Doubly-Charged Negative Ions as Novel Tunable Catalysts: Graphene and Fullerene Molecules Versus Atomic Metals


Abstract
:1. Introduction
2. Results
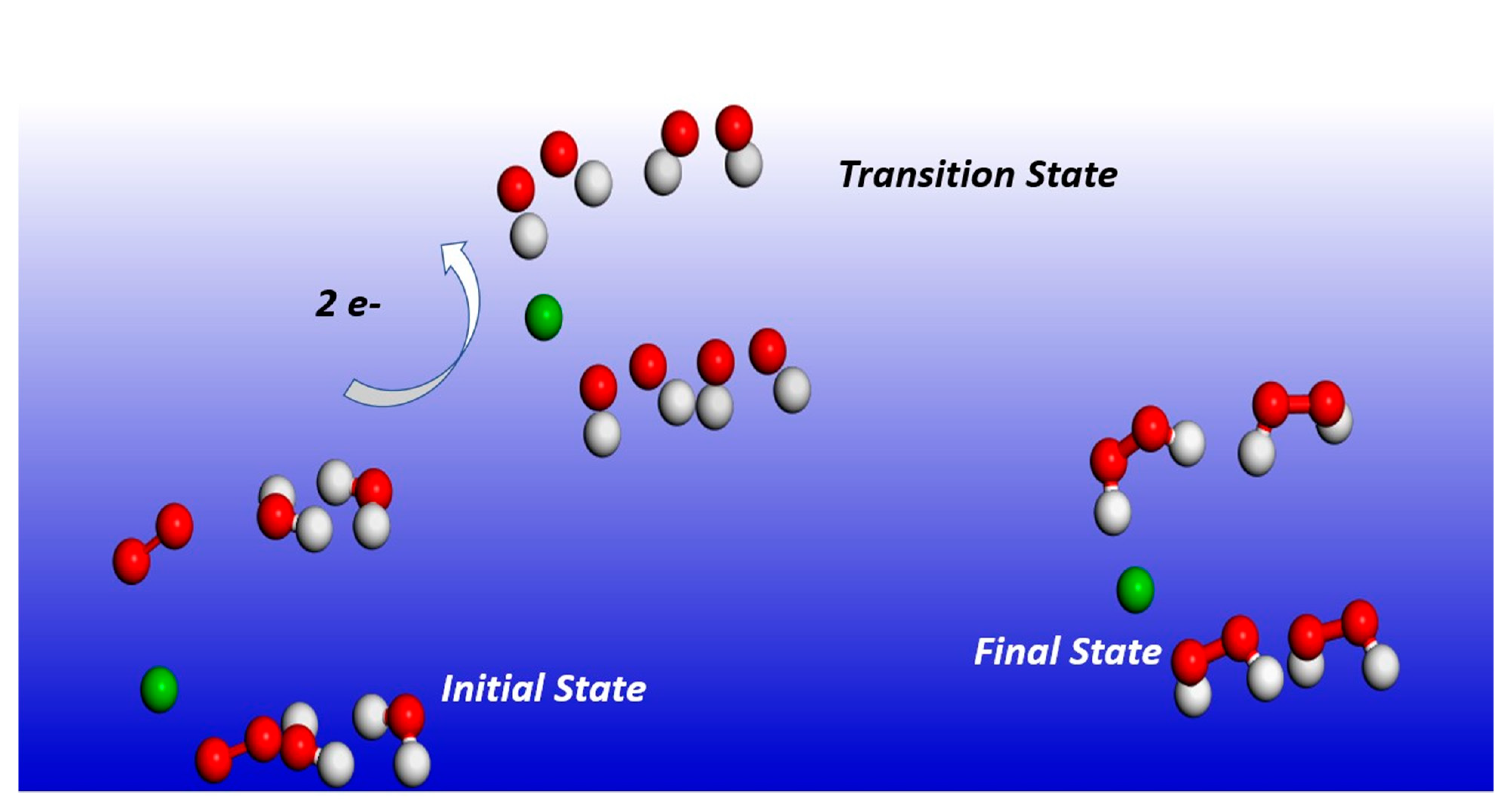
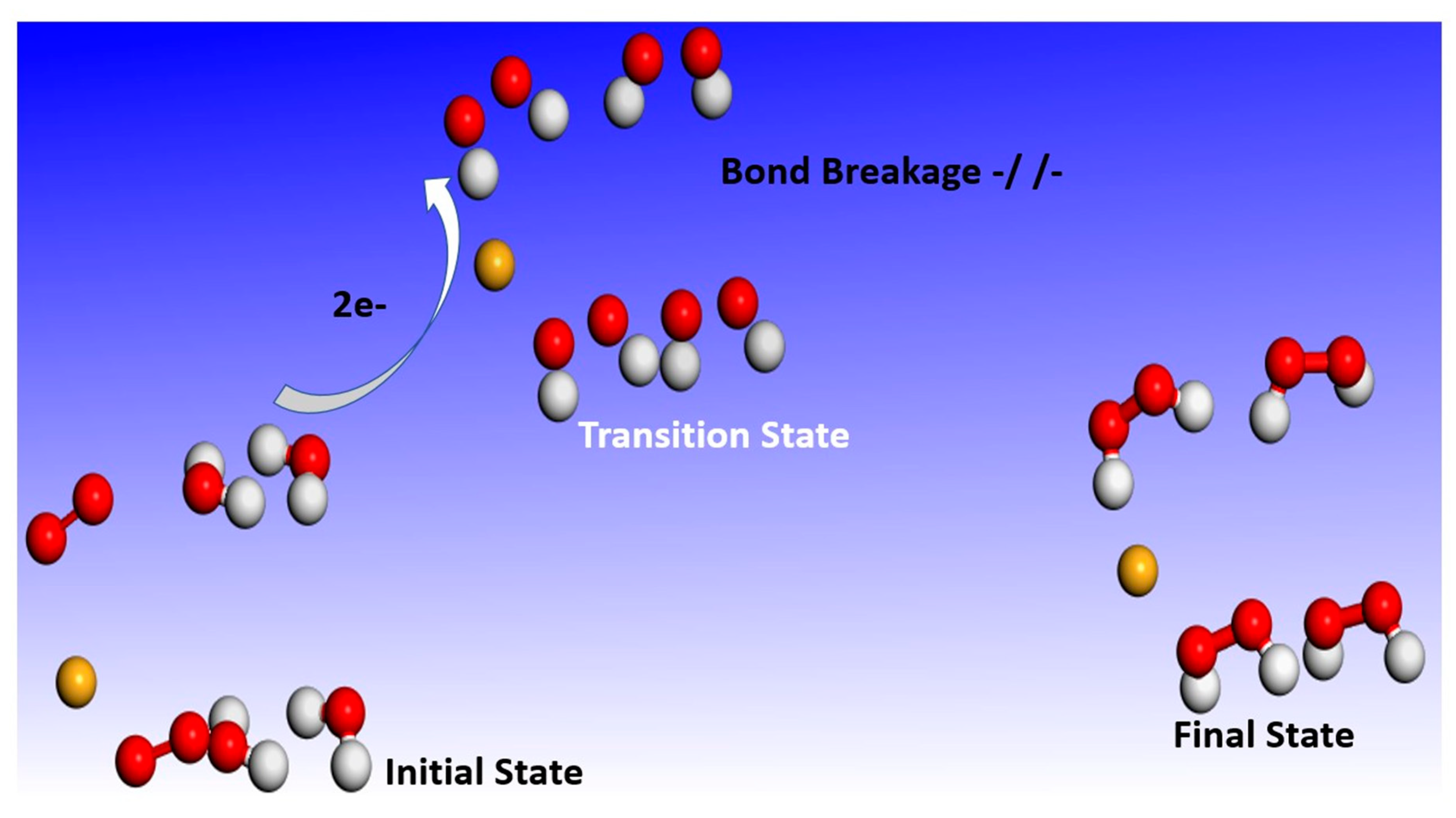
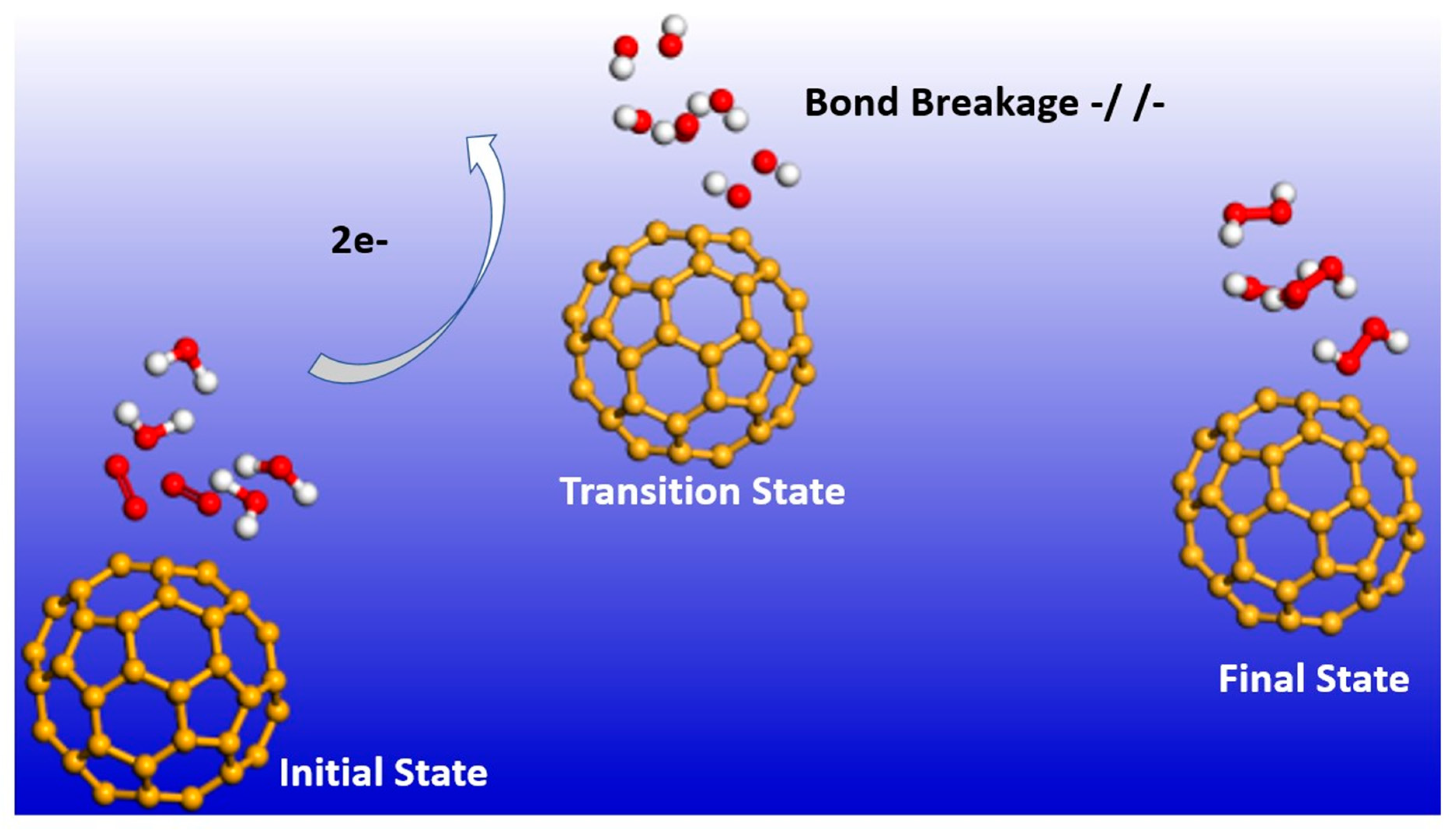
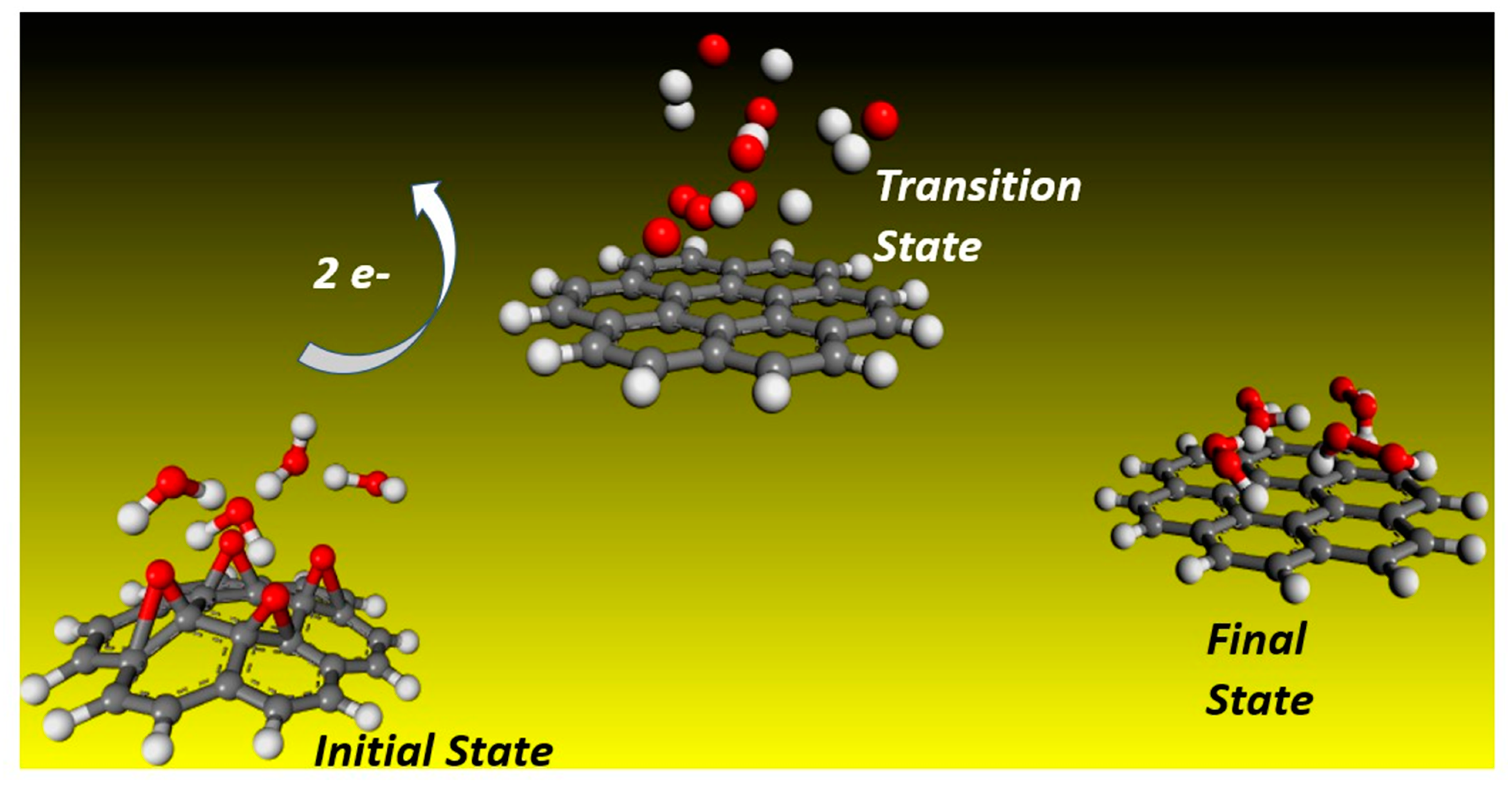
2.1. Reaction Dynamics
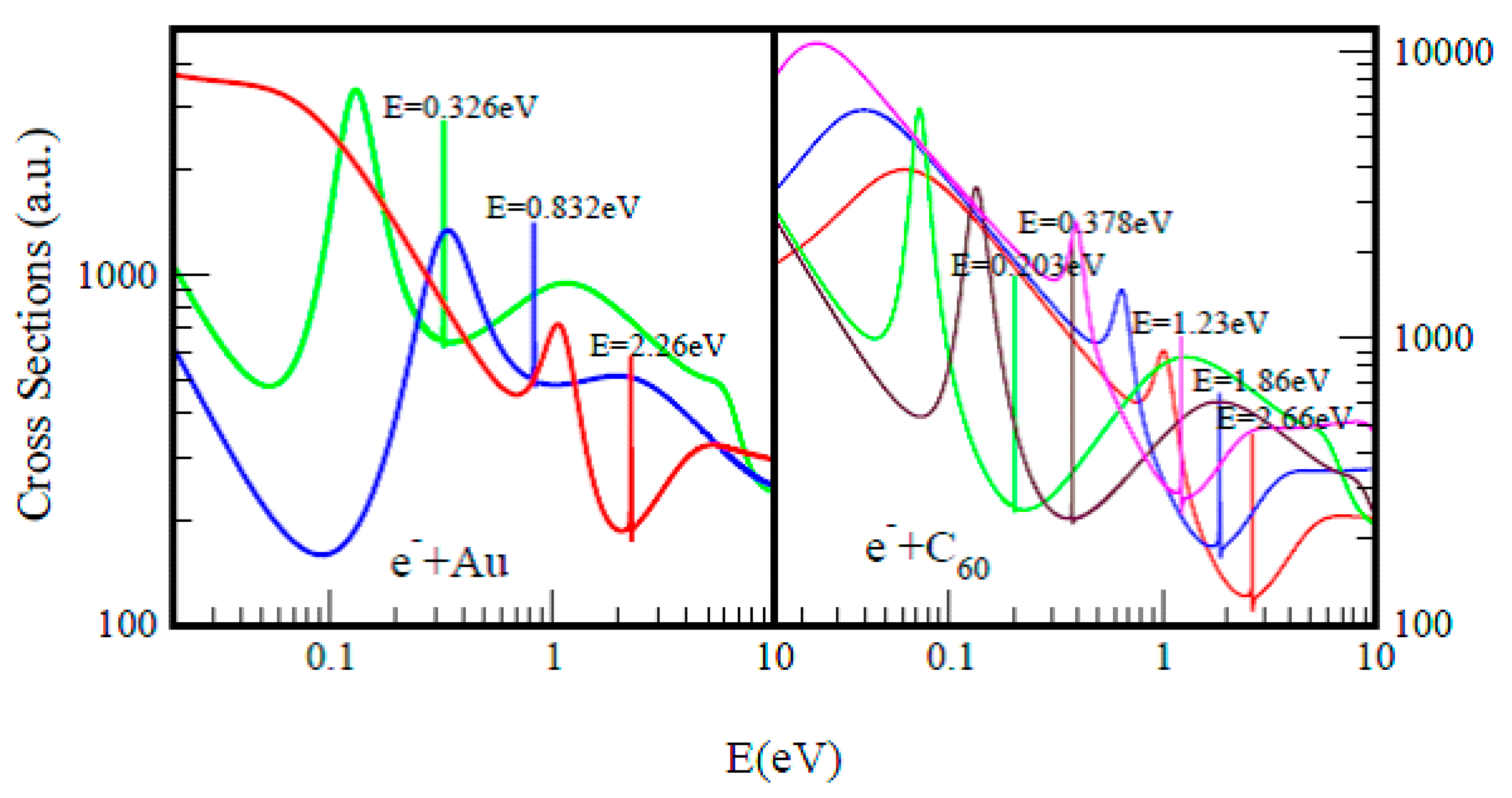
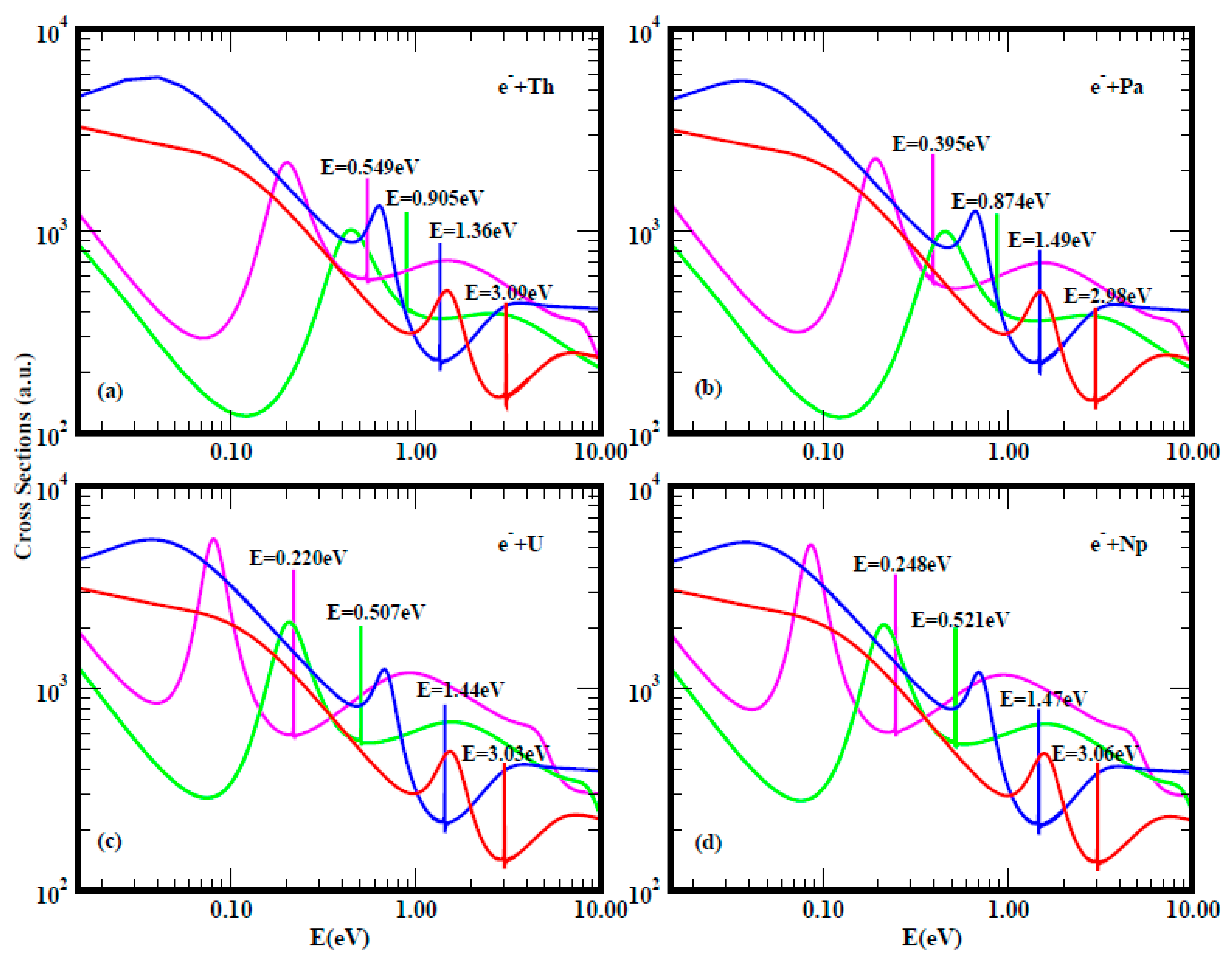
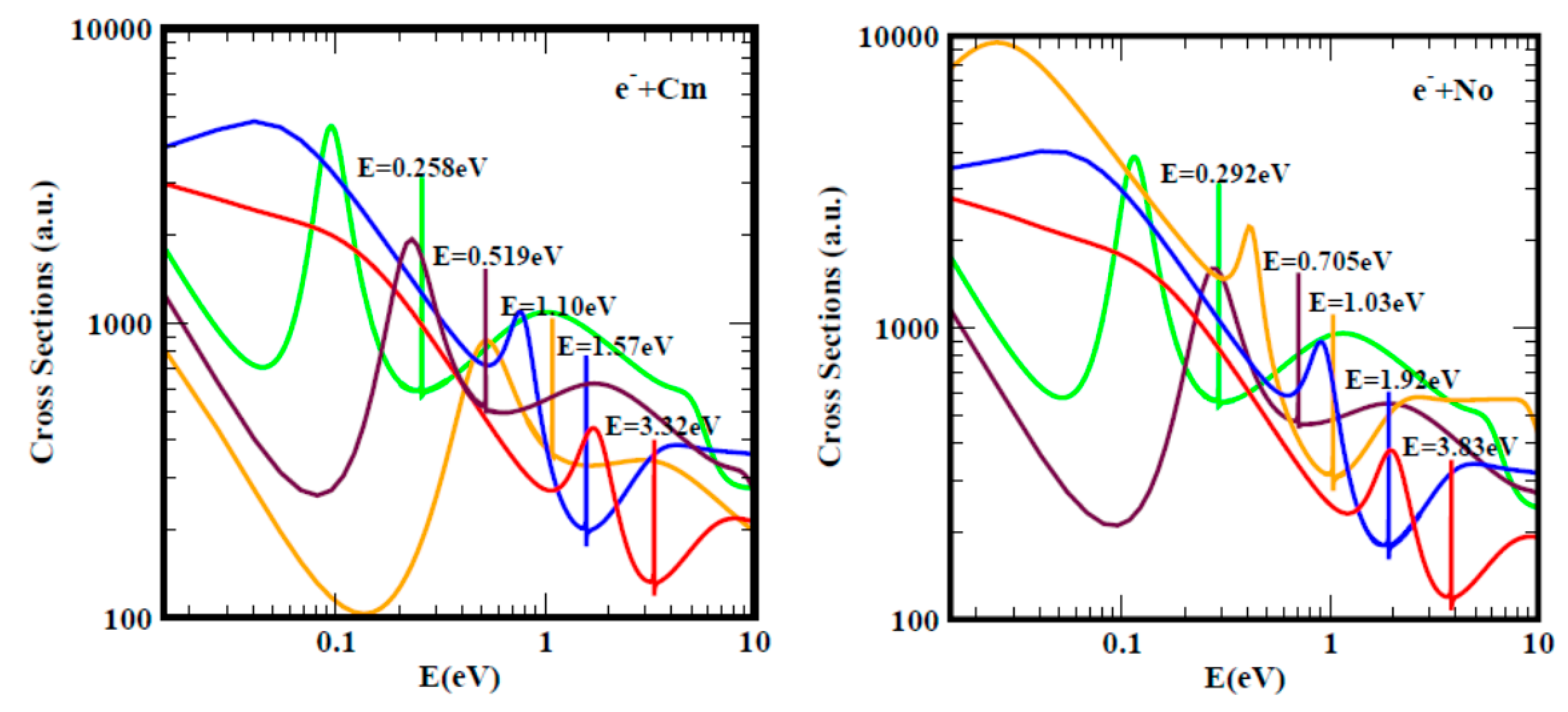
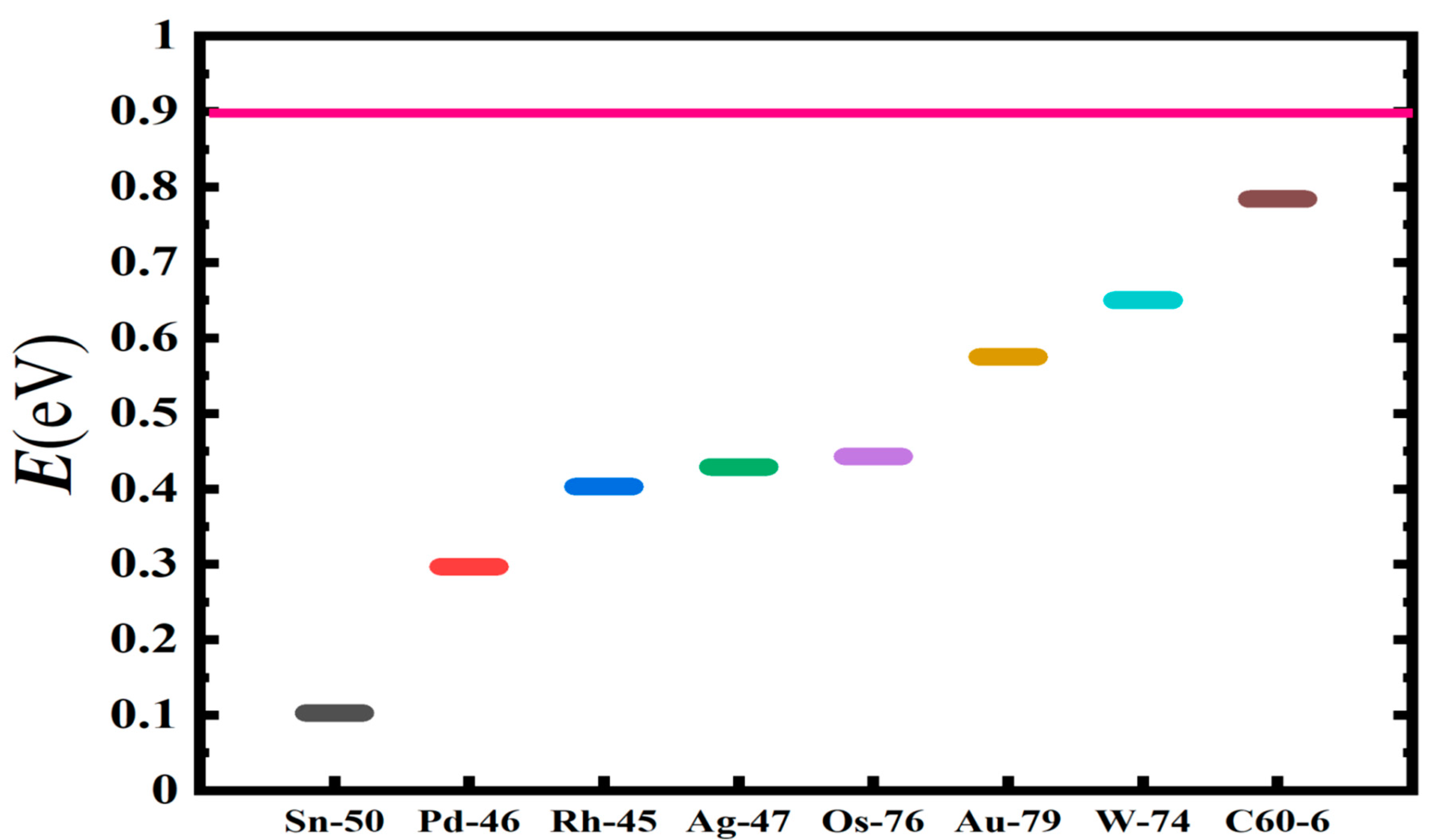
2.2. Electron Scattering Total Cross Sections and Electron Affinity Calculations
2.3. Transition State Energy Barriers Calculations
3. Method of Calculation
3.1. Total Cross Sections Calculation
3.2. Transition State Energy Barriers Calculation
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mauthe, S.; Fleischer, I.; Bernhardt, T.M.; Lang, S.M.; Barnett, R.N.; Landman, U. A Gas-Phase CanMn4–nO4+ Cluster Model for the Oxygen-Evolving Complex of Photosystem II. Angew. Chem. Int. Ed. Engl. 2019, 58, 8504. [Google Scholar] [CrossRef] [PubMed]
- Ruttinger, W.; Dismukes, G.C. Synthetic Water-Oxidation Catalysts for Artificial Photosynthetic Water Oxidation. Chem. Rev. 1997, 97, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Bozoglian, F.; Mandal, S.; Stewart, B.; Privalov, T.; Llobet, A.; Sun, L. A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem II. Nat. Chem. 2012, 4, 418–423. [Google Scholar] [CrossRef]
- Sartorel, A.; Carraro, M.; Scorrano, G.; Bonchio, M. Water oxidation catalysis by molecular metal-oxides. Energy Procedia 2012, 22, 78–87. [Google Scholar] [CrossRef]
- Du, P.; Eisenberg, R. Catalysts made of earth-abundant elements (Co, Ni, Fe) for water splitting: Recent progress and future challenges. Energy Environ. Sci. 2012, 5, 6012. [Google Scholar] [CrossRef]
- Anantharaj, S.; Ede, S.R.; Sakthikumar, K.; Karthick, K.; Mishra, S.; Kundu, S. Recent trends and perspectives in electrochemical water splitting with an emphasis on sulfide, selenide, and phosphide catalysts of Fe, Co, and Ni: A review. ACS Catal. 2016, 6, 8069–8097. [Google Scholar] [CrossRef]
- Jiao, Y.; Zheng, Y.; Jaroniec, M.; Qiao, S.Z. Design of electrocatalysts for oxygen-and hydrogen-involving energy conversion reactions. Chem. Soc. Rev. 2015, 44, 2060–2086. [Google Scholar] [CrossRef]
- Duan, J.; Chen, S.; Jaroniec, M.; Qiao, S.Z. Heteroatom-doped graphene-based materials for energy-relevant electrocatalytic processes. ACS Catal. 2015, 5, 5207–5234. [Google Scholar] [CrossRef]
- Sayler, R.I.; Hunter, B.M.; Fu, W.; Gray, H.B.; Britt, R.D. EPR spectroscopy of iron-and nickel-doped [ZnAl]-layered double hydroxides: Modeling active sites in heterogeneous water oxidation catalysts. J. Am. Chem. Soc. 2020, 142, 1838–1845. [Google Scholar] [CrossRef] [Green Version]
- Vogiatzis, K.D.; Polynski, M.V.; Kirkland, J.K.; Townsend, J.; Hashemi, A.; Liu, C.; Pidko, E.A. Computational approach to molecular catalysis by 3d transition metals: Challenges and opportunities. Chem. Rev. 2019, 119, 2453–2523. [Google Scholar] [CrossRef] [Green Version]
- Mehrabani, S.; Bikas, R.; Zand, Z.; Suleyman, I. Water splitting by a pentanuclear iron complex. Int. J. Hydrogen Energy 2020, 45, 17434. [Google Scholar] [CrossRef]
- Askerka, M.; Brudvig, G.W.; Batista, V.S. The O2-evolving complex of photosystem II: Recent insights from quantum mechanics/molecular mechanics (QM/MM), extended X-ray absorption fine structure (EXAFS), and femtosecond X-ray crystallography data. Acc. Chem. Res. 2017, 50, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, B.; Sun, L. Iron-based molecular water oxidation catalysts: Abundant, cheap, and promising. Chem. Asian J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Shylin, S.I.; Pavliuk, M.V.; D’Amario, L.; Mamedov, F.; Sá, J.; Berggren, G.; Fritsky, I.O. Efficient visible light-driven water oxidation catalysed by an iron (iv) clathrochelate complex. Chem. Commun. 2019, 55, 3335–3338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Takatsuka, K. Charge separation and successive reconfigurations of electronic and protonic states in a water-splitting catalytic cycle with the Mn 4 CaO 5 cluster. On the mechanism of water splitting in PSII. Phys. Chem. Chem. Phys. 2020, 22, 7912–7934. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Masuzaki, D.; Mafuné, F. Hydrophilicity and oxophilicity of the isolated CaMn 4 O 5 cationic cluster modeling inorganic core of the oxygen-evolving complex. Chem. Commun. 2019, 55, 14327–14330. [Google Scholar] [CrossRef]
- Moitra, P.; Alafeef, M.; Dighe, K.; Frieman, M.B.; Pan, D. Selective naked-eye detection of SARS-CoV-2 mediated by N Gene targeted antisense oligonucleotide capped plasmonic nanoparticles. ACS Nano 2020, 14, 7617. [Google Scholar] [CrossRef]
- Iglesias-Mayor, A.; Amor-Gutiérrez, O.; Novelli, A.; Fernández-Sánchez, M.-T.; Costa-García, A.; de la Escosura-Muñiz, A. Bifunctional Au@ Pt/Au core@ shell nanoparticles as novel electrocatalytic tags in immunosensing: Application for Alzheimer’s disease biomarker detection. Anal. Chem. 2020, 92, 7209. [Google Scholar] [CrossRef]
- Speller, E.M. The significance of fullerene electron acceptors in organic solar cell photo-oxidation. Mater. Sci. Technol. 2017, 33, 924–933. [Google Scholar] [CrossRef] [Green Version]
- Armour, E.A.G. Muon, positron and antiproton interactions with atoms and molecules. J. Phys. Conf. Ser. 2010, 225, 012002. [Google Scholar] [CrossRef] [Green Version]
- Leimbach, D.; Sundberg, J.; Guo, Y.; Ahmed, R.; Ballof, J.; Bengtsson, L.; Pamies, F.B.; Borschevsky, A.; Chrysalidis, K.; Eliav, E.; et al. The electron affinity of astatine. arXiv 2002, arXiv:2002.11418. [Google Scholar]
- International Year of the Periodic Table: Single Atoms as Active Catalysts. Available online: https://0-pubs-rsc-org.brum.beds.ac.uk/en/journals/articlecollectionlanding?sercode=nr&themeid=1fc90a67-e081-4265-99eb-2201eb17c286 (accessed on 12 January 2017).
- Felfli, Z.; Suggs, K.; Nicholas, N.; Msezane, A.Z. Fullerene negative ions: Formation and catalysis. Int. J. Mol. Sci. 2020, 21, 3159. [Google Scholar] [CrossRef] [PubMed]
- Msezane, A.Z.; Suggs, K. Open Access Government. Available online: www.openaccessgovernment.org/www.pbctoday.co.uk (accessed on 17 June 2020).
- Edwards, J.K.; Carley, A.F.; Herzing, A.A.; Kiely, C.J.; Hutchings, G.J. Direct synthesis of hydrogen peroxide from H 2 and O 2 using supported Au–Pd catalysts. Chem. Soc. Faraday Discuss 2008, 138, 225. [Google Scholar] [CrossRef]
- Edwards, J.K.; Solsona, B.; Landon, P.; Carley, A.F.; Herzing, A.; Watanabe, M.; Kiely, C.J.; Hutchings, G.J. Direct synthesis of hydrogen peroxide from H2 and O2 using Au–Pd/Fe 2O3 catalysts. J. Mater. Chem. 2005, 15, 4595–4600. [Google Scholar] [CrossRef]
- Freakley, S.J.; He, Q.; Harrhy, J.H.; Lu, L.; Crole, D.A.; Morgan, D.J.; Ntainjua, E.N.; Edwards, J.K.; Carley, A.F.; Borisevich, A.Y.; et al. Palladium-tin catalysts for the direct synthesis of H2O2 with high selectivity. Science 2016, 351, 959–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesfamichael, A.; Suggs, K.; Felfli, Z.; Wang, X.-Q.; Msezane, A.Z. Atomic gold and palladium negative ion-catalysis of water to peroxide: Fundamental mechanism. J. Nanoparticles Res. 2013, 15, 1333. [Google Scholar] [CrossRef]
- Gao, Y.; Huang, W.; Woodford, J.; Wang, L.-S.; Zeng, X.C. Detecting weak interactions between Au− and gas molecules: A photoelectron spectroscopic and ab initio study. J. Am. Chem. Soc. 2009, 131, 9484–9485. [Google Scholar] [CrossRef]
- Zheng, W.; Li, X.; Eustis, S.; Grubisic, A.; Thomas, O.; De Clercq, H.; Bowen, K. Anion photoelectron spectroscopy of Au−(H2O) 1, 2, Au2-(D2O) 1–4, and AuOH−. Chem. Phys. Lett. 2007, 444, 232–236. [Google Scholar] [CrossRef]
- Msezane, A.Z.; Felfli, Z.; Sokolovski, D. Novel mechanism for nanoscale catalysis. J. Phys. B 2010, 43, 201001. [Google Scholar] [CrossRef]
- Felfli, Z.; Msezane, A.Z. Negative ion formation in low-energy electron collisions with the actinide atoms Th, Pa, U, Np and Pu. Appl. Phys. Res. 2019, 11, 52. [Google Scholar] [CrossRef] [Green Version]
- Hotop, H.; Lineberger, W.C. Dye-laser photodetachment studies of Au−, Pt−, PtN−, and Ag−. J. Chem. Phys. 2003, 58, 2379–2387. [Google Scholar] [CrossRef]
- Andersen, T.; Haugen, H.K.; Hotop, H. Binding energies in atomic negative ions: III. J. Phys. Chem. Ref. Data 1999, 28, 1511–1533. [Google Scholar] [CrossRef]
- Huang, D.-L.; Dau, P.D.; Liu, H.T.; Wang, L.-S. High-resolution photoelectron imaging of cold C 60− anions and accurate determination of the electron affinity of C60. J. Chem. Phys. 2014, 140, 224315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brink, C.; Andersen, L.H.; Hvelplund, P.; Mathur, D.; Voldstad, J.D. Laser photodetachment of C60− and C70− ions cooled in a storage ring. Chem. Phys. Lett. 1995, 233, 52. [Google Scholar] [CrossRef]
- Støchkel, K.; Andersen, J.U. Photo excitation and laser detachment of C60− anions in a storage ring. J. Chem. Phys. 2013, 139, 164304. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Si, R.; Fei, Z.; Fu, X.; Lu, Y.; Brage, T.; Liu, H.; Chen, C.; Ning, C. Candidate for laser cooling of a negative ion: High-resolution photoelectron imaging of Th−. Phys. Rev. Lett. 2019, 123, 203002. [Google Scholar] [CrossRef] [PubMed]
- Felfli, Z.; Msezane, A.Z. Low-energy electron elastic total cross sections for Ho, Er, Tm, Yb, Lu, and Hf atoms. Atoms 2020, 8, 17. [Google Scholar] [CrossRef]
- Msezane, A.Z.; Felfli, Z. New insights in low-energy electron-fullerene interactions. Chem. Phys. 2018, 503, 50–55. [Google Scholar] [CrossRef]
- Frautschi, S.C. Regge Poles and S-Matrix Theory; Benjamin: New York, NY, USA, 1963; Chapter X. [Google Scholar]
- D’Alfaro, V.; Regge, T.E. Potential Scattering; North-Holland: Amsterdam, The Netherlands, 1965. [Google Scholar]
- Omnes, R.; Froissart, M. Mandelstam Theory and Regge Poles; Benjamin: New York, NY, USA, 1963; Chapter 2. [Google Scholar]
- Hiscox, A.; Brown, B.M.; Marletta, M. On the low energy behavior of Regge poles. J. Math. Phys. 2010, 51, 102104. [Google Scholar] [CrossRef]
- Mulholland, H.P. An Asymptotic Expansion for. In Mathematical Proceedings of the Cambridge Philosophical Society; Cambridge University Press: London, UK, 1928; Volume 24, pp. 280–289. [Google Scholar] [CrossRef]
- Macek, J.H.; Krstic, P.S.; Ovchinnikov, S.Y. Regge oscillations in integral cross sections for proton impact on atomic hydrogen. Phys. Rev. Lett. 2004, 93, 183203. [Google Scholar] [CrossRef] [PubMed]
- Sokolovski, D.; Felfli, Z.; Ovchinnikov, S.Y.; Macek, J.H.; Msezane, A.Z. Regge oscillations in electron-atom elastic cross sections. Phys. Rev. A 2007, 76, 012705. [Google Scholar] [CrossRef]
- Felfli, Z.; Msezane, A.Z.; Sokolovski, D. Resonances in low-energy electron elastic cross sections for lanthanide atoms. Phys. Rev. A 2009, 79, 012714. [Google Scholar] [CrossRef] [Green Version]
- Dolmatov, V.K.; Amusia, M.Y.; Chernysheva, L.V. Electron elastic scattering off A@C60: The role of atomic polarization under confinement. Phys. Rev. A 2017, 95, 012709. [Google Scholar] [CrossRef] [Green Version]
- Felfli, Z.; Belov, S.; Avdonina, N.B.; Marletta, M.; Msezane, A.Z.; Naboko, S.N. Regge poles trajectories for nonsingular potentials: The Thomas-Fermi potentials. In Proceedings of the Third International Workshop on Contemporary Problems in Mathematical Physics, Cotonou, Benin, 1–7 November 2003; Govaerts, J., Hounkonnou, M.N., Msezane, A.Z., Eds.; World Scientific: Singapore, 2004; pp. 217–232. [Google Scholar]
- Belov, S.; Thylwe, K.-E.; Marletta, M.; Msezane, A.Z.; Naboko, S.N. On Regge pole trajectories for a rational function approximation of Thomas–Fermi potentials. J. Phys. A 2010, 43, 365301. [Google Scholar] [CrossRef]
- Burke, P.G.; Tate, C. A program for calculating Regge trajectories in potential scattering. Comp. Phys. Commun. 1969, 1, 97. [Google Scholar] [CrossRef]
- Thylwe, K.W. On relativistic shifts of negative-ion resonances. Eur. Phys. J. D 2012, 66, 7. [Google Scholar] [CrossRef]
- Tkatchenko, A.; Scheffler, M. Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102, 73005. [Google Scholar] [CrossRef] [Green Version]
- DMOL3 GUIDE. Available online: moz-extension://75baf89d-92ea-4fcb-86cb-05280d756e86/pdf-viewer/web/viewer.html?file=http%3A%2F%2Fnees.sci.upc.edu.cn%2F_upload%2Farticle%2Ffiles%2F39%2Ff5%2F5460e8894554bd75148145ba414e%2F188a6221-e993-431c-bf9e-28e3051fd772.pdf (accessed on 10 September 2020).
- Suggs, K.; Kiros, F.; Tesfamichael, A.; Felfli, Z.; Msezane, A.Z. Charge modification of metal atoms: Catalysis of water to peroxide. J. Phys. Conf. Ser. 2015, 635, 052018. [Google Scholar] [CrossRef]
- Hu, Y.-L.; Zhu, H.-R.; Wei, S.H. Single-doped charged gold cluster with highly selective catalytic activity for the reduction of SO2 by CO: First-principles study. Chin. Phys. B 2019, 28, 113101. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
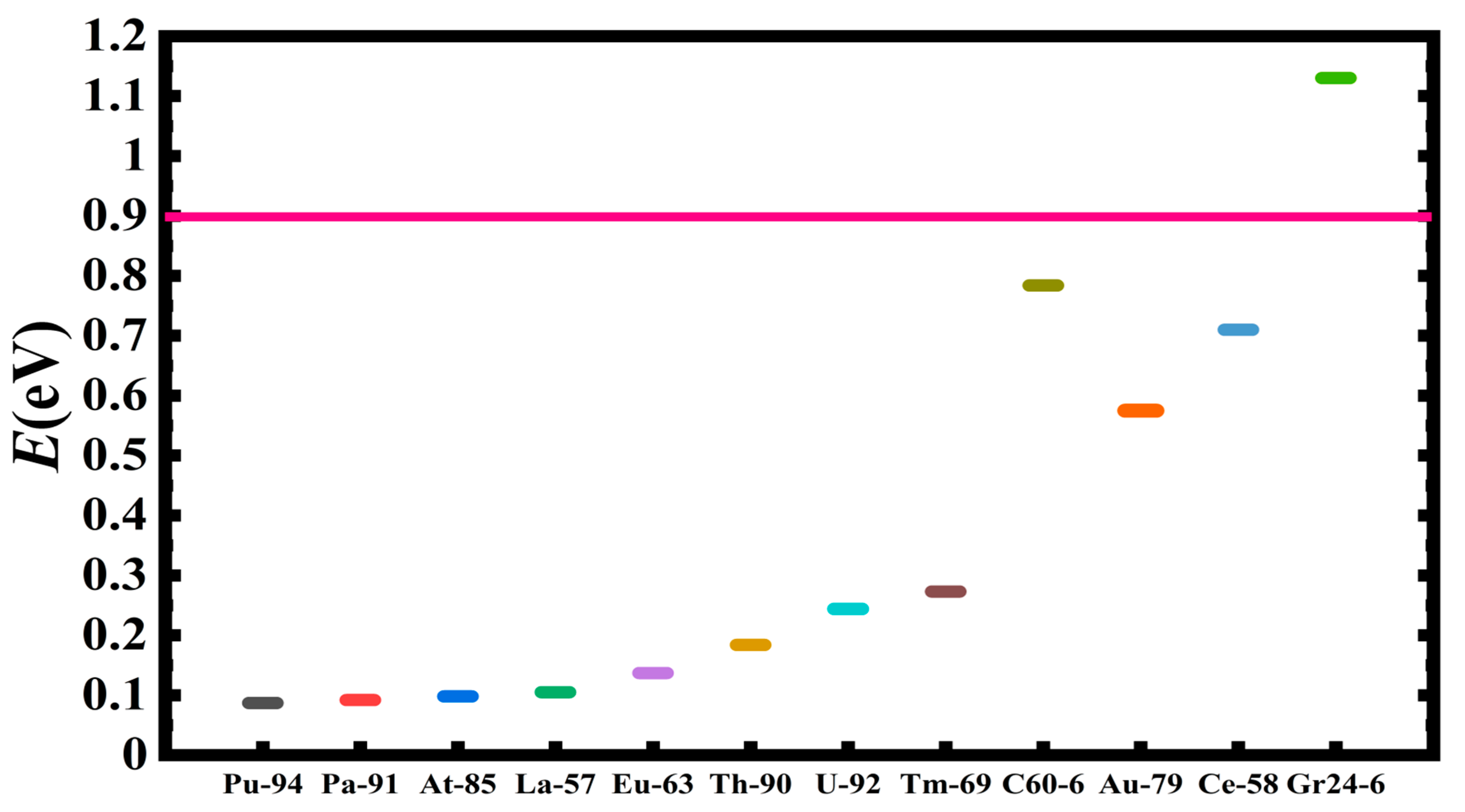
| System | DCS(-2) E(eV) | System | DCS(-2) E(eV) | System | DCS(-2) E(eV) |
|---|---|---|---|---|---|
| Si | 0.030 | Pt | 0.283 | Ti | 1.074 |
| Pu | 0.087 | Pd | 0.297 | V | 1.100 |
| Pa | 0.092 | Ni | 0.342 | Gr | 1.130 |
| - | - | Rh | 0.403 | C136 | 1.440 |
| At | 0.098 | Ag | 0.429 | C20 | 2.070 |
| Sn | 0.103 | Os | 0.443 | ||
| La | 0.105 | Co | 0.541 | ||
| Zn | 0.127 | Au | 0.575 | ||
| Eu | 0.137 | Ca | 0.621 | ||
| Th | 0.184 | Cr | 0.627 | ||
| U | 0.244 | Ce | 0.710 | ||
| K | 0.248 | C60 | 0.784 | ||
| Tm | 0.273 | Fe | 0.879 | ||
| Cu | 0.277 | Mn | 1.033 |
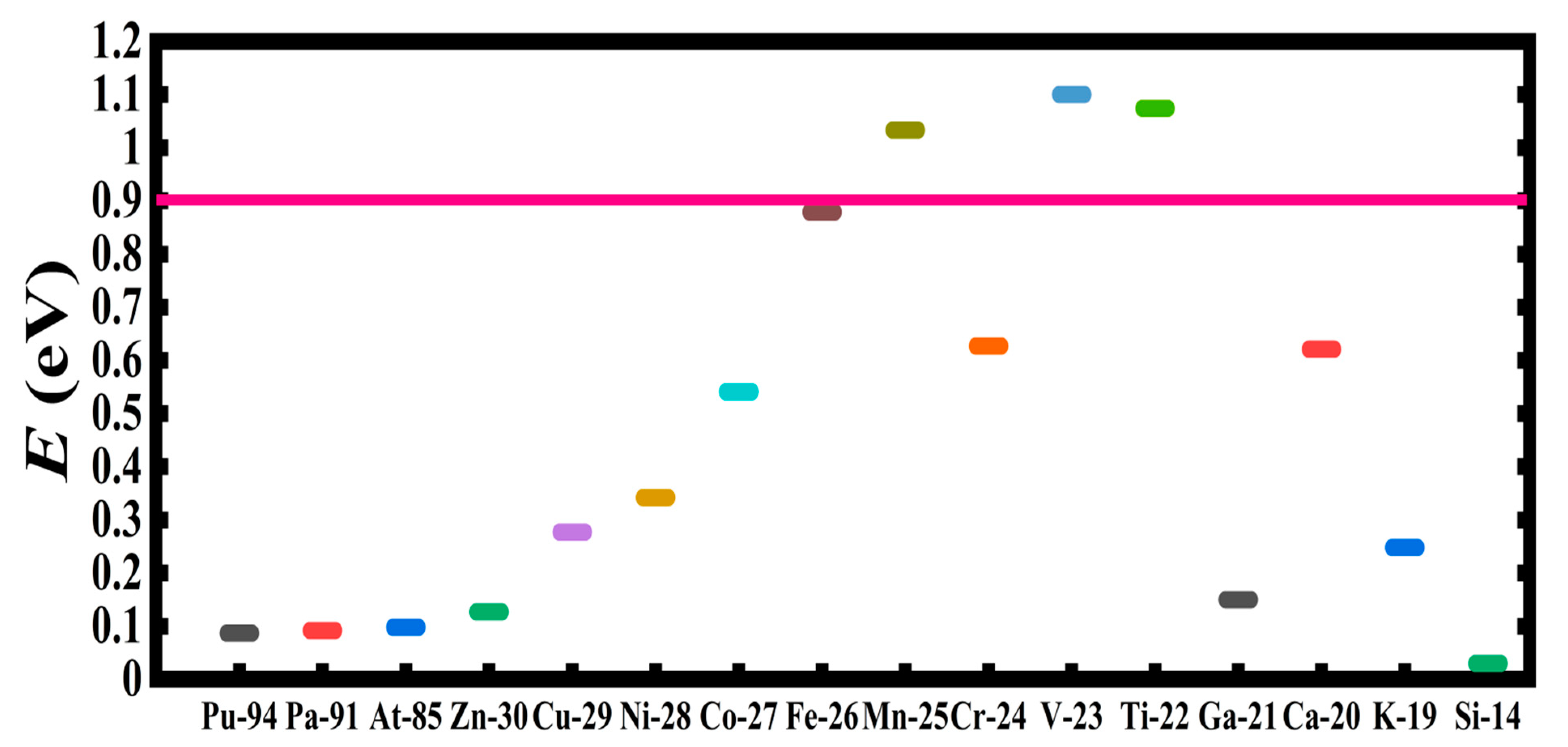
| System | DCS(-2) E(eV) | SCS(-1) E(eV) | System | DCS(-2) E(eV) | SCS(-1) E(eV) |
|---|---|---|---|---|---|
| Si | 0.030 | - | Rh | 0.403 | 0.420 |
| Pu | 0.087 | 0.084 | Ag | 0.429 | 0.420 |
| Pa | 0.092 | 0.326 | Au | 0.575 | 0.400 |
| At | 0.098 | 0.172 | Ca | 0.621 | 1.188 |
| Sn | 0.103 | 1.210 | Cr | 0.627 | - |
| La | 0.105 | 0.035 | C60 | 0.784 | 1.960 |
| K | 0.248 | - | Gr | 1.130 | 1.440 |
| Pt | 0.283 | 0.323 | C136 | 1.440 | 0.680 |
| Pd | 0.297 | 0.200 | C20 | 2.070 | 1.940 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suggs, K.; Msezane, A.Z. Doubly-Charged Negative Ions as Novel Tunable Catalysts: Graphene and Fullerene Molecules Versus Atomic Metals. Int. J. Mol. Sci. 2020, 21, 6714. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186714
Suggs K, Msezane AZ. Doubly-Charged Negative Ions as Novel Tunable Catalysts: Graphene and Fullerene Molecules Versus Atomic Metals. International Journal of Molecular Sciences. 2020; 21(18):6714. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186714
Chicago/Turabian StyleSuggs, Kelvin, and Alfred Z. Msezane. 2020. "Doubly-Charged Negative Ions as Novel Tunable Catalysts: Graphene and Fullerene Molecules Versus Atomic Metals" International Journal of Molecular Sciences 21, no. 18: 6714. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186714