization of estrogen receptor β. Proc. Natl. Acad. Sci. USA 2004, 101, 4130–4135. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, M.I.G.L.; Shearwood, A.-M.J.; Chia, T.; Davies, S.M.K.; Rackham, O.; Filipovska, A. Estrogen-Mediated Regulation of Mitochondrial Gene Expression. Mol. Endocrinol. 2014, 29, 14–27. [Google Scholar] [CrossRef] [Green Version]
- Grober, O.M.; Mutarelli, M.; Giurato, G.; Ravo, M.; Cicatiello, L.; De Filippo, M.R.; Ferraro, L.; Nassa, G.; Papa, M.F.; Paris, O.; et al. Global analysis of estrogen receptor beta binding to breast cancer cell genome reveals an extensive interplay with estrogen receptor alpha for target gene regulation. BMC Genom. 2011, 12, 36. [Google Scholar] [CrossRef]
- Velickovic, K.; Cvoro, A.; Srdić, B.; Stokić, E.; Markelić, M.B.; Golic, I.; Otasevic, V.; Stancic, A.; Jankovic, A.; Vučetić, M.; et al. Expression and Subcellular Localization of Estrogen Receptors α and β in Human Fetal Brown Adipose Tissue. J. Clin. Endocrinol. Metab. 2014, 99, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Seval, Y.; Cakmak, H.A.; Kayisli, U.; Arici, A. Estrogen-Mediated Regulation of p38 Mitogen-Activated Protein Kinase in Human Endometrium. J. Clin. Endocrinol. Metab. 2006, 91, 2349–2357. [Google Scholar] [CrossRef] [Green Version]
- Bauerfeld, C.; Talwar, H.; Zhang, K.; Liu, Y.; Samavati, L. MKP-1 Modulates Mitochondrial Transcription Factors, Oxidative Phosphorylation, and Glycolysis. ImmunoHorizons 2020, 4, 245–258. [Google Scholar] [CrossRef]
- Hoppstädter, J.; Ammit, A.J. Role of Dual-Specificity Phosphatase 1 in Glucocorticoid-Driven Anti-inflammatory Responses. Front. Immunol. 2019, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, I.; Starskaia, I.; Nagy, T.; Guo, J.; Yatkin, E.; Väänänen, K.; Watford, W.T.; Chen, Z. Estrogen receptor α contributes to T cell–mediated autoimmune inflammation by promoting T cell activation and proliferation. Sci. Signal 2018, 11, eaap9415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, V.; Drew, B.G.; Zhou, Z.; Phun, J.; Kalajian, N.Y.; Soleymani, T.; Daraei, P.; Widjaja, K.; Wanagat, J.; Vallim, T.Q.D.A.; et al. Skeletal muscle action of estrogen receptor α is critical for the maintenance of mitochondrial function and metabolic homeostasis in females. Sci. Transl. Med. 2016, 8, 334ra54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, T.-L.; Lee, Y.-C.; Tzeng, C.-R.; Wang, Y.-P.; Chang, H.-Y.; Lin, Y.-F.; Kao, S.-H. Mitochondrial translocation of estrogen receptor β affords resistance to oxidative insult-induced apoptosis and contributes to the pathogenesis of endometriosis. Free. Radic. Boil. Med. 2019, 134, 359–373. [Google Scholar] [CrossRef]
- Bauza-Thorbrügge, M.; Rodríguez-Cuenca, S.; Vidal-Puig, A.; Galmés-Pascual, B.M.; Sbert-Roig, M.; Gianotti, M.; Lladó, I.; Proenza, A.M. GPER and ERα mediate estradiol enhancement of mitochondrial function in inflamed adipocytes through a PKA dependent mechanism. J. Steroid Biochem. Mol. Boil. 2019, 185, 256–267. [Google Scholar] [CrossRef]
- Torres, M.J.; Kew, K.A.; Ryan, T.E.; Pennington, E.R.; Lin, C.-T.; Buddo, K.A.; Fix, A.M.; Smith, C.A.; Gilliam, L.A.; Karvinen, S.; et al. 17β-Estradiol Directly Lowers Mitochondrial Membrane Microviscosity and Improves Bioenergetic Function in Skeletal Muscle. Cell Metab. 2018, 27, 167–179.e7. [Google Scholar] [CrossRef] [Green Version]
- Gallart-Palau, X.; Lee, B.S.T.; Adav, S.S.; Qian, J.; Serra, A.; Park, J.E.; Lai, M.K.P.; Chen, C.P.; Kalaria, R.N.; Sze, S.K. Gender differences in white matter pathology and mitochondrial dysfunction in Alzheimer’s disease with cerebrovascular disease. Mol. Brain 2016, 9, 27. [Google Scholar] [CrossRef] [Green Version]
- Gaignard, P.; Liere, P.; Thérond, P.; Schumacher, M.; Slama, A.; Guennoun, R. Role of Sex Hormones on Brain Mitochondrial Function, with Special Reference to Aging and Neurodegenerative Diseases. Front Aging Neurosci. 2017, 9, 406. [Google Scholar] [CrossRef]
- Greising, S.M.; Baltgalvis, K.A.; Lowe, D.A.; Warren, G.L. Hormone Therapy and Skeletal Muscle Strength: A Meta-Analysis. J. Gerontol. Ser. A Boil. Sci. Med Sci. 2009, 64, 1071–1081. [Google Scholar] [CrossRef]
- Zuercher, W.; Gaillard, S.; Orband-Miller, L.A.; Chao, E.Y.H.; Shearer, B.G.; Jones, D.G.; Miller, A.B.; Collins, J.L.; McDonnell, D.P.; Willson, T.M. Identification and Structure−Activity Relationship of Phenolic Acyl Hydrazones as Selective Agonists for the Estrogen-Related Orphan Nuclear Receptors ERRβ and ERRγ. J. Med. Chem. 2005, 48, 3107–3109. [Google Scholar] [CrossRef]
- Coward, P.; Lee, D.; Hull, M.V.; Lehmann, J.M. 4-Hydroxytamoxifen binds to and deactivates the estrogen-related receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 8880–8884. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Dufour, C.R.; Giguère, V. ERRα as a Bridge Between Transcription and Function: Role in Liver Metabolism and Disease. Front. Endocrinol. 2019, 10, 206. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, S.N.; Knutti, D.; Brogli, K.; Uhlmann, T.; Kralli, A. The Transcriptional Coactivator PGC-1 Regulates the Expression and Activity of the Orphan Nuclear Receptor Estrogen-Related Receptor α (ERRα). J. Boil. Chem. 2003, 278, 9013–9018. [Google Scholar] [CrossRef] [Green Version]
- Chaveroux, C.; Eichner, L.J.; Dufour, C.R.; Shatnawi, A.; Khoutorsky, A.; Bourque, G.; Sonenberg, N.; Giguère, V. Molecular and Genetic Crosstalks between mTOR and ERRα Are Key Determinants of Rapamycin-Induced Nonalcoholic Fatty Liver. Cell Metab. 2013, 17, 586–598. [Google Scholar] [CrossRef] [Green Version]
- Gantner, M.L.; Hazen, B.C.; Conkright, J.; Kralli, A. GADD45 regulates the thermogenic capacity of brown adipose tissue. Proc. Natl. Acad. Sci. USA 2014, 111, 11870–11875. [Google Scholar] [CrossRef] [Green Version]
- Charest-Marcotte, A.; Dufour, C.R.; Wilson, B.J.; Tremblay, A.M.; Eichner, L.J.; Arlow, D.H.; Mootha, V.K.; Giguère, V. The homeobox protein Prox1 is a negative modulator of ERR /PGC-1 bioenergetic functions. Genes Dev. 2010, 24, 537–542. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Schindler, J.; Summermatter, S.; Salatino, S.; Zorzato, F.; Beer, M.; Balwierz, P.J.; Van Nimwegen, E.; Feige, J.N.; Auwerx, J.; Handschin, C. The Corepressor NCoR1 Antagonizes PGC-1α and Estrogen-Related Receptor α in the Regulation of Skeletal Muscle Function and Oxidative Metabolism. Mol. Cell. Boil. 2012, 32, 4913–4924. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; McDonald, C.; Petrenko, N.B.; Leblanc, M.; Wang, T.; Giguère, V.; Evans, R.M.; Patel, V.V.; Pei, L. Estrogen-Related Receptor α (ERRα) and ERRγ Are Essential Coordinators of Cardiac Metabolism and Function. Mol. Cell. Boil. 2015, 35, 1281–1298. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.L.; Hazen, B.C.; Eury, E.; Wattez, J.-S.; Gantner, M.L.; Albert, V.; Chau, S.; Sanchez-Alavez, M.; Conti, B.; Kralli, A. Estrogen-Related Receptors Mediate the Adaptive Response of Brown Adipose Tissue to Adrenergic Stimulation. iScience 2018, 2, 221–237. [Google Scholar] [CrossRef] [Green Version]
- Gantner, M.L.; Hazen, B.C.; Eury, E.; Brown, E.L.; Kralli, A. Complementary Roles of Estrogen-Related Receptors in Brown Adipocyte Thermogenic Function. Endocrinology 2016, 157, 4770–4781. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Wang, Y.; Hunter, R.G.; Wei, Y.; Blumenthal, R.; Falke, C.; Khairova, R.; Zhou, R.; Yuan, P.; Machado-Vieira, R.; et al. Dynamic regulation of mitochondrial function by glucocorticoids. Proc. Natl. Acad. Sci. USA 2009, 106, 3543–3548. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-R.; Kim, H.-K.; Song, I.-S.; Youm, J.; Dizon, L.A.; Jeong, S.-H.; Ko, T.-H.; Heo, H.-J.; Ko, K.S.; Rhee, B.D.; et al. Glucocorticoids and their receptors: Insights into specific roles in mitochondria. Prog. Biophys. Mol. Boil. 2013, 112, 44–54. [Google Scholar] [CrossRef]
- Doig, C.L.; Fletcher, R.S.; Morgan, S.A.; McCabe, E.L.; Larner, D.P.; Tomlinson, J.W.; Stewart, P.M.; Philp, A.; Lavery, G.G. 11β-HSD1 Modulates the Set Point of Brown Adipose Tissue Response to Glucocorticoids in Male Mice. Endocrinology 2017, 158, 1964–1976. [Google Scholar] [CrossRef] [Green Version]
- Glantschnig, C.; Mattijssen, F.; Vogl, E.S.; Khan, A.A.; Garcia, M.R.; Fischer, K.; Müller, T.; Uhlenhaut, H.; Nawroth, P.; Scheideler, M.; et al. The glucocorticoid receptor in brown adipocytes is dispensable for control of energy homeostasis. EMBO Rep. 2019, 20, e48552. [Google Scholar] [CrossRef]
- Chen, Q.M.; Alexander, D.; Sun, H.; Xie, L.; Lin, Y.; Terrand, J.; Morrissy, S.; Purdom, S. Corticosteroids Inhibit Cell Death Induced by Doxorubicin in Cardiomyocytes: Induction of Antiapoptosis, Antioxidant, and Detoxification Genes. Mol. Pharmacol. 2005, 67, 1861–1873. [Google Scholar] [CrossRef] [Green Version]
- Muzikar, K.A.; Nickols, N.G.; Dervan, P.B. Repression of DNA-binding dependent glucocorticoid receptor-mediated gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 16598–16603. [Google Scholar] [CrossRef] [Green Version]
- André, F.; Trinh, A.; Balayssac, S.; Maboudou, P.; Dekiouk, S.; Malet-Martino, M.; Quesnel, B.; Idziorek, T.; Kluza, J.; Marchetti, P. Metabolic rewiring in cancer cells overexpressing the glucocorticoid-induced leucine zipper protein (GILZ): Activation of mitochondrial oxidative phosphorylation and sensitization to oxidative cell death induced by mitochondrial targeted drugs. Int. J. Biochem. Cell Boil. 2017, 85, 166–174. [Google Scholar] [CrossRef]
- Hunter, R.G.; Seligsohn, M.; Rubin, T.G.; Griffiths, B.B.; Ozdemir, Y.; Pfaff, N.W.; Datson, N.A.; McEwen, B. Stress and corticosteroids regulate rat hippocampal mitochondrial DNA gene expression via the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 2016, 113, 9099–9104. [Google Scholar] [CrossRef] [Green Version]
- Desquiret-Dumas, V.; Gueguen, N.; Malthièry, Y.; Ritz, P.; Simard, G. Mitochondrial effects of dexamethasone imply both membrane and cytosolic-initiated pathways in HepG2 cells. Int. J. Biochem. Cell Boil. 2008, 40, 1629–1641. [Google Scholar] [CrossRef] [Green Version]
- Fuller, P.J.; Young, M.J. Mechanisms of Mineralocorticoid Action. Hypertension 2005, 46, 1227–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The Effect of Spironolactone on Morbidity and Mortality in Patients with Severe Heart Failure. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Zannad, F.; McMurray, J.J.; Krum, H.; Van Veldhuisen, D.J.; Swedberg, K.; Shi, H.; Vincent, J.; Pocock, S.J.; Pitt, B. Eplerenone in Patients with Systolic Heart Failure and Mild Symptoms. N. Engl. J. Med. 2011, 364, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Fuller, P.J.; Yang, J.; Young, M.J. Mechanisms of Mineralocorticoid Receptor Signaling. Pancreat. Beta Cell 2019, 109, 37–68. [Google Scholar] [CrossRef]
- Van Weert, L.; Buurstede, R.; Mahfouz, A.; Braakhuis, P.S.; Polman, J.A.E.; Sips, H.C.; Roozendaal, B.; Balog, J.; De Kloet, E.R.; Datson, N.A.; et al. NeuroD Factors Discriminate Mineralocorticoid From Glucocorticoid Receptor DNA Binding in the Male Rat Brain. Endocrinology 2017, 158, 1511–1522. [Google Scholar] [CrossRef]
- Le Billan, F.; Khan, J.A.; Lamribet, K.; Viengchareun, S.; Bouligand, J.; Fagart, J.; Lombes, M. Cistrome of the aldosterone-activated mineralocorticoid receptor in human renal cells. FASEB J. 2015, 29, 3977–3989. [Google Scholar] [CrossRef] [Green Version]
- Caldiz, C.I.; Díaz, R.G.; Nolly, M.B.; De Cingolani, G.E.C.; Ennis, I.L.; Cingolani, H.E.; Pérez, N.G. Mineralocorticoid receptor activation is crucial in the signalling pathway leading to the Anrep effect. J. Physiol. 2011, 589, 6051–6061. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Dong, R.; Zhang, Y.; Yang, Y.; Chen, Z.; Tang, Y.; Fu, M.; Xu, X.; Tu, L. Age-related changes in mineralocorticoid receptors in rat hearts. Mol. Med. Rep. 2020, 22, 1859–1867. [Google Scholar] [CrossRef]
- Jie, Q.-Q.; Li, G.; Duan, J.-B.; Li, X.-B.; Yang, W.; Chu, Y.-P.; Yu, S.-D.; Liu, X.-Y.; Wang, C.-Y.; Liu, F.-F.; et al. Remodeling of myocardial energy and metabolic homeostasis in a sheep model of persistent atrial fibrillation. Biochem. Biophys. Res. Commun. 2019, 517, 8–14. [Google Scholar] [CrossRef]
- Ibarrola, J.; Sadaba, R.; Martinez-Martinez, E.; Garcia-Peña, A.; Arrieta, V.; Álvarez, V.; Fernandez-Celis, A.; Gainza, A.; Cachofeiro, V.; Santamaría, E.; et al. Aldosterone Impairs Mitochondrial Function in Human Cardiac Fibroblasts via A-Kinase Anchor Protein 12. Sci. Rep. 2018, 8, 6801. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Martinez, E.; Ibarrola, J.; Lachén-Montes, M.; Fernández-Celis, A.; Jaisser, F.; Santamaría, E.; Fernández-Irigoyen, J.; López-Andrés, N. Differential proteomics reveals S100-A11 as a key factor in aldosterone-induced collagen expression in human cardiac fibroblasts. J. Proteom. 2017, 166, 93–100. [Google Scholar] [CrossRef]
- Azuma, K.; Ikeda, K.; Inoue, S. Functional Mechanisms of Mitochondrial Respiratory Chain Supercomplex Assembly Factors and Their Involvement in Muscle Quality. Int. J. Mol. Sci. 2020, 21, 3182. [Google Scholar] [CrossRef] [PubMed]
- Kastner, P.; Krust, A.; Turcotte, B.; Stropp, U.; Tora, L.; Gronemeyer, H.; Chambon, P. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. EMBO J. 1990, 9, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, B.M.; Horwitz, K.B. Progesterone receptors, their isoforms and progesterone regulated transcription. Mol. Cell. Endocrinol. 2012, 357, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Vegeto, E.; Shahbaz, M.M.; Wen, D.X.; Goldman, M.E.; O’Malley, B.W.; McDonnell, D.P. Human progesterone receptor A form is a cell- and promoter-specific repressor of human progesterone receptor B function. Mol. Endocrinol. 1993, 7, 1244–1255. [Google Scholar] [PubMed]
- Dinh, D.T.; Breen, J.; Akison, L.K.; DeMayo, F.J.; Brown, H.M.; Robker, R.L.; Russell, D.L. Tissue-specific progesterone receptor-chromatin binding and the regulation of progesterone-dependent gene expression. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chi, R.-P.A.; Wang, T.; Adams, N.; Wu, S.-P.; Young, S.L.; Spencer, T.E.; DeMayo, F.J. Human Endometrial Transcriptome and Progesterone Receptor Cistrome Reveal Important Pathways and Epithelial Regulators. J. Clin. Endocrinol. Metab. 2019, 105, 1419–1439. [Google Scholar] [CrossRef]
- Boonyaratanakornkit, V.; Scott, M.P.; Ribon, V.; Sherman, L.; Anderson, S.M.; Maller, J.L.; Miller, W.; Edwards, D.P. Progesterone Receptor Contains a Proline-Rich Motif that Directly Interacts with SH3 Domains and Activates c-Src Family Tyrosine Kinases. Mol. Cell 2001, 8, 269–280. [Google Scholar] [CrossRef]
- Rodríguez, A.M.; Monjo, M.; Roca, P.; Palou, A. Opposite actions of testosterone and progesterone on UCP1 mRNA expression in cultured brown adipocytes. Cell. Mol. Life Sci. 2002, 59, 1714–1723. [Google Scholar] [CrossRef]
- Rodriguez-Cuenca, S.; Monjo, M.; Gianotti, M.; Proenza, A.M.; Roca, P. Expression of mitochondrial biogenesis-signaling factors in brown adipocytes is influenced specifically by 17β-estradiol, testosterone, and progesterone. Am. J. Physiol. Metab. 2007, 292, E340–E346. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Price, T.M. The Role of a Mitochondrial Progesterone Receptor (PR-M) in Progesterone Action. Semin. Reprod. Med. 2015, 33, 185–194. [Google Scholar] [CrossRef]
- Feng, Q.; Crochet, J.R.; Dai, Q.; Leppert, P.C.; Price, T.M. Expression of a Mitochondrial Progesterone Receptor (PR-M) in Leiomyomata and Association With Increased Mitochondrial Membrane Potential. J. Clin. Endocrinol. Metab. 2014, 99, E390–E399. [Google Scholar] [CrossRef] [Green Version]
- Behera, M.A.; Dai, Q.; Garde, R.; Saner, C.; Jungheim, E.; Price, T.M. Progesterone stimulates mitochondrial activity with subsequent inhibition of apoptosis in MCF-10A benign breast epithelial cells. Am. J. Physiol. Metab. 2009, 297, E1089–E1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Q.; Provost, M.P.; Raburn, D.J.; Price, T.M. Progesterone Increases Mitochondria Membrane Potential in Non-human Primate Oocytes and Embryos. Reprod. Sci. 2020, 27, 1206–1214. [Google Scholar] [CrossRef]
- Wang, F.; Yang, J.; Sun, J.; Dong, Y.; Zhao, H.; Shi, H.; Fu, L. Testosterone replacement attenuates mitochondrial damage in a rat model of myocardial infarction. J. Endocrinol. 2015, 225, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hioki, T.; Suzuki, S.; Morimoto, M.; Masaki, T.; Tozawa, R.; Morita, S.; Horiguchi, T. Brain Testosterone Deficiency Leads to Down-Regulation of Mitochondrial Gene Expression in Rat Hippocampus Accompanied by a Decline in Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α Expression. J. Mol. Neurosci. 2013, 52, 531–537. [Google Scholar] [CrossRef]
- Yan, W.; Kang, Y.; Ji, X.; Li, S.; Li, Y.; Zhang, G.; Cui, H.; Shi, G. Testosterone Upregulates the Expression of Mitochondrial ND1 and ND4 and Alleviates the Oxidative Damage to the Nigrostriatal Dopaminergic System in Orchiectomized Rats. Oxidative Med. Cell. Longev. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- Pourshafie, N.; Masati, E.; Bunker, E.; Nickolls, A.R.; Thepmankorn, P.; Johnson, K.; Feng, X.; Ekins, T.; Grunseich, C.; Fischbeck, K.H. Linking epigenetic dysregulation, mitochondrial impairment, and metabolic dysfunction in SBMA motor neurons. JCI Insight 2020, 5, 136539. [Google Scholar] [CrossRef]
- Sen, A.; Hammes, S.R. Granulosa cell-specific androgen receptors are critical regulators of ovarian. Mol. Endocrinol. 2010, 24, 1393–1403. [Google Scholar] [CrossRef]
- Wang, R.-S.; Chang, H.-Y.; Kao, S.-H.; Kao, C.-H.; Wu, Y.-C.; Yeh, S.; Tzeng, C.-R.; Chang, C. Abnormal Mitochondrial Function and Impaired Granulosa Cell Differentiation in Androgen Receptor Knockout Mice. Int. J. Mol. Sci. 2015, 16, 9831–9849. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, P.; Koc, E.; Sonpavde, G.; Singh, R.; Singh, K.K. Mitochondrial localization, import, and mitochondrial function of the androgen receptor. J. Boil. Chem. 2019, 294, 6621–6634. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobayashi, A.; Azuma, K.; Ikeda, K.; Inoue, S. Mechanisms Underlying the Regulation of Mitochondrial Respiratory Chain Complexes by Nuclear Steroid Receptors. Int. J. Mol. Sci. 2020, 21, 6683. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186683
Kobayashi A, Azuma K, Ikeda K, Inoue S. Mechanisms Underlying the Regulation of Mitochondrial Respiratory Chain Complexes by Nuclear Steroid Receptors. International Journal of Molecular Sciences. 2020; 21(18):6683. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186683
Chicago/Turabian StyleKobayashi, Ami, Kotaro Azuma, Kazuhiro Ikeda, and Satoshi Inoue. 2020. "Mechanisms Underlying the Regulation of Mitochondrial Respiratory Chain Complexes by Nuclear Steroid Receptors" International Journal of Molecular Sciences 21, no. 18: 6683. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186683