Adenovirus Receptor Expression in Cancer and Its Multifaceted Role in Oncolytic Adenovirus Therapy



Abstract
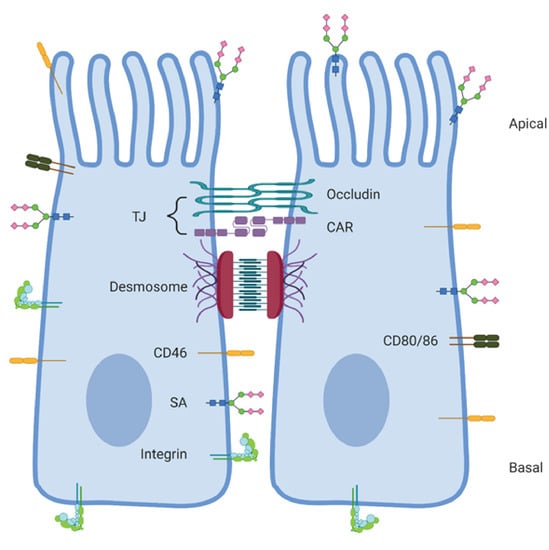
:
1. Introduction
2. Adenovirus Receptor Expression in Normal Cells and Cancer Cells
2.1. Coxsackie and Adenovirus Receptor
2.2. CD46
2.3. Desmoglein-2
2.4. CD80 and CD86
2.5. Sialic Acid
2.6. Integrins
2.7. Other Receptors
3. Adenovirus Receptor Expression after Therapy
4. The Multifaceted Role of Adenovirus Receptors
4.1. Viral Spread
4.2. Broad Tropism
5. Future Directions
Funding
Conflicts of Interest
Abbreviations
| OVT | Oncolytic virus therapy |
| Ads | Adenoviruses |
| HAdV | Human Adenovirus |
| nAbs | Neutralizing antibodies |
| TME | Tumor microenvironment |
| CAR | Coxsackie and Adenovirus receptor |
| DSG-2 | Desmoglein-2 |
| HSPG | Heparan sulphate proteoglycan |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| MHC-I α2 | Major histocompatibility complex-1 α2 domain |
| MARCO | Macrophage receptor with collagenous structure |
| SA | Sialic acid |
| TJ | Tight junction |
| IRS | Immunoreactive score |
| HIF-1 α | Hypoxia inducible factor-1α |
| EMT | Epithelial to mesenchymal transition |
| STAT3 | Signal transducers and activators of transcription 3 |
| APC | Antigen presenting cell |
| CMP | Cytidine-monophosphate |
| TAM | Tumor associated macrophage |
| FX | Factor X |
| DPPC | Dipalmitoyl phosphatidylcholine |
| HSV | Herpes Simplex Virus |
References
- Kelly, E.; Russell, S.J. History of oncolytic viruses: Genesis to genetic engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef]
- De Graaf, J.F.; De Vor, L.; Fouchier, R.A.M.; Hoogen, B.G. Van Den Armed oncolytic viruses: A kick-start for anti-tumor immunity. Cytokine Growth Factor Rev. 2018, 41, 28–39. [Google Scholar] [CrossRef]
- de Matos, A.L.; Franco, L.S.; McFadden, G. Oncolytic Viruses and the Immune System: The Dynamic Duo. Mol. Ther. Methods Clin. Dev. 2020, 17, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Seto, D.; Chodosh, J.; Brister, J.R.; Jones, M.S. Using the Whole-Genome Sequence to Characterize and Name Human Adenoviruses. J. Virol. 2011, 85, 5701–5702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, A.T.; Aguirre-Hernández, C.; Halldén, G.; Parker, A.L. Designer oncolytic adenovirus: Coming of age. Cancers 2018, 10, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Water Recreation and Disease: Viruses; IWA Publishing: London, UK, 2005; ISBN 1843390663. [Google Scholar]
- Zhang, Y.; Bergelson, J.M. Adenovirus receptors. J. Virol. 2005, 79, 12125–12131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnberg, N. Adenovirus receptors: Implications for targeting of viral vectors. Trends Pharmacol. Sci. 2012, 33, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, E.C.; Storm, R.J.; Bauer, J.; Johansson, S.M.C.; Lookene, A.; Ångström, J.; Hedenström, M.; Eriksson, T.L.; Frängsmyr, L.; Rinaldi, S.; et al. The GD1a glycan is a cellular receptor for adenoviruses causing epidemic keratoconjunctivitis. Nat. Med. 2011, 17, 105–109. [Google Scholar] [CrossRef]
- Vogels, R.; Zuijdgeest, D.; van Rijnsoever, R.; Hartkoorn, E.; Damen, I.; de Béthune, M.-P.; Kostense, S.; Penders, G.; Helmus, N.; Koudstaal, W.; et al. Replication-Deficient Human Adenovirus Type 35 Vectors for Gene Transfer and Vaccination: Efficient Human Cell Infection and Bypass of Preexisting Adenovirus Immunity. J. Virol. 2003, 77, 8263–8271. [Google Scholar] [CrossRef] [Green Version]
- Fausther-Bovendo, H.; Kobinger, G.P. Pre-existing immunity against Ad vectors: Humoral, cellular, and innate response, what’s important? Hum. Vaccines Immunother. 2014, 10, 2875–2884. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, P.; Li, H.; Du, X.; Liu, M.; Huang, Q.; Wang, Y.; Wang, S. The efficacy of oncolytic adenovirus is mediated by T-cell responses against virus and tumor in Syrian hamster model. Clin. Cancer Res. 2017, 23, 239–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaiss, A.K.; Machado, H.B.; Herschman, H.R. The influence of innate and pre-existing immunity on adenovirus therapy. J. Cell. Biochem. 2009, 108, 778–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mennechet, F.J.D.; Paris, O.; Ouoba, A.R.; Salazar Arenas, S.; Sirima, S.B.; Takoudjou Dzomo, G.R.; Diarra, A.; Traore, I.T.; Kania, D.; Eichholz, K.; et al. A review of 65 years of human adenovirus seroprevalence. Expert Rev. Vaccines 2019, 18, 597–613. [Google Scholar] [CrossRef] [PubMed]
- Bots, S.T.F.; Hoeben, R.C. Non-Human Primate-Derived Adenoviruses for Future Use as Oncolytic Agents? Int. J. Mol. Sci. 2020, 21, 4821. [Google Scholar] [CrossRef]
- Hong, S.S.; Karayan, L.; Tournier, J.; Curiel, D.T.; Boulanger, P.A. Adenovirus type 5 fiber knob binds to MHC class I α2 domain at the surface of human epithelial and B lymphoblastoid cells. EMBO J. 1997, 16, 2294–2306. [Google Scholar] [CrossRef] [Green Version]
- Maler, M.D.; Nielsen, P.J.; Stichling, N.; Cohen, I.; Ruzsics, Z.; Wood, C.; Engelhard, P.; Suomalainen, M.; Gyory, I.; Huber, M.; et al. Key role of the scavenger receptor MARCO in mediating adenovirus infection and subsequent innate responses of macrophages. mBio 2017, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Lenman, A.; Liaci, A.M.; Liu, Y.; Årdahl, C.; Rajan, A.; Nilsson, E.; Bradford, W.; Kaeshammer, L.; Jones, M.S.; Frängsmyr, L.; et al. Human Adenovirus 52 Uses Sialic Acid-containing Glycoproteins and the Coxsackie and Adenovirus Receptor for Binding to Target Cells. PLoS Pathog. 2015, 11, e1004657. [Google Scholar] [CrossRef]
- Lenman, A.; Manuel Liaci, A.; Liu, Y.; Frängsmyr, L.; Frank, M.; Blaum, B.S.; Chai, W.; Podgorski, I.I.; Harrach, B.; Benko, M.; et al. Polysialic acid is a cellular receptor for human adenovirus 52. Proc. Natl. Acad. Sci. USA 2018, 115, E4264–E4273. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Vélez, N.; Garcia-Moure, M.; Marigil, M.; González-Huarriz, M.; Puigdelloses, M.; Gallego Pérez-Larraya, J.; Zalacaín, M.; Marrodán, L.; Varela-Guruceaga, M.; Laspidea, V.; et al. The oncolytic virus Delta-24-RGD elicits an antitumor effect in pediatric glioma and DIPG mouse models. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Cheng, P.H.; Wechman, S.L.; McMasters, K.M.; Zhou, H.S. Oncolytic replication of E1b-deleted adenoviruses. Viruses 2015, 7, 5767–5779. [Google Scholar] [CrossRef] [Green Version]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a Common Receptor for Coxsackie B Viruses and Adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Farkas, T.; Yang, K.; Le Pendu, J.; Baines, J.D.; Cardin, R.D. The Coxsackievirus and Adenovirus Receptor, a Required Host Factor for Recovirus Infection, Is a Putative Enteric Calicivirus Receptor. J. Virol. 2019, 93, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Roelvink, P.W.; Lizonova, A.; Lee, J.G.M.; Li, Y.; Bergelson, J.M.; Finberg, R.W.; Brough, D.E.; Kovesdi, I.; Wickham, T.J. The Coxsackievirus-Adenovirus Receptor Protein Can Function as a Cellular Attachment Protein for Adenovirus Serotypes from Subgroups A, C, D, E, and F. J. Virol. 1998, 72, 7909–7915. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Sato, K.; Hamada, H. Reduction of Natural Adenovirus Tropism to the Liver by both Ablation of Fiber-Coxsackievirus and Adenovirus Receptor Interaction and Use of Replaceable Short Fiber. J. Virol. 2003, 77, 2512–2521. [Google Scholar] [CrossRef] [Green Version]
- Cohen, C.J.; Shieh, J.T.C.; Pickles, R.J.; Okegawa, T.; Hsieh, J.T.; Bergelson, J.M. The coxsackievirus and adenovirus receptor is a transmembrane component of the tight junction. Proc. Natl. Acad. Sci. USA 2001, 98, 15191–15196. [Google Scholar] [CrossRef] [Green Version]
- Reeh, M.; Bockhorn, M.; Görgens, D.; Vieth, M.; Hoffmann, T.; Simon, R.; Izbicki, J.R.; Sauter, G.; Schumacher, U.; Anders, M. Presence of the Coxsackievirus and Adenovirus Receptor (CAR) in human neoplasms: A multitumour array analysis. Br. J. Cancer 2013, 109, 1848–1858. [Google Scholar] [CrossRef]
- Smith, T.A.G.; Iverson, W.O.; Sherer, A.D.; Markovits, J.E.; Lyons, R.M.; Kaleko, M.; Stevenson, S.C.; Idamakanti, N.; Marshall-Neff, J.; Rollence, M.L.; et al. Receptor Interactions Involved in Adenoviral-Mediated Gene Delivery after Systemic Administration in Non-Human Primates. Hum. Gene Ther. 2003, 14, 1595–1604. [Google Scholar] [CrossRef]
- Persson, A.; Fan, X.; Widegren, B.; Englund, E. Cell type- and region- dependent coxsackie adenovirus receptor expression in the central nervous system. J. Neurooncol. 2006, 78, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Sachs, M.D.; Rauen, K.A.; Ramamurthy, M.; Dodson, J.L.; De Marzo, A.M.; Putzi, M.J.; Schoenberg, M.P.; Rodriguez, R. Integrin αv and coxsackie adenovirus receptor expression in clinical bladder cancer. Urology 2002, 60, 531–536. [Google Scholar] [CrossRef]
- Ma, Y.Y.; Wang, X.J.; Han, Y.; Li, G.; Wang, H.J.; Wang, S.B.; Chen, X.Y.; Liu, F.L.; He, X.L.; Tong, X.M.; et al. Loss of coxsackie and adenovirus receptor expression in human colorectal cancer: A potential impact on the efficacy of adenovirus-mediated gene therapy in Chinese Han population. Mol. Med. Rep. 2016, 14, 2541–2547. [Google Scholar] [CrossRef] [PubMed]
- Stecker, K.; Vieth, M.; Koschel, A.; Wiedenmann, B.; Röcken, C.; Anders, M. Impact of the coxsackievirus and adenovirus receptor on the adenoma-carcinoma sequence of colon cancer. Br. J. Cancer 2011, 104, 1426–1433. [Google Scholar] [CrossRef] [PubMed]
- Küster, K.; Koschel, A.; Rohwer, N.; Fischer, A.; Wiedenmann, B.; Anders, M. Downregulation of the coxsackie and adenovirus receptor in cancer cells by hypoxia depends on HIF-1α. Cancer Gene Ther. 2010, 17, 141–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21. [Google Scholar] [CrossRef]
- Lacher, M.D.; Shiina, M.; Chang, P.; Keller, D.; Tiirikainen, M.I.; Korn, W.M. ZEB1 limits adenoviral infectability by transcriptionally repressing the Coxsackie virus and Adenovirus Receptor. Mol. Cancer 2011, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarakanova, V.L.; Wold, W.S.M. Transforming Growth Factor β1 Receptor II Is Downregulated by E1A in Adenovirus-Infected Cells. J. Virol. 2003, 77, 9324–9336. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Li, Z.Y.; Liu, Y.; Persson, J.; Beyer, I.; Möller, T.; Koyuncu, D.; Drescher, M.R.; Strauss, R.; Zhang, X.B.; et al. Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat. Med. 2011, 17, 96–104. [Google Scholar] [CrossRef]
- Gaggar, A.; Shayakhmetov, D.M.; Lieber, A. CD46 is a cellular receptor for group B adenoviruses. Nat. Med. 2003, 9, 1408–1412. [Google Scholar] [CrossRef]
- Liu, J.; Boehme, P.; Zhang, W.; Fu, J.; Yumul, R.; Mese, K.; Tsoukas, R.; Solanki, M.; Kaufmann, M.; Lu, R.; et al. Human adenovirus type 17 from species D transduces endothelial cells and human CD46 is involved in cell entry. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Wu, E.; Trauger, S.A.; Pache, L.; Mullen, T.-M.; Von Seggern, D.J.; Siuzdak, G.; Nemerow, G.R. Membrane Cofactor Protein Is a Receptor for Adenoviruses Associated with Epidemic Keratoconjunctivitis. J. Virol. 2004, 78, 3897–3905. [Google Scholar] [CrossRef] [Green Version]
- Seya, T.; Hirano, A.; Matsumoto, M.; Nomura, M.; Ueda, S. Human membrane cofactor protein (MCP, CD46): Multiple isoforms and functions. Int. J. Biochem. Cell Biol. 1999, 31, 1255–1260. [Google Scholar] [CrossRef]
- Do, M.H.; To, P.K.; Cho, Y.S.; Kwon, S.Y.; Hwang, E.C.; Choi, C.; Cho, S.H.; Lee, S.J.; Hemmi, S.; Jung, C. Targeting CD46 enhances anti-tumoral activity of adenovirus type 5 for bladder cancer. Int. J. Mol. Sci. 2018, 19, 2694. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Liu, Y.; Behrens, C.R.; Bidlingmaier, S.; Lee, N.K.; Aggarwal, R.; Sherbenou, D.W.; Burlingame, A.L.; Hann, B.C.; Simko, J.P.; et al. Targeting CD46 for both adenocarcinoma and neuroendocrine prostate cancer. JCI Insight 2018, 3, e121497. [Google Scholar] [CrossRef] [Green Version]
- Geller, A.; Yan, J. The role of membrane bound complement regulatory proteins in tumor development and cancer immunotherapy. Front. Immunol. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.S.; Do, M.H.; Kwon, S.Y.; Moon, C.; Kim, K.; Lee, K.; Lee, S.J.; Hemmi, S.; Joo, Y.E.; Kim, M.S.; et al. Efficacy of CD46-targeting chimeric Ad5/35 adenoviral gene therapy for colorectal cancers. Oncotarget 2016, 7, 38210–38223. [Google Scholar] [CrossRef] [Green Version]
- Buettner, R.; Huang, M.; Gritsko, T.; Karras, J.; Enkemann, S.; Mesa, T.; Nam, S.; Yu, H.; Jove, R. Activated signal transducers and activators of transcription 3 signaling induces CD46 expression and protects human cancer cells from complement-dependent cytotoxicity. Mol. Cancer Res. 2007, 5, 823–832. [Google Scholar] [CrossRef] [Green Version]
- Lok, A.; Descamps, G.; Tessoulin, B.; Chiron, D.; Eveillard, M.; Godon, C.; Le Bris, Y.; Vabret, A.; Bellanger, C.; Maillet, L.; et al. P53 regulates CD46 expression and measles virus infection in myeloma cells. Blood Adv. 2018, 2, 3492–3505. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, S.; Koch, P.J.; Franke, W.W. Identification of the ubiquitous human desmoglein, Dsg2, and the expression catalogue of the desmoglein subfamily of desmosomal cadherins. Exp. Cell Res. 1994, 211, 391–399. [Google Scholar] [CrossRef]
- Ad, H.A.; Wang, H.; Ducournau, C.; Saydaminova, K.; Richter, M.; Yumul, R.; Ho, M.; Carter, D.; Zubieta, C.; Fender, P.; et al. Intracellular Signaling and Desmoglein 2 Shedding Triggered by Human Adenoviruses Ad3, Ad14, and Ad14P1. J. Virol. 2015, 89, 10841–10859. [Google Scholar] [CrossRef]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Ulasov, I.V.; Rivera, A.A.; Han, Y.; Curiel, D.T.; Zhu, Z.B.; Lesniak, M.S. Targeting adenovirus to CD80 and CD86 receptors increases gene transfer efficiency to malignant glioma cells. J. Neurosurg. 2007, 107, 617–627. [Google Scholar] [CrossRef]
- Marchiori, C.; Scarpa, M.; Kotsafti, A.; Morgan, S.; Fassan, M.; Guzzardo, V.; Porzionato, A.; Angriman, I.; Ruffolo, C.; Sut, S.; et al. Epithelial CD80 promotes immune surveillance of colonic preneoplastic lesions and its expression is increased by oxidative stress through STAT3 in colon cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Yang, G.; Guan, F. Biological Functions and Analytical Strategies of Sialic Acids in Tumor. Cells 2020, 9, 273. [Google Scholar] [CrossRef] [Green Version]
- Nemerow, G.R.; Stewart, P.L. Role of αv Integrins in Adenovirus Cell Entry and Gene Delivery. Microbiol. Mol. Biol. Rev. 1999, 63, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Brennan, D.; Mahoney, M.G. Increased expression of Dsg2 in malignant skin carcinomas: A tissue-microarray based study. Cell Adhes. Migr. 2009, 3, 148–154. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.X.; Li, Y. Significance of desmoglein-2 on cell malignant behaviors via mediating MAPK signaling in cervical cancer. Kaohsiung J. Med. Sci. 2020, 36, 336–343. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.Y.; Mintoff, C.; Johan, M.Z.; Ebert, B.W.; Fedele, C.; Zhang, Y.F.; Szeto, P.; Sheppard, K.E.; McArthur, G.A.; Foster-Smith, E.; et al. Desmoglein 2 promotes vasculogenic mimicry in melanoma and is associated with poor clinical outcome. Oncotarget 2016, 7, 46492–46508. [Google Scholar] [CrossRef]
- Cai, F.; Zhu, Q.; Miao, Y.; Shen, S.; Su, X.; Shi, Y. Desmoglein-2 is overexpressed in non-small cell lung cancer tissues and its knockdown suppresses NSCLC growth by regulation of p27 and CDK2. J. Cancer Res. Clin. Oncol. 2017, 143, 59–69. [Google Scholar] [CrossRef]
- Sun, R.; Ma, C.; Wang, W.; Yang, S. Upregulation of desmoglein 2 and its clinical value in lung adenocarcinoma: A comprehensive analysis by multiple bioinformatics methods. PeerJ 2020, 2020, 1–21. [Google Scholar] [CrossRef]
- Kamekura, R.; Kolegraff, K.N.; Nava, P.; Hilgarth, R.S.; Feng, M.; Parkos, C.A.; Nusrat, A. Loss of the desmosomal cadherin desmoglein-2 suppresses colon cancer cell proliferation through EGFR signaling. Oncogene 2014, 33, 4531–4536. [Google Scholar] [CrossRef] [Green Version]
- Han, C.P.; Yu, Y.H.; Wang, A.G.; Tian, Y.; Zhang, H.T.; Zheng, Z.M.; Liu, Y.S. Desmoglein-2 overexpression predicts poor prognosis in hepatocellular carcinoma patients. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5481–5489. [Google Scholar] [CrossRef]
- Barber, A.G.; Castillo-Martin, M.; Bonal, D.M.; Rybicki, B.A.; Christiano, A.M.; Cordon-Cardo, C. Characterization of desmoglein expression in the normal prostatic gland. Desmoglein 2 is an independent prognostic factor for aggressive prostate cancer. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Biedermann, K.; Vogelsang, H.; Becker, I.; Plaschke, S.; Siewert, J.R.; Höfler, H.; Keller, G. Desmoglein 2 is expressed abnormally rather than mutated in familial and sporadic gastric cancer. J. Pathol. 2005, 207, 199–206. [Google Scholar] [CrossRef]
- Chen, L.; Liu, X.; Zhang, J.; Liu, Y.; Gao, A.; Xu, Y.; Lin, Y.; Du, Q.; Zhu, Z.; Hu, Y.; et al. Characterization of desmoglein 2 expression in ovarian serous tumors and its prognostic significance in high-grade serous carcinoma. Int. J. Clin. Exp. Pathol. 2018, 11, 4977–4986. [Google Scholar]
- Ramani, V.C.; Hennings, L.; Haun, R.S. Desmoglein 2 is a substrate of kallikrein 7 in pancreatic cancer. BMC Cancer 2008, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Hütz, K.; Zeiler, J.; Sachs, L.; Ormanns, S.; Spindler, V. Loss of desmoglein 2 promotes tumorigenic behavior in pancreatic cancer cells. Mol. Carcinog. 2017, 56, 1884–1895. [Google Scholar] [CrossRef]
- Short, J.J.; Vasu, C.; Holterman, M.J.; Curiel, D.T.; Pereboev, A. Members of adenovirus species B utilize CD80 and CD86 as cellular attachment receptors. Virus Res. 2006, 122, 144–153. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.K.; Dmitriev, I.P.; Kashentseva, E.A.; Raes, G.; Li, L.; Kim, S.W.; Lu, Z.-H.; Arbeit, J.M.; Fleming, T.P.; Kaliberov, S.A.; et al. Development of an adenovirus vector vaccine platform for targeting dendritic cells. Cancer Gene Ther. 2018, 25, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Ertl, H.C. Viral vectors as vaccine carriers. Curr. Opin. Virol. 2016, 21, 1–8. [Google Scholar] [CrossRef]
- Varki, A. Sialic acids in human health and disease. Trends Mol. Med. 2008, 14, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, E.; Macauley, M.S. Hypersialylation in cancer: Modulation of inflammation and therapeutic opportunities. Cancers 2018, 10, 207. [Google Scholar] [CrossRef] [Green Version]
- Rajan, A.; Persson, B.D.; Frängsmyr, L.; Olofsson, A.; Sandblad, L.; Heino, J.; Takada, Y.; Mould, A.P.; Schnapp, L.M.; Gall, J.; et al. Enteric Species F Human Adenoviruses use Laminin-Binding Integrins as Co-Receptors for Infection of Ht-29 Cells. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Nestić, D.; Uil, T.G.; Ma, J.; Roy, S.; Vellinga, J.; Baker, A.H.; Custers, J.; Majhen, D. αvβ3 Integrin Is Required for Efficient Infection of Epithelial Cells with Human Adenovirus Type 26. J. Virol. 2018, 93, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Lyle, C.; McCormick, F. Integrin αvβ5 is a primary receptor for adenovirus in CAR-negative cells. Virol. J. 2010, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Weis, S.M.; Cheresh, D.A. αv Integrins in Angiogenesis and Cancer. Cold Spring Harb. Perspect. Med. 2011, 1. [Google Scholar] [CrossRef] [Green Version]
- Merchan, J.; Toro Bejarano, M. Targeting tumor vasculature through oncolytic virotherapy: Recent advances. Oncolytic Virotherapy 2015, 169. [Google Scholar] [CrossRef] [Green Version]
- Man, Y.K.S.; Davies, J.A.; Coughlan, L.; Pantelidou, C.; Blázquez-Moreno, A.; Marshall, J.F.; Parker, A.L.; Halldén, G. The novel oncolytic adenoviral mutant Ad5-3Δ-A20T retargeted to αvβ6 integrins efficiently eliminates pancreatic cancer cells. Mol. Cancer Ther. 2018, 17, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Stichling, N.; Suomalainen, M.; Flatt, J.W.; Schmid, M.; Pacesa, M.; Hemmi, S.; Jungraithmayr, W.; Maler, M.D.; Freudenberg, M.A.; Plückthun, A.; et al. Lung Macrophage Scavenger Receptor SR-A6 (MARCO) is an Adenovirus Type-Specific Virus Entry Receptor. PLoS Pathog. 2018, 14. [Google Scholar] [CrossRef] [Green Version]
- Laviron, M.; Boissonnas, A. Ontogeny of Tumor-Associated Macrophages. Front. Immunol. 2019, 10, 1799. [Google Scholar] [CrossRef] [Green Version]
- La Fleur, L.; Boura, V.F.; Alexeyenko, A.; Berglund, A.; Pontén, V.; Mattsson, J.S.M.; Djureinovic, D.; Persson, J.; Brunnström, H.; Isaksson, J.; et al. Expression of scavenger receptor MARCO defines a targetable tumor-associated macrophage subset in non-small cell lung cancer. Int. J. Cancer 2018, 143, 1741–1752. [Google Scholar] [CrossRef] [Green Version]
- Waddington, S.N.; McVey, J.H.; Bhella, D.; Parker, A.L.; Barker, K.; Atoda, H.; Pink, R.; Buckley, S.M.K.; Greig, J.A.; Denby, L.; et al. Adenovirus Serotype 5 Hexon Mediates Liver Gene Transfer. Cell 2008, 132, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Duffy, M.R.; Doszpoly, A.; Turner, G.; Nicklin, S.A.; Baker, A.H. The relevance of coagulation factor X protection of adenoviruses in human sera. Gene Ther. 2016, 23, 592–596. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Feng, J.; Fu, H.; Liu, C.; Yu, Z.; Sun, Y.; She, X.; Li, P.; Zhao, C.; Liu, Y.; et al. Coagulation factor X regulated by CASC2c recruited macrophages and induced M2 polarization in glioblastoma multiforme. Front. Immunol. 2018, 9, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Sierko, E.; Wojtukiewicz, M.Z.; Zimnoch, L.; Tokajuk, P.; Ostrowska-Cichocka, K.; Kisiel, W. Co-localization of Protein Z, Protein Z-dependent protease inhibitor and coagulation factor X in human colon cancer tissue: Implications for coagulation regulation on tumor cells. Thromb. Res. 2012, 129, e112–e118. [Google Scholar] [CrossRef]
- Balakireva, L.; Schoehn, G.; Thouvenin, E.; Chroboczek, J. Binding of Adenovirus Capsid to Dipalmitoyl Phosphatidylcholine Provides a Novel Pathway for Virus Entry. J. Virol. 2003, 77, 4858–4866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jianyong, Z.; Jianjun, X.; Haiyan, L.; Jianhua, D.; Dongliang, Y.; Penghui, L.; Jianwen, X.; Xingxing, L.; Huanwen, C.; Yiping, W. Altered phosphatidylcholines expression in sputum for diagnosis of non-small cell lung cancer. Oncotarget 2016, 7, 63158–63165. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.; Heistad, D.D.; Cybulsky, M.I.; Davidson, B.L. Vascular cell adhesion molecule-1 augments adenovirus-mediated gene transfer. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 238–242. [Google Scholar] [CrossRef] [Green Version]
- Schlesinger, M.; Bendas, G. Vascular cell adhesion molecule-1 (VCAM-1)—An increasing insight into its role in tumorigenicity and metastasis. Int. J. Cancer 2015, 136, 2504–2514. [Google Scholar] [CrossRef]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eissa, I.R.; Bustos-Villalobos, I.; Ichinose, T.; Matsumura, S.; Naoe, Y.; Miyajima, N.; Morimoto, D.; Mukoyama, N.; Zhiwen, W.; Tanaka, M.; et al. The current status and future prospects of oncolytic viruses in clinical trials against melanoma, glioma, pancreatic, and breast cancers. Cancers 2018, 10, 365. [Google Scholar] [CrossRef] [Green Version]
- Garber, K. China Approves World’s First Oncolytic. J. Natl. Cancer Inst. 2006, 98, 298–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.C.; Wang, Q.; Dong, Z.M.; Chu, E.; Roberson, R.S.; Ivanova, I.C.; Wu, D.Y. Expression of coxsackie and adenovirus receptor distinguishes transitional cancer states in therapy-induced cellular senescence. Cell Death Dis. 2010, 1, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakhawat, A.; Ma, L.; Muhammad, T.; Khan, A.A.; Chen, X.; Huang, Y. A tumor targeting oncolytic adenovirus can improve therapeutic outcomes in chemotherapy resistant metastatic human breast carcinoma. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- O’Cathail, S.M.; Pokrovska, T.D.; Maughan, T.S.; Fisher, K.D.; Seymour, L.W.; Hawkins, M.A. Combining oncolytic adenovirus with radiation-a paradigm for the future of radiosensitization. Front. Oncol. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Touchefeu, Y.; Vassaux, G.; Harrington, K.J. Oncolytic viruses in radiation oncology. Radiother. Oncol. 2011, 99, 262–270. [Google Scholar] [CrossRef]
- Liu, C.; Sarkaria, J.N.; Petell, C.A.; Paraskevakou, G.; Zollman, P.J.; Schroeder, M.; Carlson, B.; Decker, P.A.; Wu, W.; James, C.D.; et al. Combination of measles virus virotherapy and radiation therapy has synergistic activity in the treatment of glioblastoma multiforme. Clin. Cancer Res. 2007, 13, 7155–7165. [Google Scholar] [CrossRef] [Green Version]
- Stasiak, A.C.; Stehle, T. Human adenovirus binding to host cell receptors: A structural view. Med. Microbiol. Immunol. 2020, 209, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Maginnis, M.S. Virus–Receptor Interactions: The Key to Cellular Invasion. J. Mol. Biol. 2018, 430, 2590–2611. [Google Scholar] [CrossRef]
- Walters, R.W.; Freimuth, P.; Moninger, T.O.; Ganske, I.; Zabner, J.; Welsh, M.J. Adenovirus fiber disrupts CAR-mediated intercellular adhesion allowing virus escape. Cell 2002, 110, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Spear, P.G. Herpes simplex virus: Receptors and ligands for cell entry. Cell. Microbiol. 2004, 6, 401–410. [Google Scholar] [CrossRef]
- Sharma, P.; Martis, P.C.; Excoffon, K.J.D.A. Adenovirus transduction: More complicated than receptor expression. Virology 2017, 502, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Del Papa, J.; Petryk, J.; Bell, J.C.; Parks, R.J. An Oncolytic Adenovirus Vector Expressing p14 FAST Protein Induces Widespread Syncytium Formation and Reduces Tumor Growth Rate In Vivo. Mol. Ther. Oncolytics 2019, 14, 107–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wing, A.; Fajardo, C.A.; Posey, A.D.; Shaw, C.; Da, T.; Young, R.M.; Alemany, R.; June, C.H.; Guedan, S. Improving CART-Cell Therapy of Solid Tumors with Oncolytic Virus–Driven Production of a Bispecific T-cell Engager. Cancer Immunol. Res. 2018, 6, 605–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchino, J.; Curiel, D.T.; Ugai, H. Species D human adenovirus type 9 exhibits better virus-spread ability for antitumor efficacy among alternative serotypes. PLoS ONE 2014, 9, e0087342. [Google Scholar] [CrossRef] [Green Version]
- Shao, W.; Li, X.; Goraya, M.U.; Wang, S.; Chen, J.L. Evolution of influenza a virus by mutation and re-assortment. Int. J. Mol. Sci. 2017, 18, 1650. [Google Scholar] [CrossRef] [Green Version]
- Kuryk, L.; Møller, A.S.W. Chimeric oncolytic Ad5/3 virus replicates and lyses ovarian cancer cells through desmoglein-2 cell entry receptor. J. Med. Virol. 2020, 92, 1309–1315. [Google Scholar] [CrossRef] [Green Version]
- Plan, D.; Van Den Eede, G. The EU Legislation on GMOs—An overview; Publications Office of the European Union: Luxembourg, 2010; ISBN 9789279152245. [Google Scholar]


{kind=link}
{kind=link}
| Species | Types | Receptors |
|---|---|---|
| A | 12, 31 | CAR 1 |
| B:1 | 3, 7, 16, 21, 50 | CD46, CD80, CD86, DSG-2 |
| B:2 | 11, 14, 35 | CD46, CD80, CD86, DSG-2 |
| C | 1, 2, 5 | CAR, αvβ5 integrin, HSPG, VCAM-1, MHC-Iα2 [16], MARCO [17] |
| D | 8, 9, 17, 19, 37, 48 | CAR, SA, CD46 |
| E | 4 | CAR |
| F | 40, 41 2 | CAR |
| G | 52 | CAR [18], poly-SA [19] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hensen, L.C.M.; Hoeben, R.C.; Bots, S.T.F. Adenovirus Receptor Expression in Cancer and Its Multifaceted Role in Oncolytic Adenovirus Therapy. Int. J. Mol. Sci. 2020, 21, 6828. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186828
Hensen LCM, Hoeben RC, Bots STF. Adenovirus Receptor Expression in Cancer and Its Multifaceted Role in Oncolytic Adenovirus Therapy. International Journal of Molecular Sciences. 2020; 21(18):6828. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186828
Chicago/Turabian StyleHensen, Lobke C.M., Rob C. Hoeben, and Selas T.F. Bots. 2020. "Adenovirus Receptor Expression in Cancer and Its Multifaceted Role in Oncolytic Adenovirus Therapy" International Journal of Molecular Sciences 21, no. 18: 6828. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186828