Role of Glutaredoxin-1 and Glutathionylation in Cardiovascular Diseases

 , , ,
, , ,
Abstract
:1. Introduction
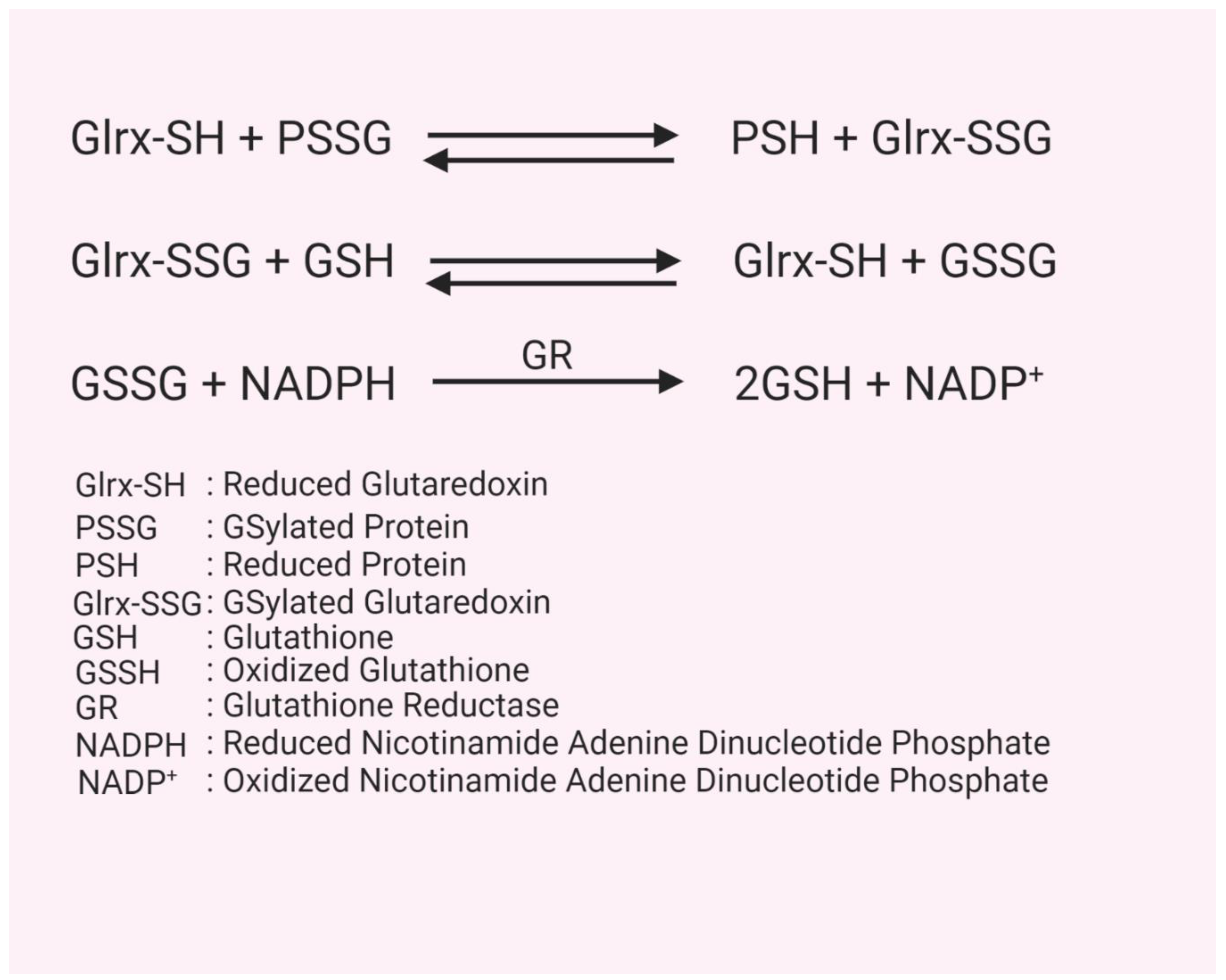
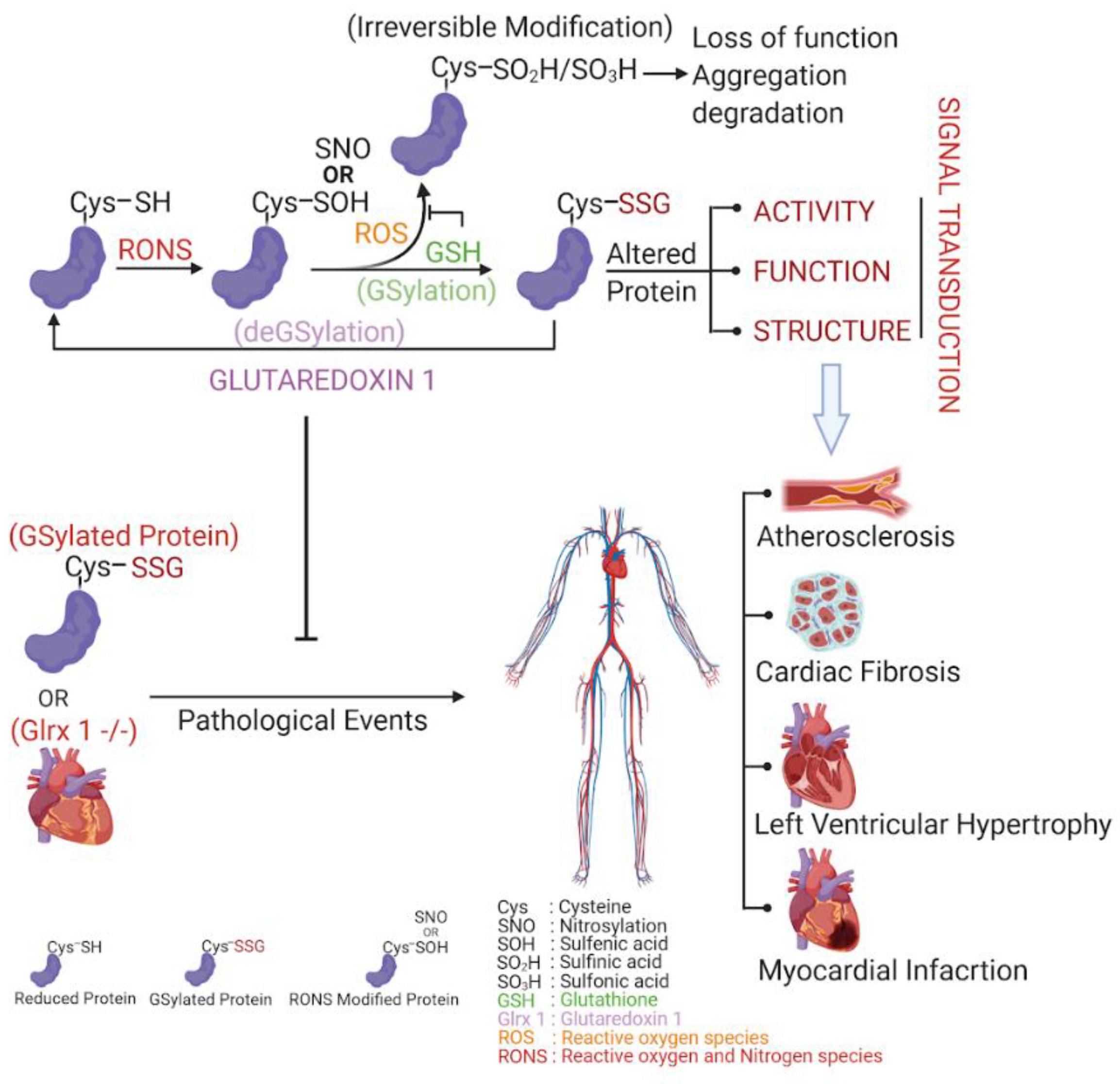
2. Glutathion(GS)ylation
3. Glutaredoxin Structure, Function, and Mechanism
4. Cardiovascular Disease
4.1. Myocardial Ischemia and Reperfusion
4.2. Cardiac Hypertrophy
4.3. Peripheral Arterial Disease
4.4. Atherosclerosis
4.5. Lipid and Glucose Metabolism
5. Summary and Translational Application
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GSylation | glutathionylation |
| GSH | glutathione |
| CVD | cardiovascular disease |
| Glrx | glutaredoxin |
References
- Agha, M.; Agha, R. The rising prevalence of obesity. Int. J. Surg. Oncol. 2017, 2, e17. [Google Scholar]
- Sturm, R.; Hattori, A. Morbid obesity rates continue to rise rapidly in the United States. Int. J. Obes. 2013, 37, 889–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagidipati, N.J.; Gaziano, T.A. Estimating Deaths from Cardiovascular Disease: A Review of Global Methodologies of Mortality Measurement. Circulation 2013, 127, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Csányi, G.F., Jr. Oxidative Stress in Cardiovascular Disease. Int. J. Mol. Sci. 2014, 15, 6002–6008. [Google Scholar] [PubMed] [Green Version]
- Elahi, M.M.; Kong, Y.X.; Matata, B.M. Oxidative Stress as a Mediator of Cardiovascular Disease. Oxidative Med. Cell. Longev. 2009, 2, 259–269. [Google Scholar] [CrossRef] [Green Version]
- Mashamaite, L.N.; Rohwer, J.M.; Pillay, C.S. The glutaredoxin mono- and di-thiol mechanisms for deglutathionylation are functionally equivalent: Implications for redox systems biology. Biosci. Rep. 2015, 35, e00173. [Google Scholar] [CrossRef]
- Madamanchi, N.R.; Vendrov, A.; Runge, M.S. Oxidative Stress and Vascular Disease. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar]
- Miller, O.G.; Behring, J.B.; Siedlak, S.L.; Jiang, S.; Matsui, R.; Bachschmid, M.M.; Zhu, X.; Mieyal, J.J. Upregulation of Glutaredoxin-1 Activates Microglia and Promotes Neurodegeneration: Implications for Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 967–982. [Google Scholar] [CrossRef] [Green Version]
- Kenchappa, R.S.; Diwakar, L.; Annepu, J.; Ravindranath, V. Estrogen and neuroprotection: Higher constitutive expression of glutaredoxin in female mice offers protection against MPTP-mediated neurodegeneration. FASEB J. 2004, 18, 1102–1104. [Google Scholar] [CrossRef]
- Durgadoss, L.; Nidadavolu, P.; Valli, R.K.; Saeed, U.; Mishra, M.; Seth, P.; Ravindranath, V. Redox modification of Akt mediated by the dopaminergic neurotoxin MPTP, in mouse midbrain, leads to down-regulation of pAkt. FASEB J. 2012, 26, 1473–1483. [Google Scholar] [CrossRef] [PubMed]
- Lou, M.F. Redox regulation in the lens. Prog. Retin. Eye Res. 2003, 22, 657–682. [Google Scholar] [CrossRef]
- Liu, X.; Jann, J.; Xavier, C.; Wu, H. Glutaredoxin 1 (Grx1) Protects Human Retinal Pigment Epithelial Cells From Oxidative Damage by Preventing AKT Glutathionylation. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2821–2832. [Google Scholar] [CrossRef] [PubMed]
- Shelton, M.D.; Kern, T.S.; Mieyal, J.J. Glutaredoxin regulates nuclear factor kappa-B and intercellular adhesion molecule in Müller cells: Model of diabetic retinopathy. J. Biol. Chem. 2007, 282, 12467–12474. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Fry, J.L.; Han, J.; Hou, X.; Pimentel, D.R.; Matsui, R.; Cohen, R.A.; Bachschmid, M.M. A redox-resistant sirtuin-1 mutant protects against hepatic metabolic and oxidant stress. J. Biol. Chem. 2014, 289, 7293–7306. [Google Scholar] [CrossRef] [Green Version]
- Zee, R.S.; Yoo, C.B.; Pimentel, D.R.; Perlman, D.H.; Burgoyne, J.R.; Hou, X.; McComb, M.E.; Costello, C.E.; Cohen, R.A.; Bachschmid, M.M. Redox regulation of sirtuin-1 by S-glutathiolation. Antioxid. Redox Signal. 2010, 13, 1023–1032. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Han, J.; Hou, X.; Fry, J.; Behring, J.B.; Seta, F.; Long, M.T.; Roy, H.K.; Cohen, R.A.; Matsui, R.; et al. Glutaredoxin-1 Deficiency Causes Fatty Liver and Dyslipidemia by Inhibiting Sirtuin-1. Antioxid. Redox Signal. 2017, 27, 313–327. [Google Scholar] [CrossRef] [Green Version]
- Anathy, V.; Lahue, K.G.; Chapman, D.G.; Chia, S.B.; Casey, D.T.; Aboushousha, R.; van der Velden, J.L.J.; Elko, E.; Hoffman, S.M.; McMillan, D.H.; et al. Reducing protein oxidation reverses lung fibrosis. Nat. Med. 2018, 24, 1128–1135. [Google Scholar] [CrossRef]
- Anathy, V.; Aesif, S.W.; Hoffman, S.M.; Bement, J.L.; Guala, A.S.; Lahue, K.G.; Leclair, L.W.; Suratt, B.T.; Cool, C.D.; Wargo, M.J.; et al. Glutaredoxin-1 attenuates S-glutathionylation of the death receptor fas and decreases resolution of Pseudomonas aeruginosa pneumonia. Am. J. Respir. Crit. Care Med. 2014, 189, 463–474. [Google Scholar] [CrossRef] [Green Version]
- Lekli, I.; Mukherjee, S.; Ray, D.; Gurusamy, N.; Kim, Y.H.; Tosaki, A.; Engelman, R.M.; Ho, Y.-S.; Das, D.K. Functional recovery of diabetic mouse hearts by glutaredoxin-1 gene therapy: Role of Akt-FoxO-signaling network. Gene Ther. 2010, 17, 478–485. [Google Scholar] [CrossRef] [Green Version]
- Inadomi, C.; Murata, H.; Ihara, Y.; Goto, S.; Urata, Y.; Yodoi, J.; Kondo, T.; Sumikawa, K. Overexpression of glutaredoxin protects cardiomyocytes against nitric oxide-induced apoptosis with suppressing the S-nitrosylation of proteins and nuclear translocation of GAPDH. Biochem. Biophys. Res. Commun. 2012, 425, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Matsui, R.; Watanabe, Y.; Murdoch, C.E. Redox regulation of ischemic limb neovascularization—What we have learned from animal studies. Redox Biol. 2017, 12, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Murdoch, C.E.; Sano, S.; Ido, Y.; Bachschmid, M.M.; Cohen, R.A.; Matsui, R. Glutathione adducts induced by ischemia and deletion of glutaredoxin-1 stabilize HIF-1α and improve limb revascularization. Proc. Natl. Acad. Sci. USA 2016, 113, 6011–6016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murdoch, C.E.; Shuler, M.; Haeussler, D.J.F.; Kikuchi, R.; Bearelly, P.; Han, J.; Watanabe, Y.; Fuster, J.J.; Walsh, K.; Ho, Y.-S.; et al. Glutaredoxin-1 up-regulation induces soluble vascular endothelial growth factor receptor 1, attenuating post-ischemia limb revascularization. J. Biol. Chem. 2014, 289, 8633–8644. [Google Scholar] [CrossRef] [Green Version]
- Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988, 263, 17205–17208. [Google Scholar]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Meister, A. Biosynthesis and functions of glutathione, an essential biofactor. J. Nutr. Sci. Vitaminol. 1992, 38, 1–6. [Google Scholar] [CrossRef]
- Dagnell, M.; Cheng, Q.; Rizvi, S.H.M.; Pace, P.E.; Boivin, B.; Winterbourn, C.C.; Arnér, E.S.J. Bicarbonate is essential for protein-tyrosine phosphatase 1B (PTP1B) oxidation and cellular signaling through EGF-triggered phosphorylation cascades. J. Biol. Chem. 2019, 294, 12330–12338. [Google Scholar] [CrossRef] [Green Version]
- Schuppe-Koistinen, I.; Moldéus, P.; Bergman, T.; Cotgreave, I.A. S-thiolation of human endothelial cell glyceraldehyde-3-phosphate dehydrogenase after hydrogen peroxide treatment. Eur. J. Biochem. 1994, 221, 1033–1037. [Google Scholar] [CrossRef]
- Niture, S.K.; Velu, C.S.; Bailey, N.I.; Srivenugopal, K.S. S-thiolation mimicry: Quantitative and kinetic analysis of redox status of protein cysteines by glutathione-affinity chromatography. Arch. Biochem. Biophys. 2005, 444, 174–184. [Google Scholar] [CrossRef]
- Coles, S.J.; Easton, P.; Sharrod, H.; Hutson, S.M.; Hancock, J.; Patel, V.B.; Conway, M.E. S-Nitrosoglutathione inactivation of the mitochondrial and cytosolic BCAT proteins: S-nitrosation and S-thiolation. Biochemistry 2009, 48, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Mohr, S.; Hallak, H.; de Boitte, A.; Lapetina, E.G.; Brüne, B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 1999, 274, 9427–9430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giustarini, D.; Milzani, A.; Aldini, G.; Carini, M.; Rossi, R.; Dalle-Donne, I. S-nitrosation versus S-glutathionylation of protein sulfhydryl groups by S-nitrosoglutathione. Antioxid. Redox Signal. 2005, 7, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol. 1997, 82, 291–295. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef]
- Tousoulis, D.; Kampoli, A.-M.; Papageorgiou, C.T.N.; Stefanadis, C. The Role of Nitric Oxide on Endothelial Function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef]
- Donald, M.; Lloyd-Jones, M.D.; Kenneth, D.; Bloch, M.D. The vascular biology of nitric oxide and its role in atherogenesis. Annu. Rev. Med. 1996, 47, 365–375. [Google Scholar]
- Sharma, J.N.; Al-Omran, A.; Parvathy, S.S. Role of nitric oxide in inflammatory diseases. Inflammopharmacology 2007, 15, 252–259. [Google Scholar] [CrossRef]
- Li, S.; Yan, T.; Yang, J.Q.; Oberley, T.D.; Oberley, L.W. The role of cellular glutathione peroxidase redox regulation in the suppression of tumor cell growth by manganese superoxide dismutase. Cancer Res. 2000, 60, 3927–3939. [Google Scholar]
- Ho, Y.-S.; Xiong, Y.; Ho, D.S.; Gao, J.; Chua, B.H.L.; Pai, H.; Mieyal, J.J. Targeted disruption of the glutaredoxin 1 gene does not sensitize adult mice to tissue injury induced by ischemia/reperfusion and hyperoxia. Free Radic. Biol. Med. 2007, 43, 1299–1312. [Google Scholar] [CrossRef] [Green Version]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Rebrin, I.; Sohal, R.S. Comparison of thiol redox state of mitochondria and homogenates of various tissues between two strains of mice with different longevities. Exp. Gerontol. 2004, 39, 1513–1519. [Google Scholar] [CrossRef] [Green Version]
- Zitka, O.; Skalickova, S.; Gumulec, J.; Masarik, M.; Adam, V.; Hubalek, J.; Trnkova, L.; Kruseova, J.; Eckschlager, T.; Kizek, R. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol. Lett. 2012, 4, 1247–1253. [Google Scholar] [CrossRef] [Green Version]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Colombo, R.; Milzani, A. S-glutathionylation in protein redox regulation. Free Radic. Biol. Med. 2007, 43, 883–898. [Google Scholar] [CrossRef]
- Ravichandran, V.; Seres, T.; Moriguchi, T.; Thomas, J.A.; Johnston, R.B., Jr. S-thiolation of glyceraldehyde-3-phosphate dehydrogenase induced by the phagocytosis-associated respiratory burst in blood monocytes. J. Biol. Chem. 1994, 269, 25010–25015. [Google Scholar] [PubMed]
- Fratelli, M.; Demol, H.; Puype, M.; Casagrande, S.; Eberini, I.; Salmona, M.; Bonetto, V.; Mengozzi, M.; Duffieux, F.; Miclet, E.; et al. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 3505–3510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.; Uys, J.D.; Tew, K.D.; Townsend, D.M. S-glutathionylation: From molecular mechanisms to health outcomes. Antioxid. Redox Signal. 2011, 15, 233–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastore, A.; Piemonte, F. Protein glutathionylation in cardiovascular diseases. Int. J. Mol. Sci. 2013, 14, 20845–20876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanHecke, G.C.; Abeywardana, M.Y.; Ahn, Y.-H. Proteomic Identification of Protein Glutathionylation in Cardiomyocytes. J. Proteome Res. 2019, 18, 1806–1818. [Google Scholar] [CrossRef] [PubMed]
- Lillig, C.H.; Berndt, C.; Holmgren, A. Glutaredoxin systems. Biochim. Biophys. Acta 2008, 1780, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Eklund, H.; Cambillau, C.; Sjöberg, B.M.; Holmgren, A.; Jörnvall, H.; Höög, J.O.; Brändén, C.I. Conformational and functional similarities between glutaredoxin and thioredoxins. EMBO J. 1984, 3, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Mieyal, J.J.; Gallogly, M.M.; Qanungo, S.; Sabens, E.A.; Shelton, M.D. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal. 2008, 10, 1941–1988. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.-H.; Zhang, W.; Chen, W.-Q.; Jin, J.; Cui, X.; Butte, N.F.; Chan, L.; Hirschi, K.D. A mammalian monothiol glutaredoxin, Grx3, is critical for cell cycle progression during embryogenesis. FEBS J. 2011, 278, 2525–2539. [Google Scholar] [CrossRef] [Green Version]
- Sagemark, J.; Elgán, T.H.; Bürglin, T.R.; Johansson, C.; Holmgren, A.; Berndt, K.D. Redox properties and evolution of human glutaredoxins. Proteins Struct. Funct. Bioinform. 2007, 68, 879–892. [Google Scholar] [CrossRef] [PubMed]
- Herrero, E.; de la Torre-Ruiz, M.A. Monothiol glutaredoxins: A common domain for multiple functions. Cell. Mol. Life Sci. 2007, 64, 1518–1530. [Google Scholar] [CrossRef] [Green Version]
- Pimentel, D.R.; Adachi, T.; Ido, Y.; Heibeck, T.; Jiang, B.; Lee, Y.; Melendez, J.A.; Cohen, R.A.; Colucci, W.S. Strain-stimulated hypertrophy in cardiac myocytes is mediated by reactive oxygen species-dependent Ras S-glutathiolation. J. Mol. Cell. Cardiol. 2006, 41, 613–622. [Google Scholar] [CrossRef]
- Han, J.; Weisbrod, R.M.; Shao, D.; Watanabe, Y.; Yin, X.; Bachschmid, M.M.; Seta, F.; Janssen-Heininger, Y.M.W.; Matsui, R.; Zang, M.; et al. The redox mechanism for vascular barrier dysfunction associated with metabolic disorders: Glutathionylation of Rac1 in endothelial cells. Redox Biol. 2016, 9, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Pan, S.; Berk, B.C. Glutaredoxin Mediates Akt and eNOS Activation by Flow in a Glutathione Reductase-Dependent Manner. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1283–1288. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J.; Xuan, J.Y.; McBride, S.; Maharsy, W.; Thorn, S.; Holterman, C.E.; Kennedy, C.R.J.; Rippstein, P.; deKemp, R.; da Silva, J.; et al. Glutaredoxin-2 is required to control oxidative phosphorylation in cardiac muscle by mediating deglutathionylation reactions. J. Biol. Chem. 2014, 289, 14812–14828. [Google Scholar] [CrossRef] [Green Version]
- Kanaan, G.N.; Ichim, B.; Gharibeh, L.; Maharsy, W.; Patten, D.A.; Xuan, J.Y.; Reunov, A.; Marshall, P.; Veinot, J.; Menzies, K.; et al. Glutaredoxin-2 controls cardiac mitochondrial dynamics and energetics in mice, and protects against human cardiac pathologies. Redox Biol. 2018, 14, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Donelson, J.; Wang, Q.; Monroe, T.O.; Jiang, X.; Zhou, J.; Yu, H.; Mo, Q.; Sun, Q.; Marini, J.C.; Wang, X.; et al. Cardiac-specific ablation of glutaredoxin 3 leads to cardiac hypertrophy and heart failure. Physiol. Rep. 2019, 7, e14071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, A.G.; Palenchar, D.J.; Wildemann, J.D.; Philpott, C.C. A Glutaredoxin·BolA Complex Serves as an Iron-Sulfur Cluster Chaperone for the Cytosolic Cluster Assembly Machinery. J. Biol. Chem. 2016, 291, 22344–22356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, C.; Roos, A.K.; Montano, S.J.; Sengupta, R.; Filippakopoulos, P.; Guo, K.; von Delft, F.; Holmgren, A.; Oppermann, U.; Kavanagh, K.L. The crystal structure of human GLRX5: Iron-sulfur cluster co-ordination, tetrameric assembly and monomer activity. Biochem. J. 2011, 433, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Haunhorst, P.; Berndt, C.; Eitner, S.; Godoy, J.R.; Lillig, C.H. Characterization of the human monothiol glutaredoxin 3 (PICOT) as iron–sulfur protein. Biochem. Biophys. Res. Commun. 2010, 394, 372–376. [Google Scholar] [CrossRef]
- Ye, H.; Rouault, T.A. Human Iron−Sulfur Cluster Assembly, Cellular Iron Homeostasis, and Disease. Biochemistry 2010, 49, 4945–4956. [Google Scholar] [CrossRef]
- Rouault, T.A. Biogenesis of iron-sulfur clusters in mammalian cells: New insights and relevance to human disease. Dis. Model. Mech. 2012, 5, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Matsui, R.; Ferran, B.; Oh, A.; Croteau, D.; Shao, D.; Han, J.; Pimentel, D.R.; Bachschmid, M.M. Redox Regulation via Glutaredoxin-1 and Protein S-Glutathionylation. Antioxid. Redox Signal. 2020, 32, 677–700. [Google Scholar] [CrossRef]
- Pai, H.V.; Starke, D.W.; Lesnefsky, E.J.; Hoppel, C.L.; Mieyal, J.J. What is the functional significance of the unique location of glutaredoxin 1 (GRx1) in the intermembrane space of mitochondria? Antioxid. Redox Signal. 2007, 9, 2027–2033. [Google Scholar] [CrossRef]
- Lind, C.; Gerdes, R.; Schuppe-Koistinen, I.; Cotgreave, I.A. Studies on the mechanism of oxidative modification of human glyceraldehyde-3-phosphate dehydrogenase by glutathione: Catalysis by glutaredoxin. Biochem. Biophys. Res. Commun. 1998, 247, 481–486. [Google Scholar] [CrossRef]
- Pineda-Molina, E.; Klatt, P.; Vázquez, J.; Marina, A.; García de Lacoba, M.; Pérez-Sala, D.; Lamas, S. Glutathionylation of the p50 subunit of NF-kappaB: A mechanism for redox-induced inhibition of DNA binding. Biochemistry 2001, 40, 14134–14142. [Google Scholar] [CrossRef] [PubMed]
- Reynaert, N.L.; van der Vliet, A.; Guala, A.S.; McGovern, T.; Hristova, M.; Pantano, C.; Heintz, N.H.; Heim, J.; Ho, Y.-S.; Matthews, D.E.; et al. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc. Natl. Acad. Sci. USA 2006, 103, 13086–13091. [Google Scholar] [CrossRef] [Green Version]
- Barrett, W.C.; DeGnore, J.P.; König, S.; Fales, H.M.; Keng, Y.F.; Zhang, Z.Y.; Yim, M.B.; Chock, P.B. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry 1999, 38, 6699–6705. [Google Scholar] [CrossRef] [PubMed]
- Klatt, P.; Molina, E.P.; Lacoba, M.G.; Alicia Padilla, C.; Martínez-Galisteo, E.; Barcena, J.; Lamas, S. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J. 1999, 13, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.; Jones, A.D.; Cross, C.E.; Wong, P.S.; Van Der Vliet, A. Inactivation of creatine kinase by S-glutathionylation of the active-site cysteine residue. Biochem. J. 2000, 347 Pt 3, 821–827. [Google Scholar] [CrossRef]
- Wang, J.; Boja, E.S.; Tan, W.; Tekle, E.; Fales, H.M.; English, S.; Mieyal, J.J.; Chock, P.B. Reversible glutathionylation regulates actin polymerization in A431 cells. J. Biol. Chem. 2001, 276, 47763–47766. [Google Scholar] [CrossRef] [Green Version]
- Davis, D.A.; Newcomb, F.M.; Starke, D.W.; Ott, D.E.; Mieyal, J.J.; Yarchoan, R. Thioltransferase (glutaredoxin) is detected within HIV-1 and can regulate the activity of glutathionylated HIV-1 protease in vitro. J. Biol. Chem. 1997, 272, 25935–25940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, H.; Ihara, Y.; Nakamura, H.; Yodoi, J.; Sumikawa, K.; Kondo, T. Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J. Biol. Chem. 2003, 278, 50226–50233. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-A.; Wang, T.-Y.; Varadharaj, S.; Reyes, L.A.; Hemann, C.; Talukder, M.A.H.; Chen, Y.-R.; Druhan, L.J.; Zweier, J.L. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 2010, 468, 1115–1118. [Google Scholar] [CrossRef] [Green Version]
- Adachi, T.; Weisbrod, R.M.; Pimentel, D.R.; Ying, J.; Sharov, V.S.; Schöneich, C.; Cohen, R.A. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 2004, 10, 1200–1207. [Google Scholar] [CrossRef]
- Eaton, P.; Wright, N.; Hearse, D.J.; Shattock, M.J. Glyceraldehyde phosphate dehydrogenase oxidation during cardiac ischemia and reperfusion. J. Mol. Cell. Cardiol. 2002, 34, 1549–1560. [Google Scholar] [CrossRef]
- Gallogly, M.M.; Starke, D.W.; Mieyal, J.J. Mechanistic and kinetic details of catalysis of thiol-disulfide exchange by glutaredoxins and potential mechanisms of regulation. Antioxid. Redox Signal. 2009, 11, 1059–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anathy, V.; Aesif, S.W.; Guala, A.S.; Havermans, M.; Reynaert, N.L.; Ho, Y.-S.; Budd, R.C.; Janssen-Heininger, Y.M.W. Redox amplification of apoptosis by caspase-dependent cleavage of glutaredoxin 1 and S-glutathionylation of Fas. J. Cell Biol. 2009, 184, 241–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.J.; Rhee, J.G.; Suntharalingam, M.; Walsh, S.A.; Spitz, D.R.; Lee, Y.J. Role of glutaredoxin in metabolic oxidative stress. Glutaredoxin as a sensor of oxidative stress mediated by H2O2. J. Biol. Chem. 2002, 277, 46566–46575. [Google Scholar] [CrossRef] [Green Version]
- Song, J.J.; Lee, Y.J. Differential role of glutaredoxin and thioredoxin in metabolic oxidative stress-induced activation of apoptosis signal-regulating kinase 1. Biochem. J. 2003, 373, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Adluri, R.S.; Thirunavukkarasu, M.; Zhan, L.; Dunna, N.R.; Akita, Y.; Selvaraju, V.; Otani, H.; Sanchez, J.A.; Ho, Y.-S.; Maulik, N. Glutaredoxin-1 overexpression enhances neovascularization and diminishes ventricular remodeling in chronic myocardial infarction. PLoS ONE 2012, 7, e34790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.C.; Ogut, O. Decline of contractility during ischemia-reperfusion injury: Actin glutathionylation and its effect on allosteric interaction with tropomyosin. Am. J. Physiol. Cell Physiol. 2006, 290, C719–C727. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, S.; Shao, D.; Pimentel, D.; Matsui, R.; Bachschmid, M. S-glutathionylation of Glyceraldehyde 3-Phosphate Dehydrogenase Regulates Sirtuin-1 Function through Trans-glutathionylation: Implication of Glutaredoxin-1. Free Radic. Biol. Med. 2019, 145, S37. [Google Scholar]
- Travasso, R.D.M.; Sampaio Dos Aidos, F.; Bayani, A.; Abranches, P.; Salvador, A. Localized redox relays as a privileged mode of cytoplasmic hydrogen peroxide signaling. Redox Biol. 2017, 12, 233–245. [Google Scholar] [CrossRef]
- Petrushanko, I.Y.; Yakushev, S.; Mitkevich, V.A.; Kamanina, Y.V.; Ziganshin, R.H.; Meng, X.; Anashkina, A.A.; Makhro, A.; Lopina, O.D.; Gassmann, M.; et al. S-Glutathionylation of the Na,K-ATPase Catalytic α Subunit Is a Determinant of the Enzyme Redox Sensitivity. J. Biol. Chem. 2012, 287, 32195–32205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poluektov, Y.M.; Petrushanko, I.Y.; Undrovinas, N.A.; Lakunina, V.A.; Khapchaev, A.Y.; Kapelko, V.I.; Abramov, A.A.; Lakomkin, V.L.; Novikov, M.S.; Shirinsky, V.P.; et al. Glutathione-related substances maintain cardiomyocyte contractile function in hypoxic conditions. Sci. Rep. 2019, 9, 4872. [Google Scholar] [CrossRef]
- Zweier, J.L.; Chen, C.-A.; Druhan, L.J. S-glutathionylation reshapes our understanding of endothelial nitric oxide synthase uncoupling and nitric oxide/reactive oxygen species-mediated signaling. Antioxid. Redox Signal. 2011, 14, 1769–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochette, L.; Lorin, J.; Zeller, M.; Guilland, J.-C.; Lorgis, L.; Cottin, Y.; Vergely, C. Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: Possible therapeutic targets? Pharmacol. Ther. 2013, 140, 239–257. [Google Scholar] [CrossRef]
- Chen, C.-A.; De Pascali, F.; Basye, A.; Hemann, C.; Zweier, J.L. Redox modulation of endothelial nitric oxide synthase by glutaredoxin-1 through reversible oxidative post-translational modification. Biochemistry 2013, 52, 6712–6723. [Google Scholar] [CrossRef]
- Evangelista, A.M.; Thompson, M.D.; Weisbrod, R.M.; Pimental, D.R.; Tong, X.; Bolotina, V.M.; Cohen, R.A. Redox regulation of SERCA2 is required for vascular endothelial growth factor-induced signaling and endothelial cell migration. Antioxid. Redox Signal. 2012, 17, 1099–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, J.; Sharov, V.; Xu, S.; Jiang, B.; Gerrity, R.; Schöneich, C.; Cohen, R.A. Cysteine-674 oxidation and degradation of sarcoplasmic reticulum Ca(2+) ATPase in diabetic pig aorta. Free Radic. Biol. Med. 2008, 45, 756–762. [Google Scholar] [CrossRef] [Green Version]
- Berenji, K.; Drazner, M.H.; Rothermel, B.A.; Hill, J.A. Does load-induced ventricular hypertrophy progress to systolic heart failure? Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H8–H16. [Google Scholar] [CrossRef]
- Haider, A.W.; Larson, M.G.; Benjamin, E.J.; Levy, D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J. Am. Coll. Cardiol. 1998, 32, 1454–1459. [Google Scholar] [CrossRef] [Green Version]
- Heineke, J.; Molkentin, J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef]
- Bachschmid, M.M.; Xu, S.; Maitland-Toolan, K.A.; Ho, Y.-S.; Cohen, R.A.; Matsui, R. Attenuated cardiovascular hypertrophy and oxidant generation in response to angiotensin II infusion in glutaredoxin-1 knockout mice. Free Radic. Biol. Med. 2010, 49, 1221–1229. [Google Scholar] [CrossRef] [Green Version]
- Jeong, D.; Kim, J.M.; Cha, H.; Oh, J.G.; Park, J.; Yun, S.-H.; Ju, E.-S.; Jeon, E.-S.; Hajjar, R.J.; Park, W.J. PICOT Attenuates Cardiac Hypertrophy by Disrupting Calcineurin–NFAT Signaling. Circ. Res. 2008, 102, 711–719. [Google Scholar] [PubMed] [Green Version]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [PubMed]
- Ross, R.; Harker, L. Hyperlipidemia and atherosclerosis. Science 1976, 193, 1094–1100. [Google Scholar] [PubMed]
- Wouters, K.; Shiri-Sverdlov, R.; van Gorp, P.J.; van Bilsen, M.; Hofker, M.H. Understanding hyperlipidemia and atherosclerosis: Lessons from genetically modified apoe and ldlr mice. Clin. Chem. Lab. Med. 2005, 43, 470–479. [Google Scholar]
- Griendling, K.K.; FitzGerald, G.A. Oxidative stress and cardiovascular injury: Part II: Animal and human studies. Circulation 2003, 108, 2034–2040. [Google Scholar]
- Fukai, T.; Galis, Z.S.; Meng, X.P.; Parthasarathy, S.; Harrison, D.G. Vascular expression of extracellular superoxide dismutase in atherosclerosis. J. Clin. Investig. 1998, 101, 2101–2111. [Google Scholar]
- Shukla, S.; Rizvi, F.; Raisuddin, S.; Kakkar, P. FoxO proteins’ nuclear retention and BH3-only protein Bim induction evoke mitochondrial dysfunction-mediated apoptosis in berberine-treated HepG2 cells. Free Radic. Biol. Med. 2014, 76, 185–199. [Google Scholar]
- Lei, K.; Davis, R.J. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 2432–2437. [Google Scholar]
- Corazza, N.; Jakob, S.; Schaer, C.; Frese, S.; Keogh, A.; Stroka, D.; Kassahn, D.; Torgler, R.; Mueller, C.; Schneider, P.; et al. TRAIL receptor-mediated JNK activation and Bim phosphorylation critically regulate Fas-mediated liver damage and lethality. J. Clin. Investig. 2006, 116, 2493–2499. [Google Scholar]
- Nigro, P.; Abe, J.-I.; Berk, B.C. Flow shear stress and atherosclerosis: A matter of site specificity. Antioxid. Redox Signal. 2011, 15, 1405–1414. [Google Scholar]
- Li, Y.; Ren, M.; Wang, X.; Cui, X.; Zhao, H.; Zhao, C.; Zhou, J.; Guo, Y.; Hu, Y.; Yan, C.; et al. Glutaredoxin 1 mediates the protective effect of steady laminar flow on endothelial cells against oxidative stress-induced apoptosis via inhibiting Bim. Sci. Rep. 2017, 7, 15539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piemonte, F.; Petrini, S.; Gaeta, L.M.; Tozzi, G.; Bertini, E.; Devito, R.; Boldrini, R.; Marcellini, M.; Ciacco, E.; Nobili, V. Protein glutathionylation increases in the liver of patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2008, 23, e457–e464. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Watanabe, K.; Fujioka, D.; Nakamura, K.; Nakamura, T.; Uematsu, M.; Bachschmid, M.M.; Matsui, R.; Kugiyama, K. Protein S-glutathionylation stimulate adipogenesis by stabilizing C/EBPβ in 3T3L1 cells. FASEB J. 2020, 34, 5827–5837. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells 2019, 8, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ban, J.-J.; Ruthenborg, R.J.; Cho, K.W.; Kim, J.-W. Regulation of obesity and insulin resistance by hypoxia-inducible factors. Hypoxia (Auckl.) 2014, 2, 171–183. [Google Scholar]
- Krishnan, J.; Suter, M.; Windak, R.; Krebs, T.; Felley, A.; Montessuit, C.; Tokarska-Schlattner, M.; Aasum, E.; Bogdanova, A.; Perriard, E.; et al. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009, 9, 512–524. [Google Scholar] [CrossRef]
- Reinbothe, T.M.; Ivarsson, R.; Li, D.-Q.; Niazi, O.; Jing, X.; Zhang, E.; Stenson, L.; Bryborn, U.; Renström, E. Glutaredoxin-1 mediates NADPH-dependent stimulation of calcium-dependent insulin secretion. Mol. Endocrinol. 2009, 23, 893–900. [Google Scholar] [CrossRef] [Green Version]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef] [Green Version]
- Klaus, A.; Zorman, S.; Berthier, A.; Polge, C.; Ramirez, S.; Michelland, S.; Sève, M.; Vertommen, D.; Rider, M.; Lentze, N.; et al. Glutathione S-transferases interact with AMP-activated protein kinase: Evidence for S-glutathionylation and activation in vitro. PLoS ONE 2013, 8, e62497. [Google Scholar] [CrossRef]
- Velu, C.S.; Niture, S.K.; Doneanu, C.E.; Pattabiraman, N.; Srivenugopal, K.S. Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry 2007, 46, 7765–7780. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Thompson, J.; Hu, Y.; Das, A.; Lesnefsky, E.J. Cardiac Specific Knockout of p53 Decreases ER Stress-Induced Mitochondrial Damage. Front. Cardiovasc. Med. 2019, 6, 10. [Google Scholar] [CrossRef]
- Wohua, Z.; Weiming, X. Glutaredoxin 2 (GRX2) deficiency exacerbates high fat diet (HFD)-induced insulin resistance, inflammation and mitochondrial dysfunction in brain injury: A mechanism involving GSK-3β. Biomed. Pharmacother. 2019, 118, 108940. [Google Scholar] [CrossRef]
- Young, A.; Gardiner, D.; Kuksal, N.; Gill, R.; O’Brien, M.; Mailloux, R.J. Deletion of the Glutaredoxin-2 Gene Protects Mice from Diet-Induced Weight Gain, Which Correlates with Increased Mitochondrial Respiration and Proton Leaks in Skeletal Muscle. Antioxid. Redox Signal. 2019, 31, 1272–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, H.; Kiyohara, Y.; Kato, I.; Kitazono, T.; Tanizaki, Y.; Kubo, M.; Ueno, H.; Ibayashi, S.; Fujishima, M.; Iida, M. Relationship Between Plasma Glutathione Levels and Cardiovascular Disease in a Defined Population. Stroke 2004, 35, 2072–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagman, M.; Ly, J.; Saing, T.; Kaur Singh, M.; Vera Tudela, E.; Morris, D.; Chi, P.-T.; Ochoa, C.; Sathananthan, A.; Venketaraman, V. Investigating the causes for decreased levels of glutathione in individuals with type II diabetes. PLoS ONE 2015, 10, e0118436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damy, T.; Kirsch, M.; Khouzami, L.; Caramelle, P.; Le Corvoisier, P.; Roudot-Thoraval, F.; Dubois-Randé, J.-L.; Hittinger, L.; Pavoine, C.; Pecker, F. Glutathione deficiency in cardiac patients is related to the functional status and structural cardiac abnormalities. PLoS ONE 2009, 4, e4871. [Google Scholar] [CrossRef]
- Redón, J.; Oliva, M.R.; Tormos, C.; Giner, V.; Chaves, J.; Iradi, A.; Sáez, G.T. Antioxidant activities and oxidative stress byproducts in human hypertension. Hypertension 2003, 41, 1096–1101. [Google Scholar] [CrossRef] [Green Version]
- Shahid, S.U.; Shabana; Humphries, S. The SNP rs10911021 is associated with oxidative stress in coronary heart disease patients from Pakistan. Lipids Health Dis. 2018, 17, 6. [Google Scholar] [CrossRef] [Green Version]
- Okuda, M.; Inoue, N.; Azumi, H.; Seno, T.; Sumi, Y.; Hirata, K.I.; Kawashima, S.; Hayashi, Y.; Itoh, H.; Yodoi, J.; et al. Expression of glutaredoxin in human coronary arteries: Its potential role in antioxidant protection against atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1483–1487. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Vaage, J.; Valen, G.; Padilla, C.A.; Björnstedt, M.; Holmgren, A. Measurements of plasma glutaredoxin and thioredoxin in healthy volunteers and during open-heart surgery. Free Radic. Biol. Med. 1998, 24, 1176–1186. [Google Scholar] [CrossRef]
- Kimura, T.; Ferran, B.; Tsukahara, Y.; Shang, Q.; Desai, S.; Fedoce, A.; Pimentel, D.R.; Luptak, I.; Adachi, T.; Ido, Y.; et al. Production of adeno-associated virus vectors for in vitro and in vivo applications. Sci. Rep. 2019, 9, 13601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Rodríguez, D.R.; Ramírez-Solís, R.; Garza-Elizondo, M.A.; Garza-Rodríguez, M.D.L.; Barrera-Saldaña, H.A. Genome editing: A perspective on the application of CRISPR/Cas9 to study human diseases (Review). Int. J. Mol. Med. 2019, 43, 1559–1574. [Google Scholar] [PubMed] [Green Version]
- Ahsan, M.K.; Lekli, I.; Ray, D.; Yodoi, J.; Das, D.K. Redox regulation of cell survival by the thioredoxin superfamily: An implication of redox gene therapy in the heart. Antioxid. Redox Signal. 2009, 11, 2741–2758. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Human Glutaredoxin | Reaction Mechanism | Primary Cellular Compartment | Primary Molecular Function | Aliases | Cardiovascular Pathologies | Active Site Motif | Mass (kDa) |
|---|---|---|---|---|---|---|---|
| Glrx | Monothiol or dithiol | Cytosol (Mitochondria, nucleus) | Glutathione oxidoreductase | Thioltransferase 1 | Ischemia [20], cardiac hypertrophy [58], atherosclerosis [59,60], peripheral arterial disease [23,24] | CPYC | 11.78 |
| Glrx2 | Monothiol or dithiol | Mitochondria | Iron-sulfur cluster assembly | Thioltransferase 2 | Cardiac hypertrophy [61,62], myocardial infarction [62] | CSYC | 18.05 |
| Glrx3 | Monothiol | Cytosol (nucleus) | Iron-sulfur cluster assembly | PICOT | Cardiac hypertrophy [63] | CGFS | 37.43 |
| Glrx5 | Monothiol | Mitochondria | Iron-sulfur cluster assembly | N/A | N/A | CGFS | 16.63 |
| Target Proteins of Glutaredoxin-1 | Glutathionylation Induced Functional Changes | Identified Glutathionylated Cysteine Residues | Mass (kDa) |
|---|---|---|---|
| GAPDH [71] | Decreased oxidoreductase and transferase activity | 150 | 36.05 |
| NF-κB [72,73] | Transcriptional inactivation | 179 (IKKβ), 62 (p50) | - |
| PTP1B [74] | Inactivation of protein tyrosine phosphatase activity | 215 | 49.97 |
| c-Jun [75] | Transcriptional inactivation | 269 | 35.68 |
| Rac-1 [59] | GTPase inactivation | 81, 157 | 21.45 |
| Creatine kinase [76] | Kinase and transferase inactivation | 283 | 43.10 |
| Actin [77] | Decreased polymerization rate and binding affinity | 374 | 41.74 |
| HIV-1 protease [78] | Decreased retroviral aspartyl protease activity | 67, 95 | 10.73 |
| Akt [79] | Inactivation of serine/threonine-protein kinase activity | 297, 311 | 55.69 |
| eNOS [80] | Uncoupling (conversion to superoxide production) | 689, 908 | 131.12 |
| Ras [58] | Increased GTPase activity | 118 | 21.30 |
| HIF-1α [23] | Stabilization and transcriptional activation | 520 | 92.67 |
| SirT1 [16] | Deacetylase inactivation | 61, 318, 613 | 81.68 |
| SERCA 2 [81] | ER Ca2+ ATPase activation | 674 | 114.14 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burns, M.; Rizvi, S.H.M.; Tsukahara, Y.; Pimentel, D.R.; Luptak, I.; Hamburg, N.M.; Matsui, R.; Bachschmid, M.M. Role of Glutaredoxin-1 and Glutathionylation in Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21, 6803. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186803
Burns M, Rizvi SHM, Tsukahara Y, Pimentel DR, Luptak I, Hamburg NM, Matsui R, Bachschmid MM. Role of Glutaredoxin-1 and Glutathionylation in Cardiovascular Diseases. International Journal of Molecular Sciences. 2020; 21(18):6803. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186803
Chicago/Turabian StyleBurns, Mannix, Syed Husain Mustafa Rizvi, Yuko Tsukahara, David R. Pimentel, Ivan Luptak, Naomi M. Hamburg, Reiko Matsui, and Markus M. Bachschmid. 2020. "Role of Glutaredoxin-1 and Glutathionylation in Cardiovascular Diseases" International Journal of Molecular Sciences 21, no. 18: 6803. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186803