Whole-Genome Sequencing Analysis of Quorum Quenching Bacterial Strain Acinetobacter lactucae QL-1 Identifies the FadY Enzyme for Degradation of the Diffusible Signal Factor

 ,
,  , and
, and
Abstract
:1. Introduction
2. Results
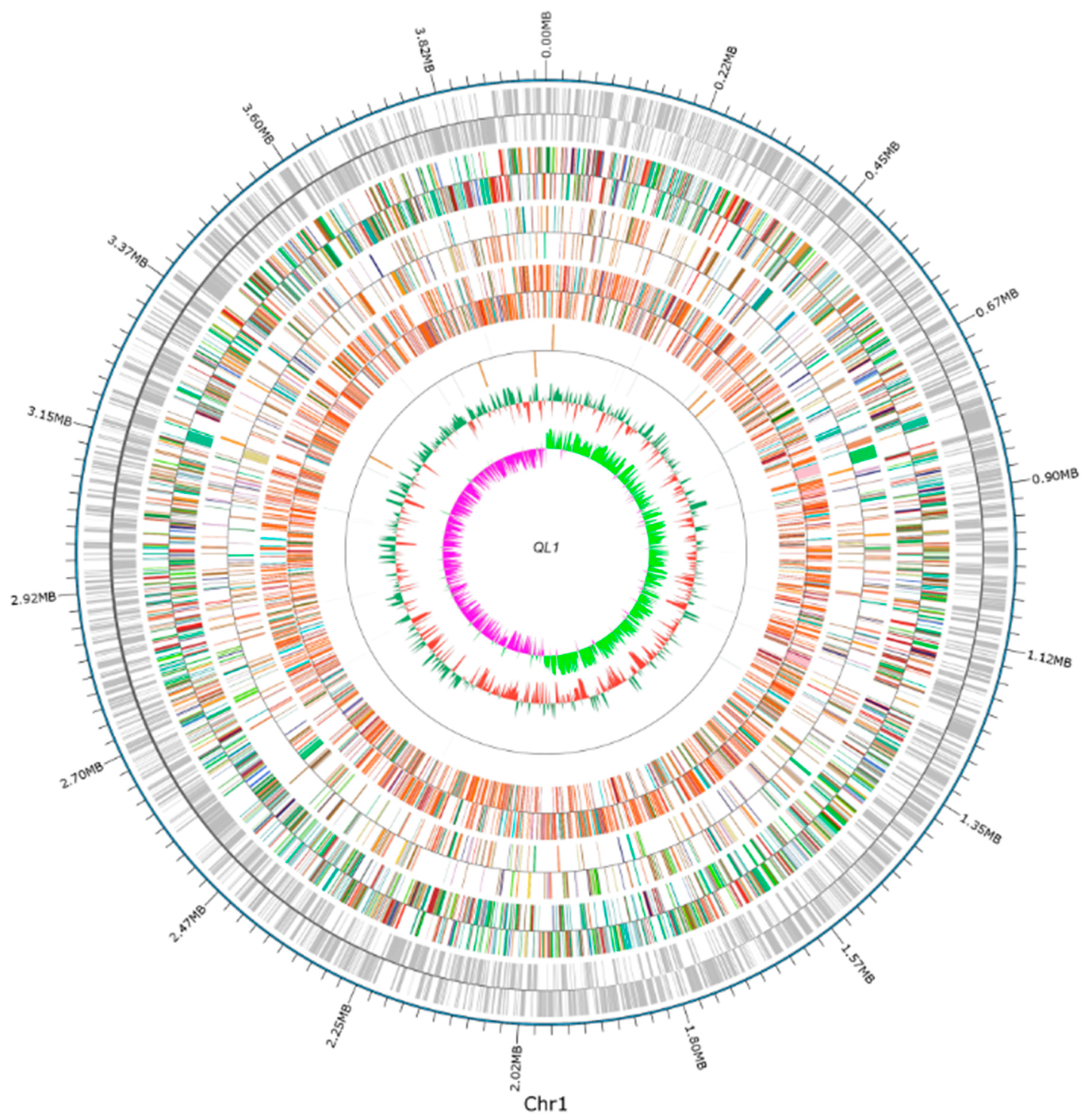
2.1. Sequencing and Analysis of the A. lactucae QL-1 Genome
2.2. Identification and Cloning of the Gene Responsible for the Degradation of DSF
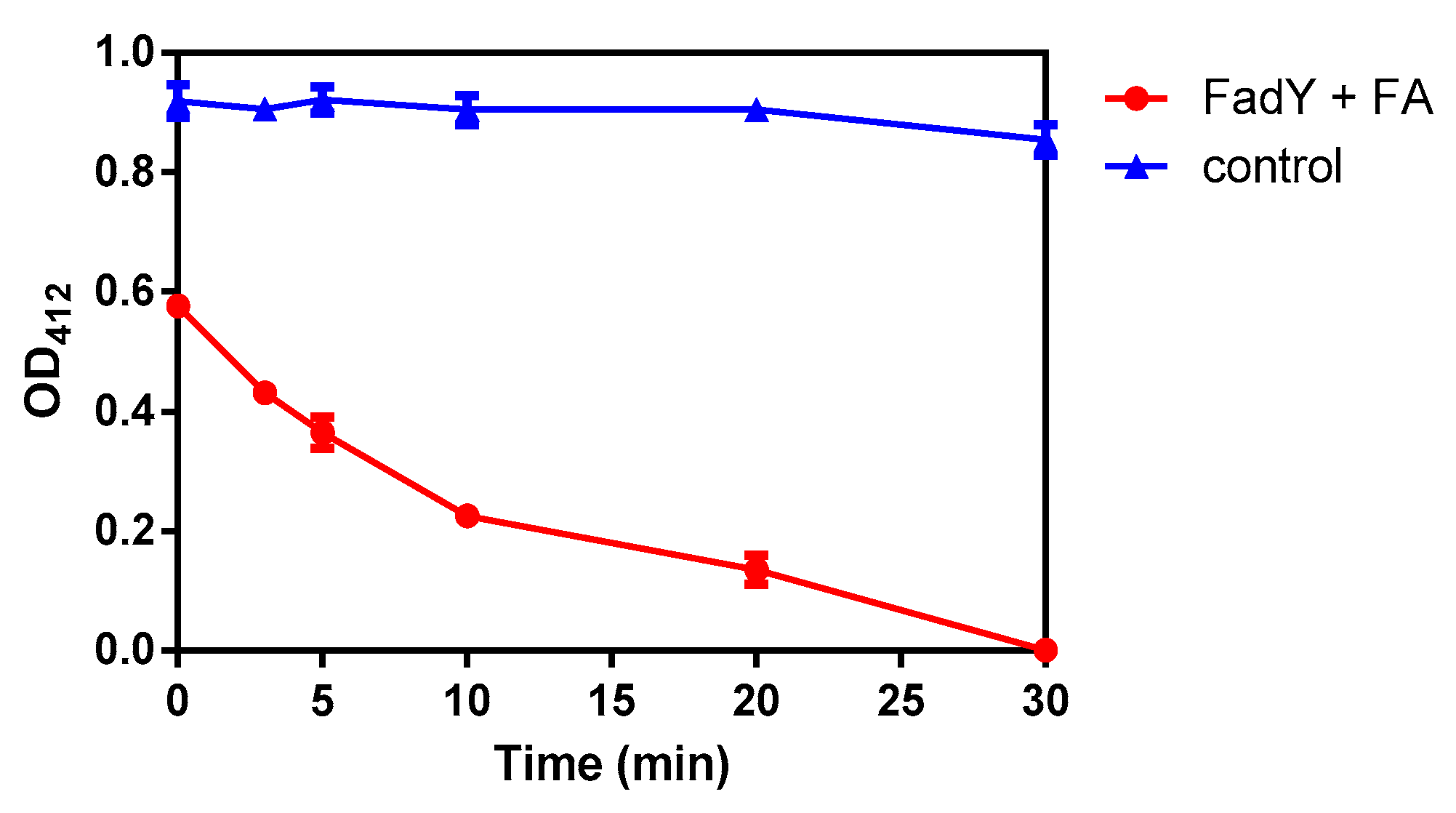
2.3. Activity Detection of FadY
2.4. Xcc Expressing FadY Loses Virulence in Planta
3. Discussion
4. Materials and Methods
4.1. Strains, Plasmids, and Media
4.2. Whole-Genome Sequencing and Assembly
4.3. Genome Component Prediction and Gene Function
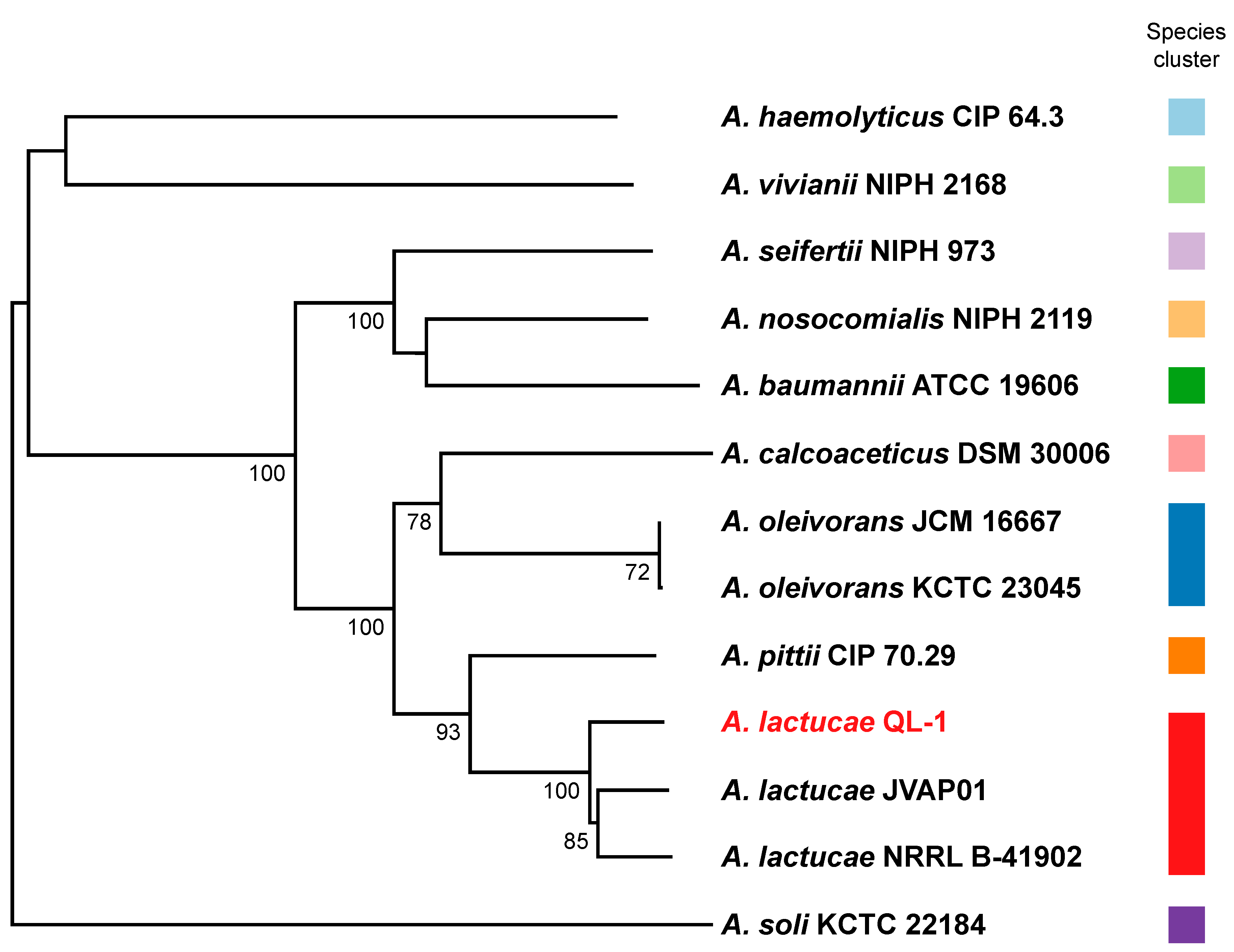
4.4. Genome-Based Taxonomic Classification Analysis
4.5. Identification of the Gene Responsible for the Inactivation of DSF
4.6. Construction of an in-Frame Deletion Mutant and Complementation
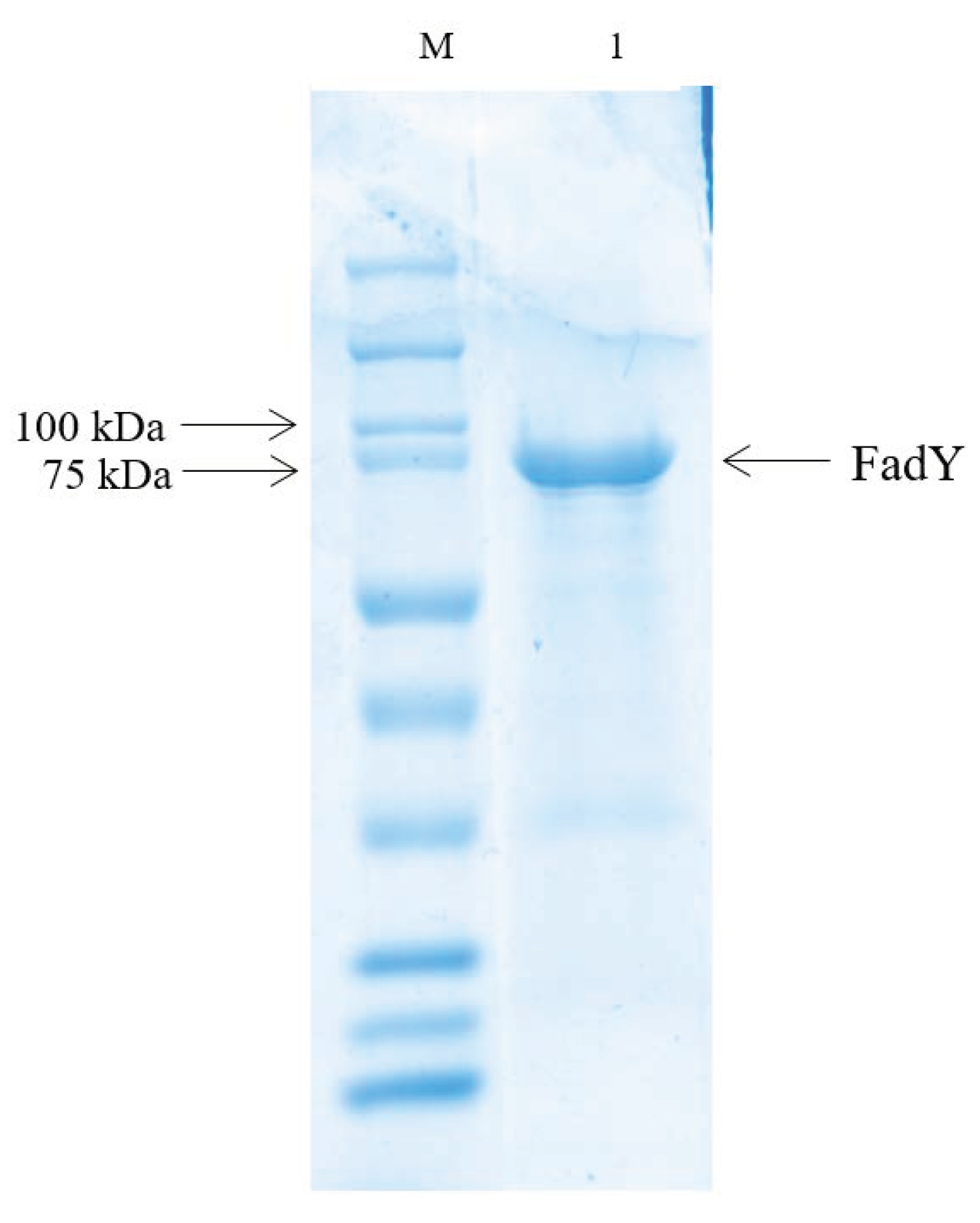
4.7. Expression and Purification of the FadY Protein
4.8. Codon Optimization of FadY
4.9. Determination of FadY Activity In Vitro
4.10. Virulence Tests
4.11. Nucleotide Accession Number
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Alvarez, A.M. Black Rot of Crucifers. In Mechanisms of Resistance to Plant Diseases; Springer: Berlin, Germany, 2000; pp. 21–52. [Google Scholar]
- Newman, K.L.; Chatterjee, S.; Ho, K.A.; Lindow, S.E. Virulence of plant pathogenic bacteria attenuated by degradation of fatty acid cell-to-cell signaling factors. Mol. Plant Microbe Interact. 2008, 21, 326–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.H.; He, Y.; Gao, Y.; Wu, J.E.; Zhang, L.H. A bacterial cell-cell communication signal with cross-kingdom structural analogues. Mol. Microbiol. 2004, 51, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, L.H.; Camara, M.; He, Y. The DSF family of quorum sensing signals: Diversity, biosynthesis, and turnover. Trends Microbiol. 2017, 25, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Agers, Y.; Bruun, M.S.; Dalsgaard, I.; Larsen, J.L. The tetracycline resistance gene tet(E) is frequently occurring and present on large horizontally transferable plasmids in Aeromonas spp. from fish farms. Aquaculture 2007, 266, 47–52. [Google Scholar] [CrossRef]
- Alsan, M.; Schoemaker, L.; Eggleston, K.; Kammili, N.; Kolli, P.; Bhattacharya, J. Out-of-pocket health expenditures and antimicrobial resistance in low-income and middle-income countries: An economic analysis. Lancet Infect. Dis. 2015, 15, 1203–1210. [Google Scholar] [CrossRef] [Green Version]
- Ge, B.; Wang, F.; Sjölund-Karlsson, M.; McDermott, P.F. Antimicrobial resistance in Campylobacter: Susceptibility testing methods and resistance trends. J. Microbiol. Methods 2013, 95, 57–67. [Google Scholar] [CrossRef]
- Uroz, S.; Dessaux, Y.; Oger, P. Quorum sensing and quorum quenching: The yin and yang of bacterial communication. ChemBioChem 2009, 10, 205–216. [Google Scholar] [CrossRef]
- Fetzner, S. Quorum quenching enzymes. J. Biotechnol. 2015, 201, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.H.; Xu, J.L.; Li, X.Z.; Zhang, L.H. AiiA, an enzyme that inactivates the acylhomoserine lactone quorum-sensing signal and attenuates the virulence of Erwinia carotovora. Proc. Natl. Acad. Sci. USA 2000, 97, 3526–3531. [Google Scholar] [CrossRef]
- Ban, H.; Chai, X.; Lin, Y.; Zhou, Y.; Peng, D.; Zhou, Y.; Zou, Y.; Yu, Z.; Sun, M. Transgenic Amorphophallus konjac expressing synthesized acyl-homoserine lactonase (aiiA) gene exhibit enhanced resistance to soft rot disease. Plant Cell Rep. 2009, 28, 1847–1855. [Google Scholar] [CrossRef]
- Cho, H.S.; Park, S.Y.; Ryu, C.M.; Kim, J.F.; Kim, J.G.; Park, S.H. Interference of quorum sensing and virulence of the rice pathogen Burkholderia glumae by an engineered endophytic bacterium. FEMS Microbiol. Ecol. 2007, 60, 14–23. [Google Scholar] [CrossRef]
- Zhang, L.; Ruan, L.; Hu, C.; Wu, H.; Chen, S.; Yu, Z.; Sun, M. Fusion of the genes for AHL-lactonase and S-layer protein in Bacillus thuringiensis increases its ability to inhibit soft rot caused by Erwinia carotovora. Appl. Microbiol. Biotechnol. 2007, 74, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, P.; Rene, E.R.; Kumar, A.J.; Zhang, W.; Chen, S. Binding interaction of allethrin with esterase: Bioremediation potential and mechanism. Bioresour. Technol. 2020, 315, 123845. [Google Scholar] [CrossRef] [PubMed]
- Zhan, H.; Wang, H.; Liao, L.; Feng, Y.; Fan, X.; Zhang, L.; Chen, S. Kinetics and novel degradation pathway of permethrin in Acinetobacter baumannii ZH-14. Front. Microbiol. 2018, 9, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nhan, D.; Cam, D.; Wille, M.; Defoirdt, T.; Bossier, P.; Sorgeloos, P. Quorum quenching bacteria protect Macrobrachium rosenbergii larvae from Vibrio harveyi infection. J. Appl. Microbiol. 2010, 109, 1007–1016. [Google Scholar] [CrossRef]
- Defoirdt, T.; Thanh, L.D.; Van Delsen, B.; De Schryver, P.; Sorgeloos, P.; Boon, N.; Bossier, P. N-acylhomoserine lactone-degrading Bacillus strains isolated from aquaculture animals. Aquaculture 2011, 311, 258–260. [Google Scholar] [CrossRef]
- Torres, M.; Rubio-Portillo, E.; Antón, J.; Ramos-Esplá, A.A.; Quesada, E.; Llamas, I. Selection of the N-acylhomoserine lactone-degrading bacterium Alteromonas stellipolaris PQQ-42 and of its potential for biocontrol in aquaculture. Front. Microbiol. 2016, 7, 646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Zhou, Z.; Cao, Y.; Bai, Y.; Yao, B. High yield expression of an AHL-lactonase from Bacillus sp. B546 in Pichia pastoris and its application to reduce Aeromonas hydrophila mortality in aquaculture. Microb. Cell Factories 2010, 9, 39. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Liu, Y.; Mao, W.; Chen, R.; He, S.; Gao, X.; Zhou, Z.; Yao, B. Effect of dietary N-acyl homoserin lactonase on the immune response and the gut microbiota of zebrafish, Danio rerio, infected with Aeromonas hydrophila. J. World Aquac. Soc. 2014, 45, 149–162. [Google Scholar] [CrossRef]
- Ye, T.; Zhou, T.; Fan, X.; Bhatt, P.; Zhang, L.; Chen, S. Acinetobacter lactucae strain QL-1, a novel quorum quenching candidate against bacterial pathogen Xanthomonas campestris pv. campestris. Front. Microbiol. 2019, 10, 2867. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreft, L.; Botzki, A.; Coppens, F.; Vandepoele, K.; Van Bel, M. PhyD3: A phylogenetic tree viewer with extended phyloXML support for functional genomics data visualization. Bioinformatics 2017, 33, 2946–2947. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Hu, M.; Li, P.; Jiang, Z.; Zhang, L.; Zhou, J. A two-component regulatory system VfmIH modulates multiple virulence traits in Dickeya zeae. Mol. Microbiol. 2019, 111, 1493–1509. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tang, Y.; Fang, X.; Qiao, T.; Han, S.; Zhu, T. Whole-genome sequence of Arthrinium phaeospermum, a globally distributed pathogenic fungus. Genomics 2020, 112, 919–929. [Google Scholar] [CrossRef]
- Galloway, W.R.J.D.; Hodgkinson, J.T.; Bowden, S.D.; Welch, M.; Spring, D.R. Quorum sensing in Gram-negative bacteria: Small-molecule modulation of AHL and AI-2 quorum sensing pathways. Chem. Rev. 2011, 111, 28–67. [Google Scholar] [CrossRef]
- Papenfort, K.; Bassler, B. Quorum sensing signal–response systems in Gram-negative bacteria. Nat. Rev. Genet. 2016, 14, 576–588. [Google Scholar] [CrossRef]
- Mahmoudi, E. Isolation and identification of N-acylhomoserin lactone degrading bacteria from potato rhizosphere. Afr. J. Microbiol. Res. 2011, 5, 1635–1642. [Google Scholar] [CrossRef]
- Devaraj, K.; Tan, G.Y.A.; Chan, K.-G. Quorum quenching properties of Actinobacteria isolated from Malaysian tropical soils. Arch. Microbiol. 2017, 199, 897–906. [Google Scholar] [CrossRef]
- Wang, H.; Liao, L.; Chen, S.; Zhang, L. A quorum quenching bacterial isolate contains multiple substrate-inducible genes conferring degradation of diffusible signal factor. Appl. Environ. Microbiol. 2020, 86, 02930-19. [Google Scholar] [CrossRef]
- Zhang, J.W.; Xuan, C.G.; Lu, C.H.; Guo, S.; Yu, J.F.; Asif, M.; Jiang, W.J.; Zhou, Z.G.; Luo, Z.Q.; Zhang, L.Q. AidB, a novel thermostable N-acylhomoserine lactonase from the bacterium Bosea sp. Appl. Environ. Microbiol. 2019, 85, 02065-19. [Google Scholar] [CrossRef]
- Caicedo, J.C.; Villamizar, S.; Ferro, M.I.T.; Kupper, K.C.; Ferro, J.A. Bacteria from the citrus phylloplane can disrupt cell-cell signalling in Xanthomonas citri and reduce citrus canker disease severity. Plant Pathol. 2015, 65, 782–791. [Google Scholar] [CrossRef]
- Ye, T.; Zhou, T.; Li, Q.; Xu, X.; Fan, X.; Zhang, L.; Chen, S. Cupriavidus sp. HN-2, a novel quorum quenching bacterial isolate, is a potent biocontrol agent against Xanthomonas campestris pv. campestris. Microorganisms 2020, 8, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Ye, T.; Li, Q.; Bhatt, P.; Zhang, L.; Chen, S. Potential of a quorum quenching bacteria isolate Ochrobactrum intermedium D-2 against soft rot pathogen Pectobacterium carotovorum subsp. carotovorum. Front. Microbiol. 2020, 11, 898. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Lin, N.; Chen, Y.; Liang, Z.; Liao, L.; Lv, M.; Chen, Y.; Tang, Y.; He, F.; Chen, S.; et al. Biocontrol of sugarcane smut disease by interference of fungal sexual mating and hyphal growth using a bacterial isolate. Front. Microbiol. 2017, 8, 778. [Google Scholar] [CrossRef]
- Clarke, S.D.; Thuillier, P.; Baillie, R.A. Peroxisome proliferator-activated receptors: A family of lipid–activated transcription factors. Am. J. Clin. Nutr. 1999, 70, 566–571. [Google Scholar] [CrossRef]
- Duplus, E.; Glorian, M.; Forest, C. Fatty acid regulation of gene transcription. J. Biol. Chem. 2000, 275, 30749–30752. [Google Scholar] [CrossRef] [Green Version]
- Leaf, A. Plasma nonesterified fatty acid concentration as a risk factor for sudden cardiac death: The Paris prospective study. Circulation 2001, 104, 744–745. [Google Scholar] [CrossRef]
- Haber, E.; Ximenes, H.; Procopio, J. Pleiotropic effects of fatty acids on pancreatic β-cells. J. Cell. Physiol. 2003, 194, 1–12. [Google Scholar] [CrossRef]
- An, S.Q.; Potnis, N.; Dow, M.; Vorhölter, F.J.; He, Y.Q.; Becker, A.; Teper, D.; Li, Y.; Wang, N.; Bleris, L.; et al. Mechanistic insights into host adaptation, virulence and epidemiology of the phytopathogen Xanthomonas. FEMS Microbiol. Rev. 2019, 44, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; He, F.; Lin, N.; Chen, Y.; Liang, Z.; Liao, L.; Lv, M.; Chen, Y.; Chen, S.; Zhou, J.; et al. Pseudomonas sp. ST 4 produces variety of active compounds to interfere fungal sexual mating and hyphal growth. Microb. Biotechnol. 2020, 13, 107–117. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.H.; Wang, L.H.; Xu, J.L.; Zhang, H.B.; Zhang, X.F.; Zhang, L.H. Quenching quorum-sensing-dependent bacterial infection by an N-acyl homoserine lactonase. Nature 2001, 411, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Luo, Q.; Zhang, Y.; Fan, X.; Ye, T.; Mishra, S.; Bhatt, P.; Zhang, L.; Chen, S. Quorum quenching in a novel Acinetobacter sp. XN-10 bacterial strain against Pectobacterium carotovorum subsp. carotovorum. Microorganisms 2020, 8, 1100. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Liu, X.; Wu, J.; Lee, J.; Chen, S.; Cheng, Y.; Zhang, C.; Zhang, L.H. The host plant metabolite glucose is the precursor of diffusible signal factor (DSF) family signals in Xanthomonas campestris. Appl. Environ. Microbiol. 2015, 81, 2861–2868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Wu, J.; Yin, W.; Li, P.; Zhou, J.; Chen, S.; He, F.; Cai, J.; Zhang, L. Diffusible signal factor family signals provide a fitness advantage to Xanthomonas campestris pv. campestris in interspecies competition. Environ. Microbiol. 2016, 18, 1534–1545. [Google Scholar] [CrossRef]
- Deng, Y.; Lim, A.; Lee, J.; Chen, S.; An, S.; Dong, Y.H.; Zhang, L. Diffusible signal factor (DSF) quorum sensing signal and structurally related molecules enhance the antimicrobial efficacy of antibiotics against some bacterial pathogens. BMC Microbiol. 2014, 14, 51. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Yang, C.; Song, S.; Fu, S.; Sun, X.; Yang, L.; He, F.; Zhang, L.; Zhang, Y.L.; Deng, Y. A novel two-component system modulates quorum sensing and pathogenicity in Burkholderia cenocepacia. Mol. Microbiol. 2018, 108, 32–44. [Google Scholar] [CrossRef] [Green Version]
- He, Y.W.; Boon, C.; Zhou, L.; Zhang, L.H. Co-regulation of Xanthomonas campestris virulence by quorum sensing and a novel two-component regulatory system RavS/RavR. Mol. Microbiol. 2009, 71, 1464–1476. [Google Scholar] [CrossRef]
- Ragan, T.J.; Ross, D.B.; Keshwani, M.M.; Harris, T.K. Expression, purification, and characterization of a structurally disordered and functional C-terminal autoinhibitory domain (AID) of the 70kDa 40S ribosomal protein S6 kinase-1 (S6K1). Protein Expr. Purif. 2008, 57, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.K.; Gan, J.H.; Yang, C.; Zuo, Y.L.; Peng, J.; Li, M.; Huo, W.P.; Xie, Y.P.; Zhang, Y.N.; Wang, T.T.; et al. The SiaA/B/C/D signaling network regulates biofilm formation in Pseudomonas aeruginosa. EMBO J. 2020, 39, 6. [Google Scholar] [CrossRef]
- Reiner, J.; Pisani, L.; Qiao, W.; Singh, R.; Yang, Y.; Shi, L.; Khan, W.A.; Sebra, R.; Cohen, N.; Babu, A.; et al. Cytogenomic identification and long-read single molecule real-time (SMRT) sequencing of a Bardet-Biedl Syndrome 9 (BBS9) deletion. NPJ Genomic Med. 2018, 3, 3. [Google Scholar] [CrossRef]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Bridges, S.; Magbanua, Z.V.; Peterson, D.G. Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 2008, 36, 2284–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, L.; Peter, H.; Rødland, E.A.; Stærfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic. Acids Res. 2007, 35, 3100–3108. [Google Scholar]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Gardner, P.P.; Daub, J.; Tate, J.G.; Nawrocki, E.P.; Kolbe, D.L.; Lindgreen, S.; Wilkinson, A.C.; Finn, R.D.; Griffiths-Jones, S.; Eddy, S.R.; et al. Rfam: Updates to the RNA families database. Nucleic Acids Res. 2009, 37, D136–D140. [Google Scholar] [CrossRef] [Green Version]
- Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R. Infernal 1.0: Inference of RNA alignments. Bioinformatics 2009, 25, 1335–1337. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, W.W.; Wan, I.; Jones, S.; Brinkman, F.S. IslandPath: Aiding detection of genomic islands in prokaryotes. Bioinformatics 2003, 19, 418–420. [Google Scholar] [CrossRef] [Green Version]
- Eichinger, V.; Nussbaumer, T.; Platzer, A.; Jehl, M.A.; Arnold, R.; Rattei, T. EffectiveDB--updates and novel features for a better annotation of bacterial secreted proteins and Type III, IV, VI secretion systems. Nucleic Acids Res. 2015, 44, D669–D674. [Google Scholar] [CrossRef]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2014, 43, D261–D269. [Google Scholar] [CrossRef]
- Li, W.; Jaroszewski, L.; Godzik, A. Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 2002, 18, 77–82. [Google Scholar] [CrossRef]
- Saier, M.H.; Reddy, V.S.; Tamang, D.G.; Västermark, A. The Transporter Classification Database. Nucleic Acids Res. 2013, 42, D251–D258. [Google Scholar] [CrossRef]
- Amos, B.; Rolf, A. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Medema, M.H.; Blin, K.; Cimermancic, P.; De Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef]
- Martin, U.; Rashmi, P.; Arathi, R.; Irvine, A.G.; Helder, P.; Hammond-Kosack, K.E. The pathogen-host interactions database (PHI-base): Additions and future developments. Nucleic Acids Res. 2015, 1, D1. [Google Scholar]
- Chen, L.; Xiong, Z.; Sun, L.; Yang, J.; Jin, Q. VFDB 2012 update: Toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 2011, 40, D641–D645. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Boil. 2016, 17, 132. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.S.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [Green Version]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Boil. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [Green Version]
- Bi, H.; Yu, Y.; Dong, H.; Wang, H.; Cronan, J.E. Xanthomonas campestris RpfB is a fatty Acyl-CoA ligase required to counteract the thioesterase activity of the RpfF diffusible signal factor (DSF) synthase. Mol. Microbiology 2014, 93, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Xu, J.; Birch, R.G. High affinity binding of albicidin phytotoxins by the AlbA protein from Klebsiella oxytoca. Microbiology 1998, 144, 555–559. [Google Scholar] [CrossRef] [Green Version]
- Groot, P.; Scholte, H.; Hülsmann, W. Fatty Acid Activation: Specificity, Localization, and Function. Skin Lipids 1976, 14, 75–126. [Google Scholar] [CrossRef]
- Bar-Tana, J.; Rose, G.; Shapiro, B. The purification and properties ofmicrosomal palmitoyl-coenzyme A synthase. Biochem. J. 1971, 122, 353–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichihara, K.; Shibasaki, Y. An enzyme-coupled assay for acyl-CoA synthase. J. Lipid Res. 1991, 32, 1709–1712. [Google Scholar]
- Wehrmann, A.; Vliet, A.V.; Opsomer, C.; Botterman, J.; Schulz, A. Thesimilarities of bar and pat gene products make them equally applicable for plantengineering. Nat. Biotechnol. 1996, 14, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zarzycki-Siek, J.; Walton, C.B.; Norris, M.S.; Hoang, T.T. Multiple FadD Acyl-CoA synthetases contribute to differential fatty acid degradation and virulence in Pseudomonas aeruginosa. PLoS ONE 2010, 5, e13557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Hu, M.; Xue, Y.; Chen, X.; Lu, G.; Zhang, L.; Zhou, J. Screening, identification and efficacy evaluation of antagonistic bacteria for biocontrol of soft rot disease caused by Dickeya zeae. Microorganisms 2020, 8, 697. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
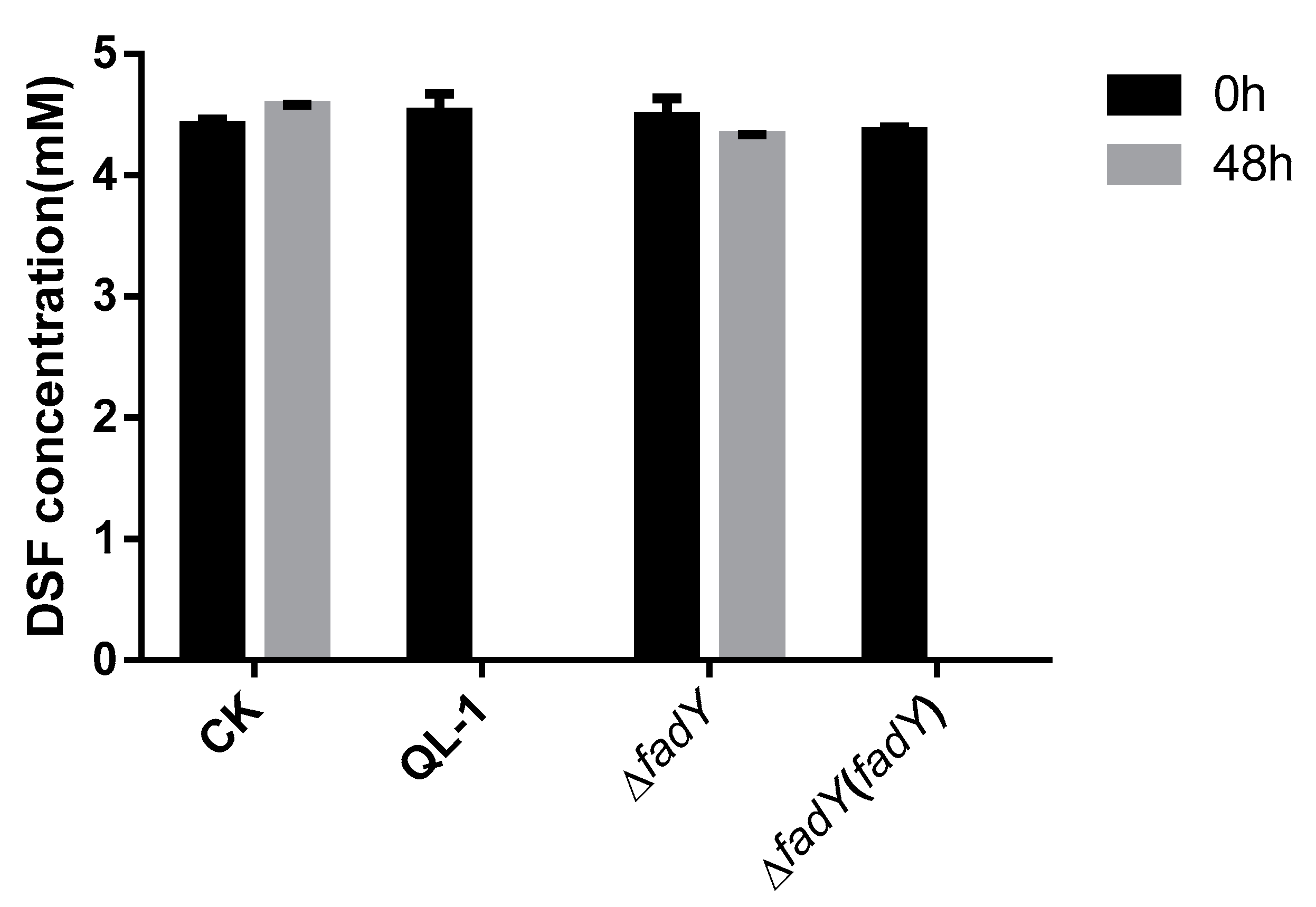{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Characteristics | |
|---|---|
| Total length (bp) | 3,973,648 |
| GC content (%) | 40.04 |
| Number of protein-coding genes | 3.707 |
| Average length of protein-coding genes (bp) | 940 |
| % of Genome (protein-coding genes) | 87.71 |
| rRNA genes | 18 |
| tRNA genes | 73 |
| Repeats | 153 |
| % of Genome (repeats) | 0.37 |
| Time (min) | FadY + FA | Control | ||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 1 | 2 | 3 | |
| 0 | 0.565 | 0.586 | 0.578 | 0.934 | 0.887 | 0.936 |
| 3 | 0.452 | 0.426 | 0.415 | 0.912 | 0.905 | 0.900 |
| 5 | 0.382 | 0.377 | 0.334 | 0.943 | 0.922 | 0.899 |
| 10 | 0.221 | 0.233 | 0.221 | 0.878 | 0.925 | 0.910 |
| 20 | 0.128 | 0.162 | 0.115 | 0.904 | 0.921 | 0.890 |
| 30 | 0.000 | 0.000 | 0.000 | 0.869 | 0.869 | 0.824 |
| Strains or Plasmids | Relevant Genotype or Phenotype | Sources |
|---|---|---|
| QL-1 strains | ||
| QL-1 | Laboratory storage | This study |
| ∆fadY | fadY deletion mutant of strain QL-1 with 1680-nt internal coding region deleted | This study |
| Xcc XC1 | Pathogenic bacteria causing black rot | Lab collection |
| Escherichia coli strains | ||
| DH5α | spuE44 ∆lacU169(φ80lacZ∆M15) hsdR17λpir recA1 endA1 gyrA96 thi-1 relA1 | Lab collection |
| BL21 | F-ompThsdS (rB−mB−) dcm+ Tetr gal (DE3) endA | Lab collection |
| pRK2013 | Tra+, Mob-, ColE1-replicon, Kanr, Sper | Lab collection |
| Plasmids | ||
| pBBR1-MCS5 | Broad host-range cloning vector; Gmr | Lab collection |
| pK18mobsacB | Broad-host-range gene replacement vector, sacB,Gmr | Lab collection |
| pBBR1-MCS5-fadY | pBBR1-MCS5 containing fadY under control of Plac | This study |
| pK18-fadY | pK18mobsacB contain flanking of fadY | This study |
| pGEX-6p-1 | GST fusion protein expression vector, Ampr | Invitrogen |
| pGEX-fadY | pGEX-6p-1 containing fadY | This study |
| Primers | Sequence (5′–3′) | Applications |
|---|---|---|
| De3487upF | GAGCTCGGTACCCGGGGATCCGGAGCGCCTGGCGATCAT | For amplification of the 5′-region of fadY |
| De3487upR | CGGAGATAATGGCATTTAAGTTAATAAAAAAGCGCCTTAGGG | |
| De3487dnF | CTTAAATGCCATTATCTCCGATTCGT | For amplification of the 3′-region of fadY |
| De3487dnR | CGACGGCCAGTGCCAAGCTTAGTTGATACAACTTGAAGCG | |
| Ts3487-F | CTGCATAGTGCCATCCATCAC | For identification of ∆fadY |
| Ts3487-R | GCCAAGGCAGGAAAAAGC | |
| cfadY-F | GTCGACGGTATCGATAAGCTTAAGCTAGCGTCGGGCAACA | For construction of pBBR1-MCS5-fadY |
| cfadY-R | CGCTCTAGAACTAGTGGATCCAAATATAGAAACAAAAAAAGCGCCC | |
| pfadY-F | CAGTCAGTCACGATGCGGCCGATGGAAAAGATTTGGTTTGCAGA | For construction of pGEX-6P-1-fadY |
| pfadY-R | CCCCTGGGATCCCCGGAATTCTTAGGTTGGTTTACGTAAGTCTTTACG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, T.; Zhou, T.; Xu, X.; Zhang, W.; Fan, X.; Mishra, S.; Zhang, L.; Zhou, X.; Chen, S. Whole-Genome Sequencing Analysis of Quorum Quenching Bacterial Strain Acinetobacter lactucae QL-1 Identifies the FadY Enzyme for Degradation of the Diffusible Signal Factor. Int. J. Mol. Sci. 2020, 21, 6729. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186729
Ye T, Zhou T, Xu X, Zhang W, Fan X, Mishra S, Zhang L, Zhou X, Chen S. Whole-Genome Sequencing Analysis of Quorum Quenching Bacterial Strain Acinetobacter lactucae QL-1 Identifies the FadY Enzyme for Degradation of the Diffusible Signal Factor. International Journal of Molecular Sciences. 2020; 21(18):6729. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186729
Chicago/Turabian StyleYe, Tian, Tian Zhou, Xudan Xu, Wenping Zhang, Xinghui Fan, Sandhya Mishra, Lianhui Zhang, Xiaofan Zhou, and Shaohua Chen. 2020. "Whole-Genome Sequencing Analysis of Quorum Quenching Bacterial Strain Acinetobacter lactucae QL-1 Identifies the FadY Enzyme for Degradation of the Diffusible Signal Factor" International Journal of Molecular Sciences 21, no. 18: 6729. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186729