Soluble Prion Peptide 107–120 Protects Neuroblastoma SH-SY5Y Cells against Oligomers Associated with Alzheimer’s Disease

 , , , and
, , , and 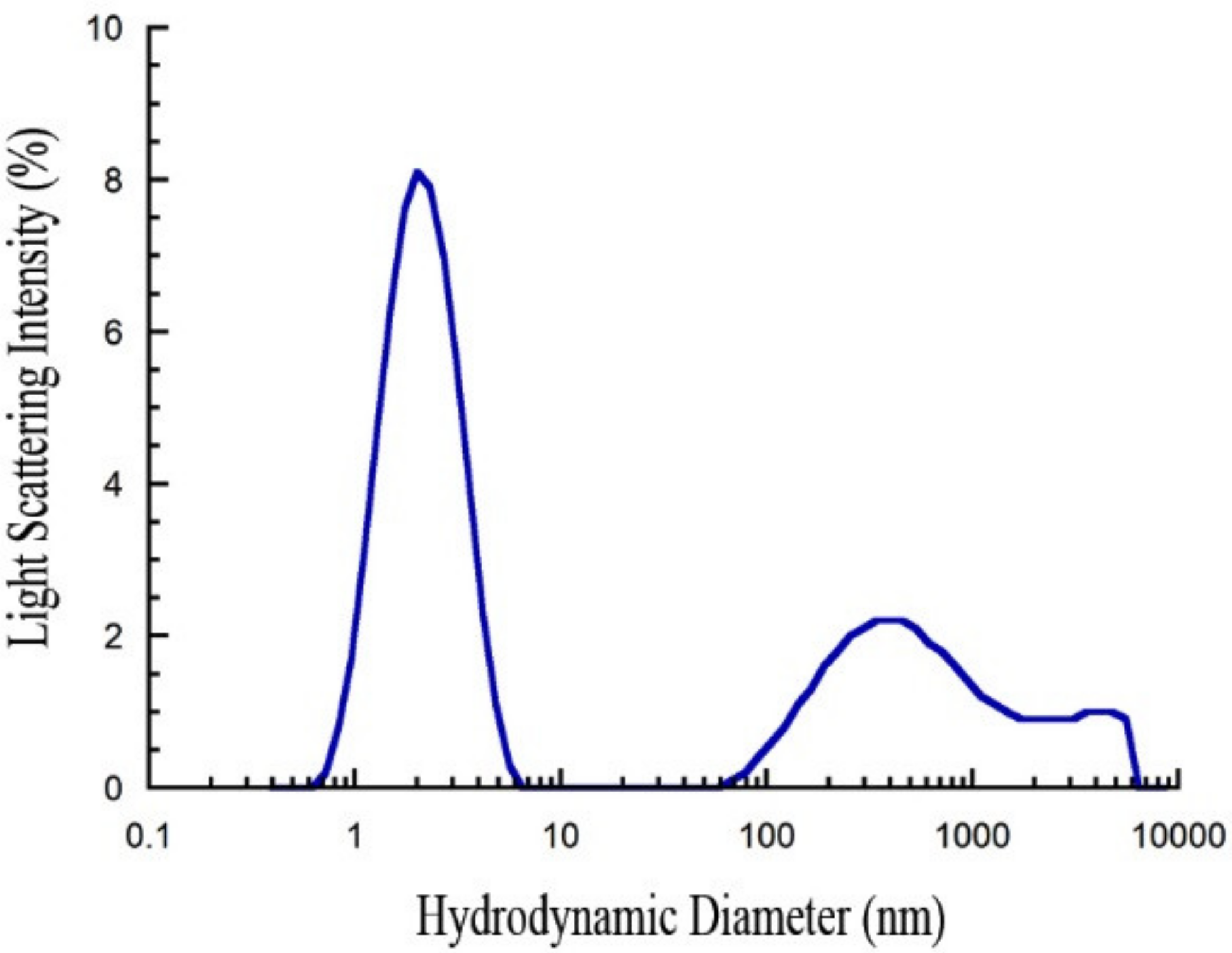{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Freshly Dissolved PrP107–120 is Monomeric
2.2. PrP107–120 Remains Monomeric and Unstructured under Different Conditions
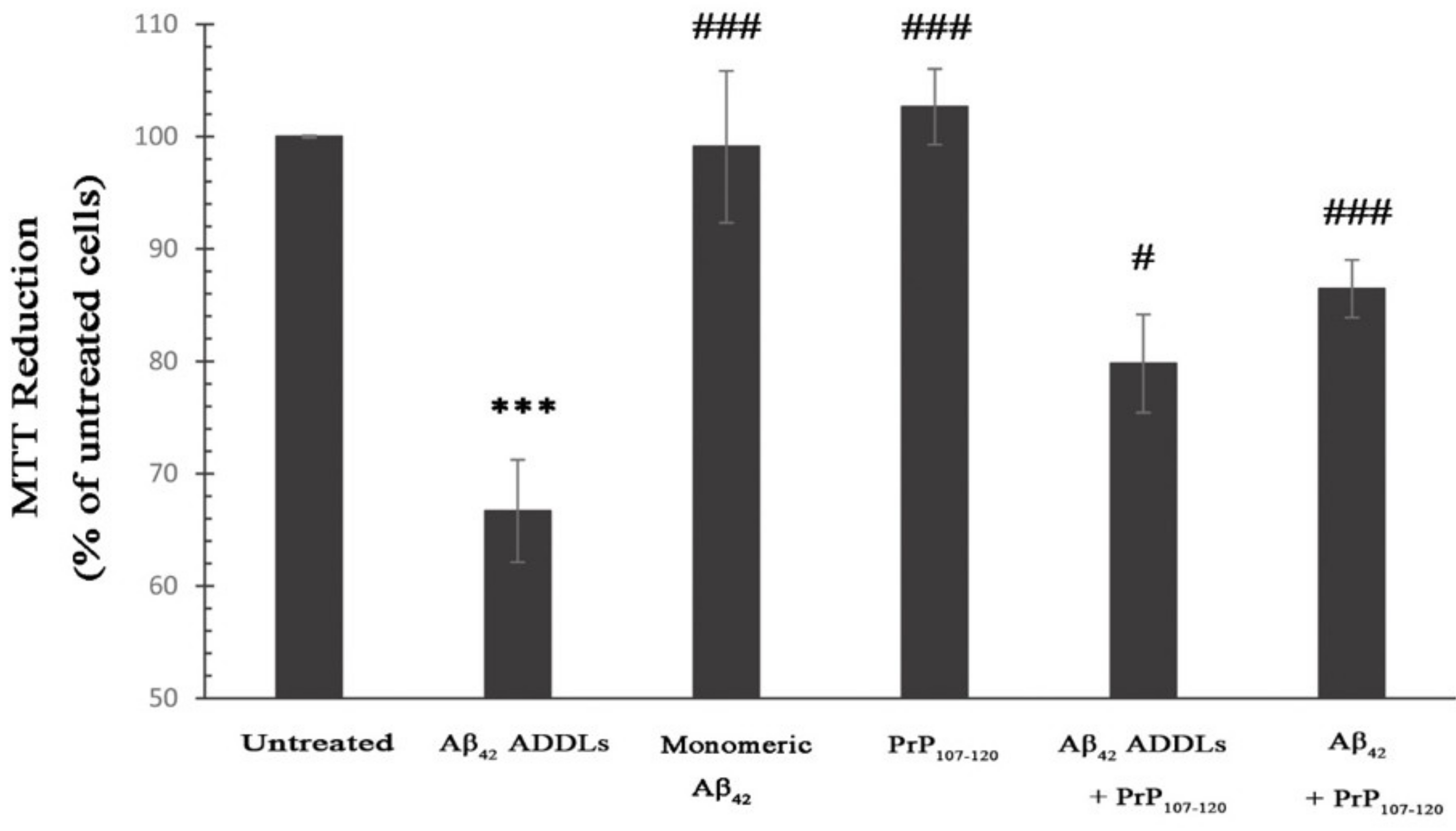
2.3. PrP107-120 Reduces Aβ42 Cytotoxicity on SH-SY5Y Cells
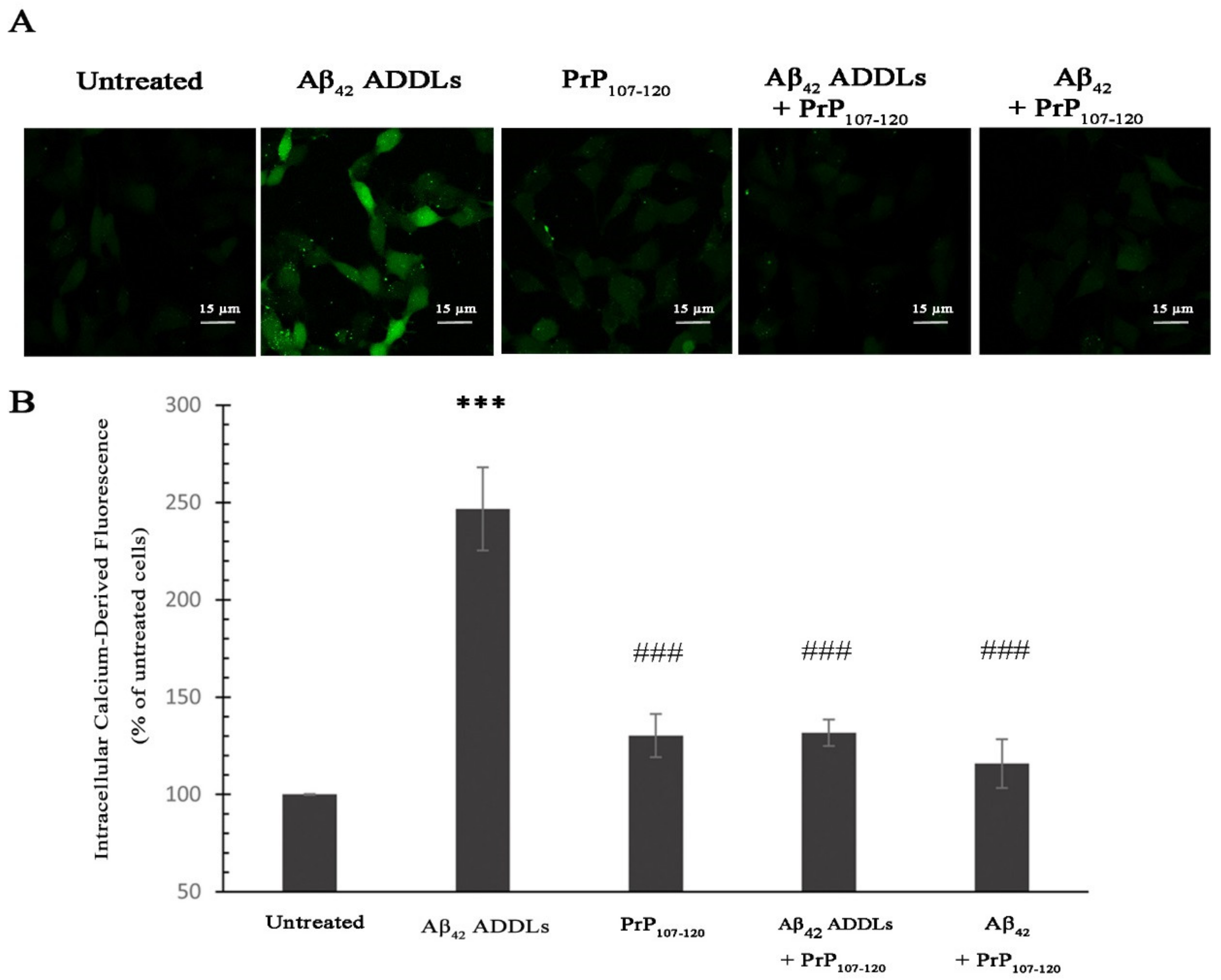
2.4. PrP107-120 Reduces Ca2+ Influx Induced by Aβ42 Oligomers
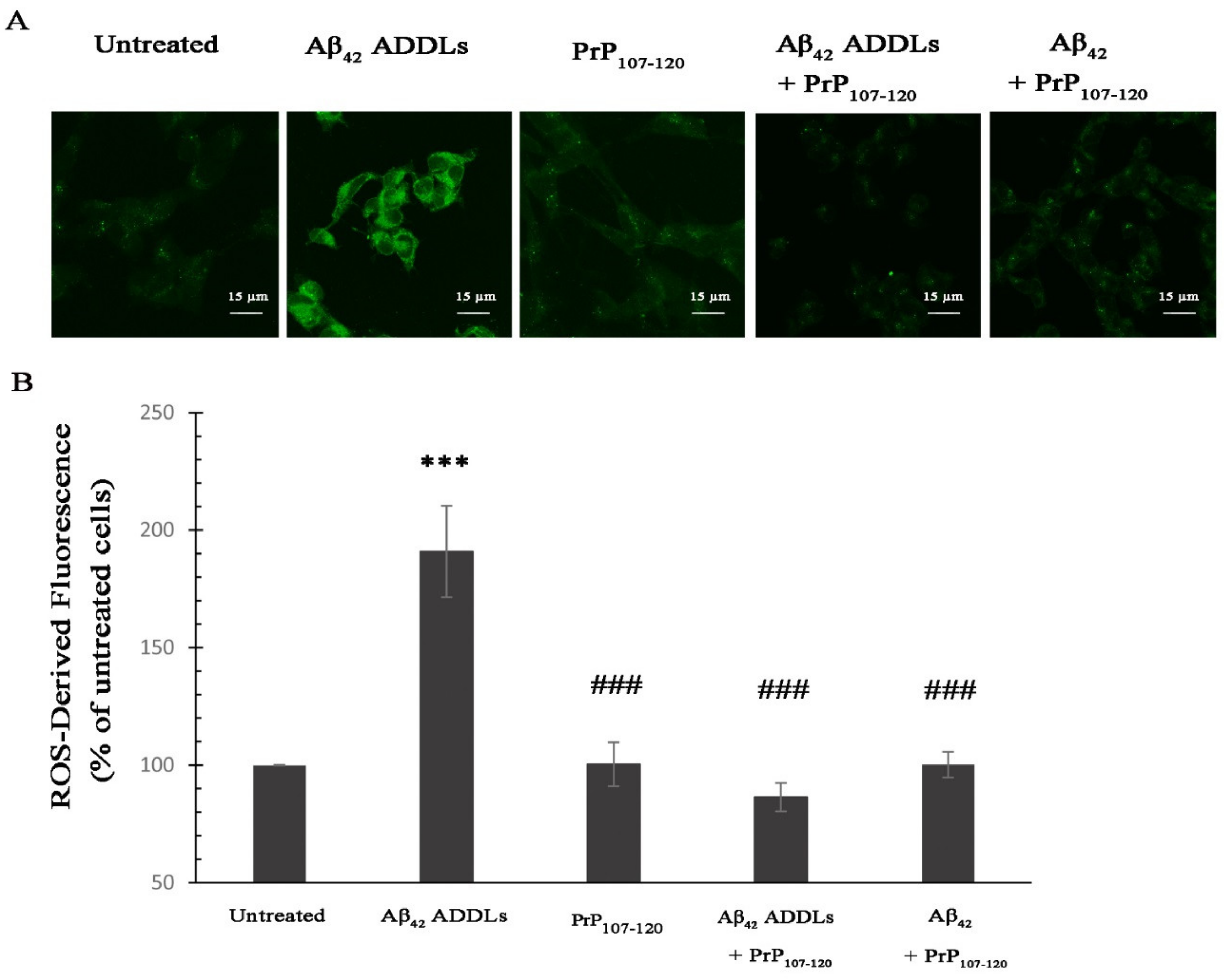
2.5. PrP107-120 Reduces Intracellular Reactive Oxygen Species (ROS) Induced by Aβ42 Oligomers
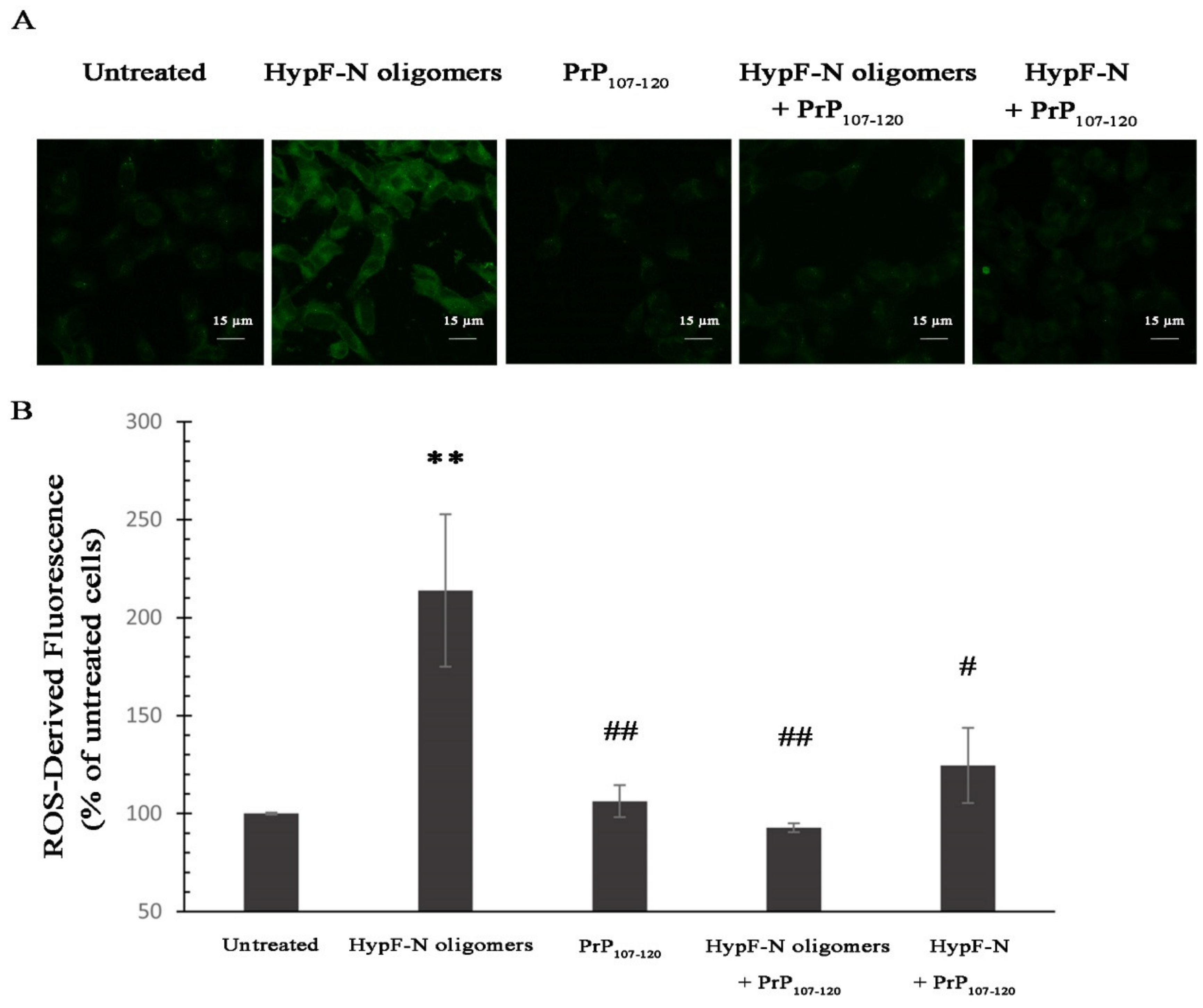
2.6. PrP107-120 Reduces the Toxicity of Other Model Oligomers
2.7. PrP107-120 Reduces Aβ42 ADDLs Internalization in SH-SY5Y Cells
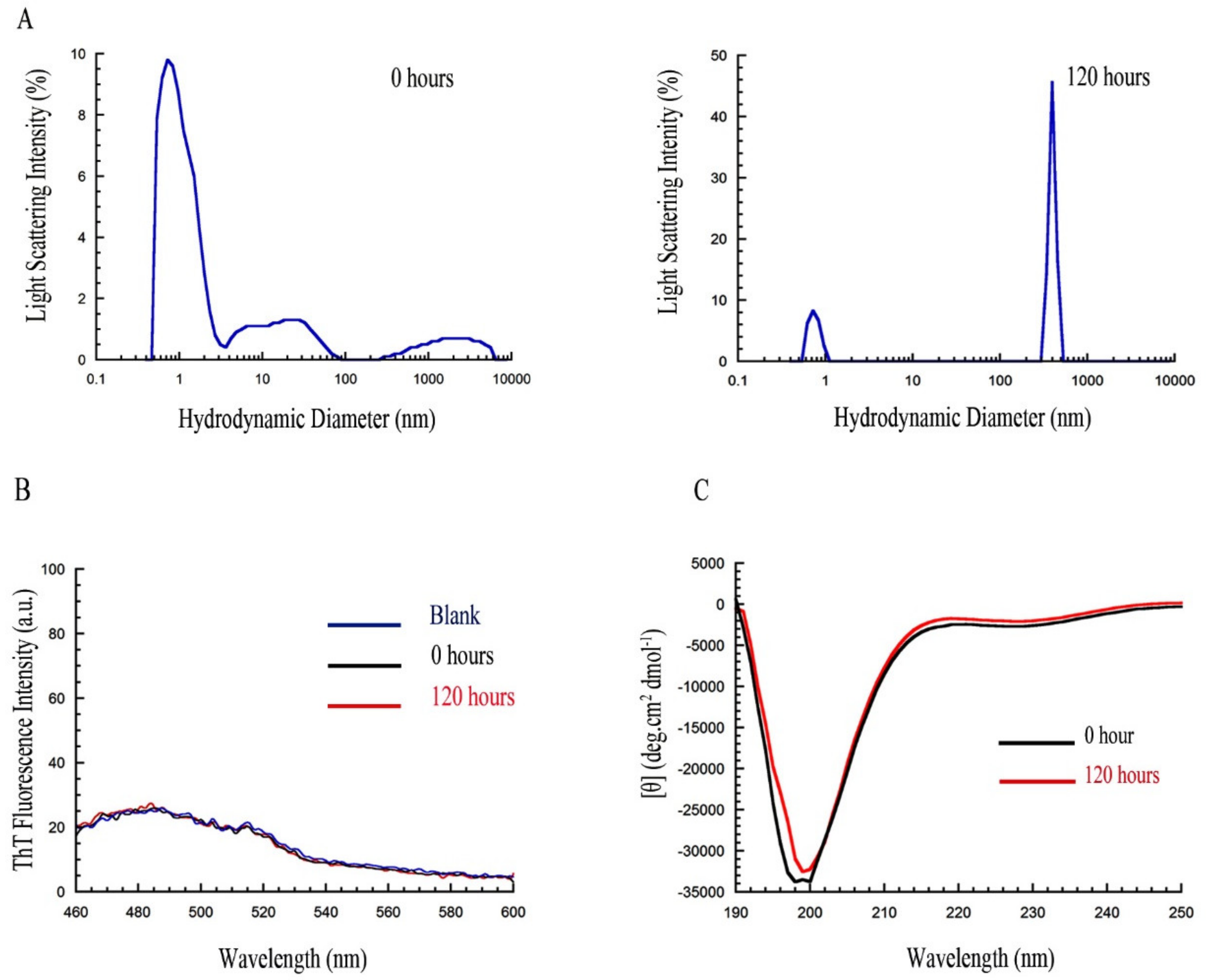
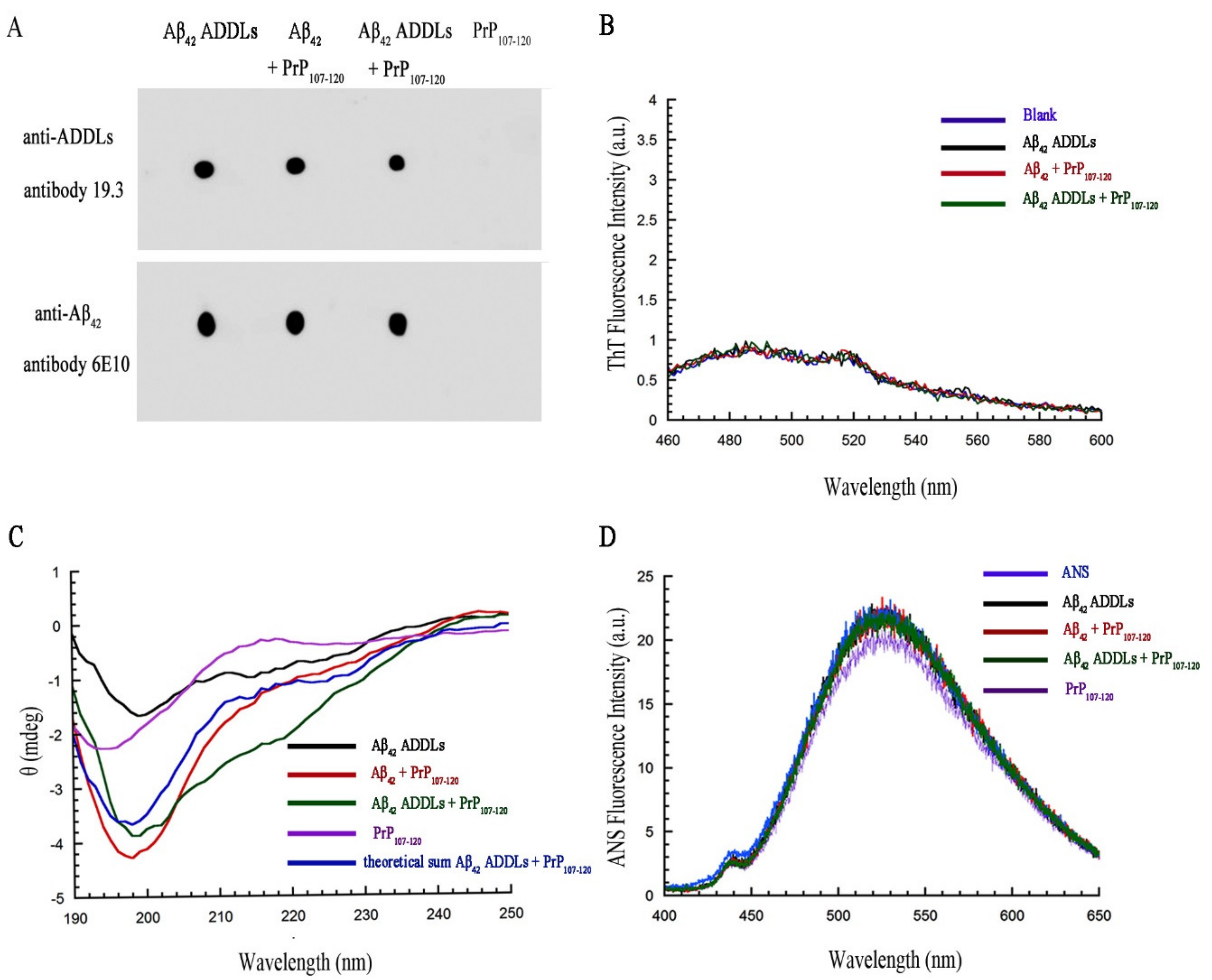
2.8. PrP107-120 Does Not Change the Structure or Aggregation State of Aβ42 ADDLs
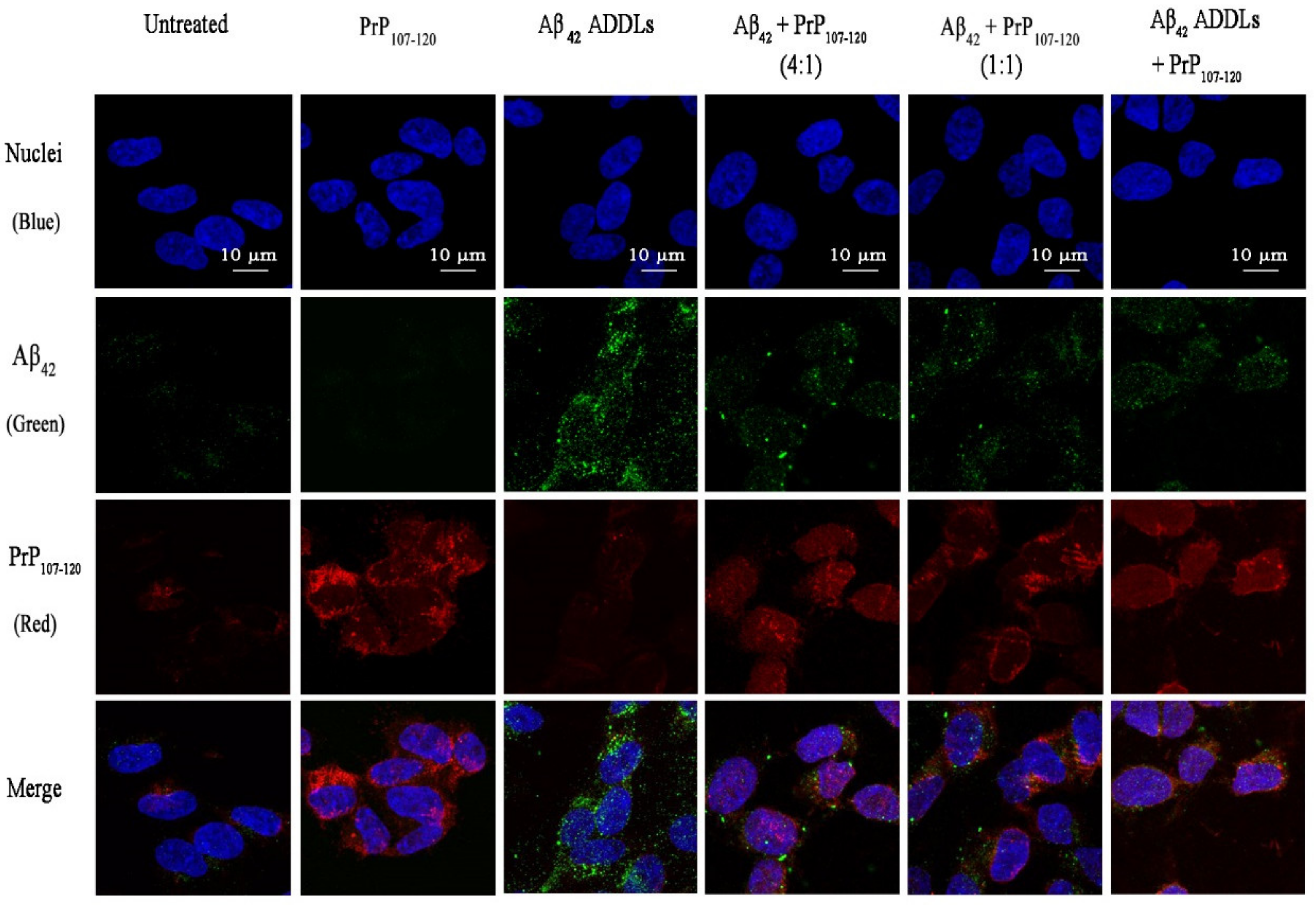
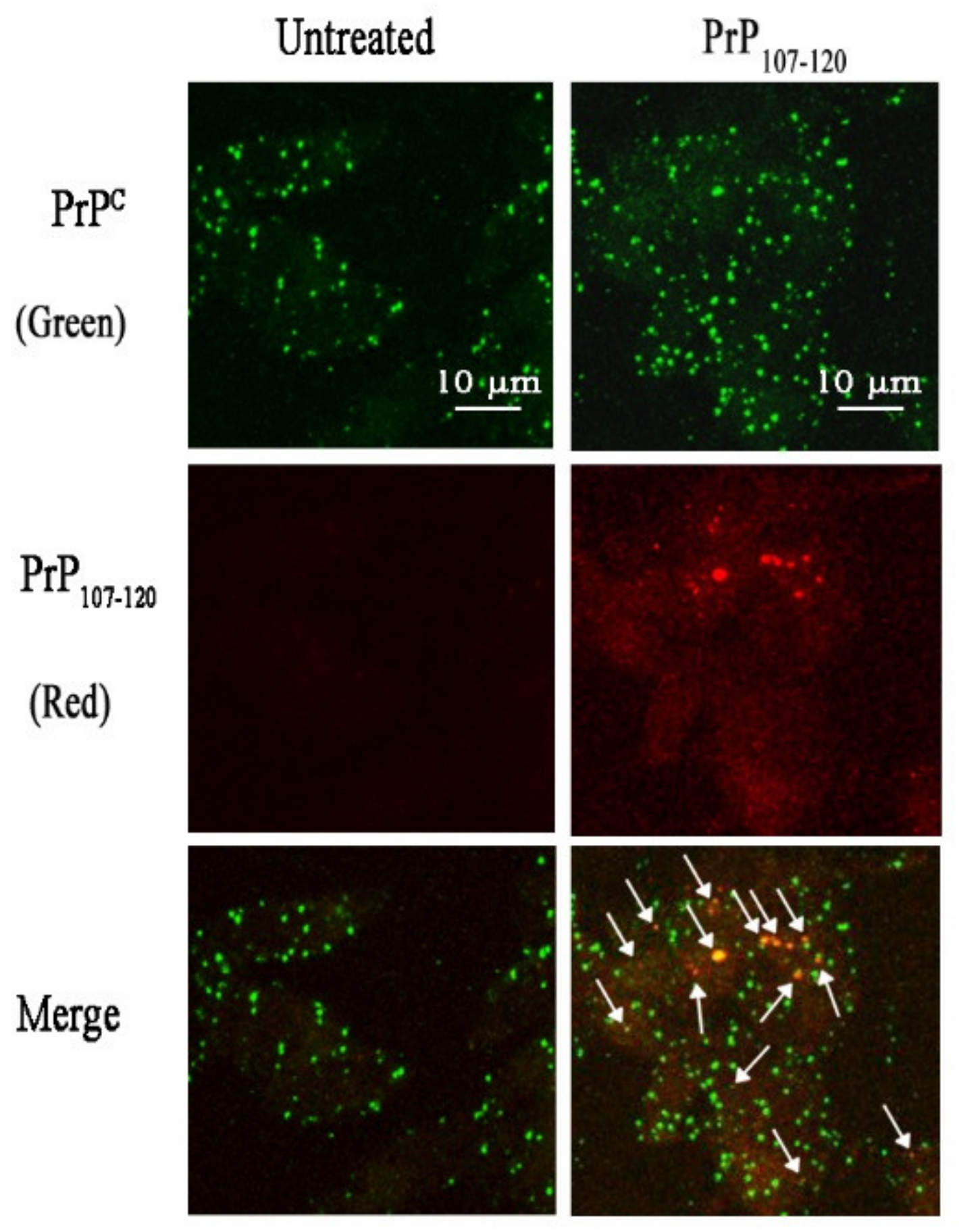
2.9. PrP107-120 Does Not Colocalise with Aβ42 ADDLs but Partially Colocalises with Cellular PrPC in SH-SY5Y Cells
3. Discussion
4. Materials and Methods
4.1. PrP107–120 Preparation
4.2. Preparation of Aβ42 ADDLs, Aβ42 ADDLs + PrP107–120 and Aβ42 + PrP107–120 Samples
4.3. Preparation of HypF-N Oligomers
4.4. Bicinchoninic Acid (BCA) Assay
4.5. Fibrillation of PrP107–120
4.6. Dynamic Light Scattering
4.7. ThT Fluorescence
4.8. Circular Dichroism (CD) Spectroscopy
4.9. Cell Culture
4.10. MTT Assay
4.11. Measurement of Intracellular Ca2+ Levels
4.12. Measurement of Intracellular Reactive Oxygen Species (ROS)
4.13. Immunofluorescence Staining
4.14. Dot Blot
4.15. ANS Binding Assay
4.16. Analysis of ADDLs Co-Localization with PrP107-120
4.17. Analysis of PrP107-120 Co-Localization with PrPC
4.18. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| PrPC | Cellular prion protein |
| Aβ | Amyloid beta |
| LTP | Long-term potentiation |
| GPI | Glycosyl-phosphatidyl-inositol |
| Rh | Hydrodynamic radius |
| Dh | Hydrodynamic diameter |
| ADDLs | Amyloid-derived diffusible ligands |
| DMSO | Dimethyl sulfoxide |
| HFIP | Hexafluoro-2-isopropanol |
| ThT | Thioflavin T |
| CD | Circular dichroism |
| DLS | Dynamic light scattering |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| FBS | Fetal bovine serum |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| ROS | Reactive oxygen species |
| RPMI | Roswell Park Memorial Institute |
| BSA | Bovine serum albumin |
| BCA | Bicinchoninic acid |
References
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienias, J.L.; Beckett, L.A.; Bennett, D.A.; Wilson, R.S.; Evans, D.A. Design of the Chicago health and aging project (CHAP). J. Alzheimer’s Dis. 2003, 5, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Kelley, B.J.; Petersen, R.C. Alzheimer’s disease and mild cognitive impairment. Neurol. Clin. 2007, 25, 577–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci. 1993, 16, 460–465. [Google Scholar] [CrossRef]
- Huang, H.C.; Jiang, Z.F. Accumulated amyloid-β peptide and hyperphosphorylated tau protein: Relationship and links in Alzheimer’s disease. J. Alzheimer’s Dis. 2009, 16, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Hu, N.W.; Smith, I.M.; Walsh, D.M.; Rowan, M.J. Soluble amyloid-β peptides potently disrupt hippocampal synaptic plasticity in the absence of cerebrovascular dysfunction in vivo. Brain 2008, 131, 2414–2424. [Google Scholar] [CrossRef]
- Li, S.; Jin, M.; Koeglsperger, T.; Shepardson, N.E.; Shankar, G.M.; Selkoe, D.J. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci. 2011, 31, 6627–6638. [Google Scholar] [CrossRef]
- Fischer, B.; Schmoll, H.; Platt, D.; Popa-Wagner, A.; Riederer, P.; Bauer, J. Complement C1q and C3 mRNA expression in the frontal cortex of Alzheimer’s patients. J. Mol. Med. 1995, 73, 465–471. [Google Scholar] [CrossRef]
- Yasojima, K.; Schwab, C.; McGeer, E.G.; McGeer, P.L. Up-regulated production and activation of the complement system in Alzheimer’s disease brain. Am. J. Pathol. 1999, 154, 927–936. [Google Scholar] [CrossRef]
- Larson, M.E.; Lesné, S.E. Soluble Aβ oligomer production and toxicity. J. Neurochem. 2012, 120, 125–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, A.E.; Klyubin, I.; Mc Donald, J.M.; Mably, A.J.; Farrell, M.A.; Scott, M.; Walsh, D.M.; Rowan, M.J. Alzheimer’s disease brain-derived amyloid-β-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J. Neurosci. 2011, 31, 7259–7263. [Google Scholar] [CrossRef] [PubMed]
- Freir, D.B.; Nicoll, A.J.; Klyubin, I.; Panico, S.; Mc Donald, J.M.; Risse, E.; Asante, E.A.; Farrow, M.A.; Sessions, R.B.; Saibil, H.R.; et al. Interaction between prion protein and toxic amyloid β assemblies can be therapeutically targeted at multiple sites. Nat. Commun. 2011, 2336. [Google Scholar] [CrossRef]
- Klyubin, I.; Nicoll, A.J.; Khalili-Shirazi, A.; Farmer, M.; Canning, S.; Mably, A.; Linehan, J.; Brown, A.; Wakeling, M.; Brandner, S.; et al. Peripheral administration of a humanized anti-PrP antibody blocks Alzheimer’s disease Aβ synaptotoxicity. J. Neurosci. 2014, 34, 6140–6145. [Google Scholar] [CrossRef] [Green Version]
- Um, J.W.; Strittmatter, S.M. Amyloid-β induced signaling by cellular prion protein and Fyn kinase in Alzheimer disease. Prion 2013, 7, 37–41. [Google Scholar] [CrossRef] [Green Version]
- Gimbel, D.A.; Nygaard, H.B.; Coffey, E.E.; Gunther, E.C.; Laurén, J.; Gimbel, Z.A.; Strittmatter, S.M. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 2010, 30, 6367–6374. [Google Scholar] [CrossRef]
- Younan, N.D.; Chen, K.F.; Rose, R.S.; Crowther, D.C.; Viles, J.H. Prion protein stabilizes amyloid-β (Aβ) oligomers and enhances Aβ neurotoxicity in a Drosophila model of Alzheimer’s disease. J. Biol. Chem. 2018, 293, 13090–13099. [Google Scholar] [CrossRef] [Green Version]
- Balducci, C.; Beeg, M.; Stravalaci, M.; Bastone, A.; Sclip, A.; Biasini, E.; Tapella, L.; Colombo, L.; Manzoni, C.; Borsello, T.; et al. Synthetic amyloid-β oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. USA 2010, 107, 2295–2300. [Google Scholar] [CrossRef] [Green Version]
- Calella, A.M.; Farinelli, M.; Nuvolone, M.; Mirante, O.; Moos, R.; Falsig, J.; Mansuy, I.M.; Aguzzi, A. Prion protein and Aβ-related synaptic toxicity impairment. EMBO Mol. Med. 2010, 2, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Kessels, H.W.; Nguyen, L.N.; Nabavi, S.; Malinow, R. The prion protein as a receptor for amyloid-β. Nature 2010, 466, E3–E4. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Selkoe, D.J. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Aβ oligomers from Alzheimer’s brain. J. Neurochem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.E.; McBride, P.A.; Farquhar, C.F. Precise targeting of the pathology of the sialoglycoprotein, PrP, and vacuolar degeneration in mouse scrapie. Neurosci. Lett. 1989, 102, 1–6. [Google Scholar] [CrossRef]
- Roucou, X.; Gains, M.; LeBlanc, A.C. Neuroprotective functions of prion protein. J. Neurosci. Res. 2004, 75, 153–161. [Google Scholar] [CrossRef]
- Westergard, L.; Christensen, H.M.; Harris, D.A. The cellular prion protein (PrPC): Its physiological function and role in disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2007, 1772, 629–644. [Google Scholar] [CrossRef] [Green Version]
- Zahn, R.; Liu, A.; Lührs, T.; Riek, R.; von Schroetter, C.; García, F.L.; Billeter, M.; Calzolai, L.; Wider, G.; Wüthrich, K. NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. USA 2000, 97, 145–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuegg, J.; Gready, J.E. Molecular dynamics simulation of human prion protein including both N-linked oligosaccharides and the GPI anchor. Glycobiology 2000, 10, 959–974. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Morantes, C.Y.; Wille, H. The structure of human prions: From biology to structural models—Considerations and pitfalls. Viruses 2014, 6, 3875–3892. [Google Scholar] [CrossRef]
- Zomosa-Signoret, V.; Arnaud, J.D.; Fontes, P.; Alvarez-Martinez, M.T.; Liautard, J.P. Physiological role of the cellular prion protein. Vet. Res. 2008, 39, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.N.A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1990, 90, 10962–10966. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collinge, J. Prion diseases of humans and animals: Their causes and molecular basis. Annu. Rev. Neurosci. 2001, 24, 519–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purro, S.A.; Nicoll, A.J.; Collinge, J. Prion protein as a toxic acceptor of amyloid-β oligomers. Biol. Psychiatry 2018, 83, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, Y.; Zhang, L.; Yu, W.; Wang, Y.; Chang, W. Cellular prion protein as a receptor of toxic amyloid-β42 oligomers is important for Alzheimer’s disease. Front. Cell. Neurosci. 2019, 13, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Yadav, S.P.; Surewicz, W.K. Interaction between human prion protein and Amyloid-β (Aβ) oligomers role of N-terminal residues. J. Biol. Chem. 2010, 285, 26377–26383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, W.; Lee, H.P.; Zou, W.Q.; Wang, X.; Perry, G.; Zhu, X.; Smith, M.A.; Petersen, R.B.; Lee, H.G. Cellular prion protein is essential for oligomeric amyloid-β-induced neuronal cell death. Hum. Mol. Genet. 2012, 21, 1138–1144. [Google Scholar] [CrossRef] [Green Version]
- Larson, M.; Sherman, M.A.; Amar, F.; Nuvolone, M.; Schneider, J.A.; Bennett, D.A.; Aguzzi, A.; Lesné, S.E. The complex PrPc-Fyn couples human oligomeric Aβ with pathological Tau changes in Alzheimer’s disease. J. Neurosci. 2012, 32, 16857–16871. [Google Scholar] [CrossRef] [Green Version]
- Cox, T.O.; Gunther, E.C.; Brody, A.H.; Chiasseu, M.T.; Stoner, A.; Smith, L.M.; Haas, L.T.; Hammersley, J.; Rees, G.; Dosanjh, B.; et al. Anti-PrPC antibody rescues cognition and synapses in transgenic alzheimer mice. Ann. Clin. Transl. Neurol. 2019, 6, 554–574. [Google Scholar] [CrossRef] [Green Version]
- Fluharty, B.R.; Biasini, E.; Stravalaci, M.; Sclip, A.; Diomede, L.; Balducci, C.; La Vitola, P.; Messa, M.; Colombo, L.; Forloni, G.; et al. An N-terminal fragment of the prion protein binds to amyloid-β oligomers and inhibits their neurotoxicity in vivo. J. Biol. Chem. 2013, 288, 7857–7866. [Google Scholar] [CrossRef] [Green Version]
- Dohler, F.; Sepulveda-Falla, D.; Krasemann, S.; Altmeppen, H.; Schlüter, H.; Hildebrand, D.; Zerr, I.; Matschke, J.; Glatzel, M. High molecular mass assemblies of amyloid-β oligomers bind prion protein in patients with Alzheimer’s disease. Brain 2014, 137, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Kostylev, M.A.; Kaufman, A.C.; Nygaard, H.B.; Patel, P.; Haas, L.T.; Gunther, E.C.; Vortmeyer, A.; Strittmatter, S.M. Prion-protein-interacting amyloid-β oligomers of high molecular weight are tightly correlated with memory impairment in multiple Alzheimer mouse models. J. Biol. Chem. 2015, 290, 17415–17438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Fujioka, H.; Mishra, R.S.; Li, R.; Singh, N. Prion peptide 106–126 modulates the aggregation of cellular prion protein and induces the synthesis of potentially neurotoxic transmembrane PrP. J. Biol. Chem. 2002, 277, 2275–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwata, K.; Matumoto, T.; Cheng, H.; Nagayama, K.; James, T.L.; Roder, H. NMR-detected hydrogen exchange and molecular dynamics simulations provide structural insight into fibril formation of prion protein fragment 106–126. Proc. Natl. Acad. Sci. USA 2003, 100, 14790–14795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norstrom, E.M.; Mastrianni, J.A. The AGAAAAGA palindrome in PrP is required to generate a productive PrPSc-PrPC complex that leads to prion propagation. J. Biol. Chem. 2005, 280, 27236–27243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergström, A.L.; Chabry, J.; Bastholm, L.; Heegaard, P.M. Oxidation reduces the fibrillation but not the neurotoxicity of the prion peptide PrP106–126. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2007, 1774, 1118–1127. [Google Scholar]
- Walsh, P.; Simonetti, K.; Sharpe, S. Core structure of amyloid fibrils formed by residues 106–126 of the human prion protein. Structure 2009, 17, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Walsh, P.; Neudecker, P.; Sharpe, S. Structural Properties and Dynamic Behavior of Nonfibrillar Oligomers Formed by PrP (106−126). J. Am. Chem. Soc. 2010, 132, 7684–7695. [Google Scholar] [CrossRef]
- Sengupta, I.; Udgaonkar, J.B. Structural mechanisms of oligomer and amyloid fibril formation by the prion protein. Chem. Commun. 2018, 54, 6230–6242. [Google Scholar] [CrossRef]
- Florio, T.; Paludi, D.; Villa, V.; Principe, D.R.; Corsaro, A.; Millo, E.; Damonte, G.; D’arrigo, C.; Russo, C.; Schettini, G.; et al. Contribution of two conserved glycine residues to fibrillogenesis of the 106–126 prion protein fragment. Evidence that a soluble variant of the 106–126 peptide is neurotoxic. J. Neurochem. 2003, 85, 62–72. [Google Scholar] [CrossRef]
- Wilkins, D.K.; Grimshaw, S.B.; Receveur, V.; Dobson, C.M.; Jones, J.A.; Smith, L.J. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry 1999, 38, 16424–16431. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Viola, K.L.; Chromy, B.A.; Chang, L.; Morgan, T.E.; Yu, J.; Venton, D.L.; Krafft, G.A.; Finch, C.E.; Klein, W.L. Vaccination with soluble Aβ oligomers generates toxicity-neutralizing antibodies. J. Neurochem. 2001, 79, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Cascella, R.; Evangelisti, E.; Bigi, A.; Becatti, M.; Fiorillo, C.; Stefani, M.; Chiti, F.; Cecchi, C. Soluble oligomers require a ganglioside to trigger neuronal calcium overload. J. Alzheimer’s Dis. 2017, 60, 923–938. [Google Scholar] [CrossRef]
- Gong, Y.; Chang, L.; Viola, K.L.; Lacor, P.N.; Lambert, M.P.; Finch, C.E.; Krafft, G.A.; Klein, W.L. Alzheimer’s disease-affected brain: Presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. USA 2003, 100, 10417–10422. [Google Scholar]
- Ghadami, S.A.; Chia, S.; Ruggeri, F.S.; Meisl, G.; Bemporad, F.; Habchi, J.; Cascella, R.; Dobson, C.M.; Vendruscolo, M.; Knowles, T.P.; et al. Transthyretin inhibits primary and secondary nucleations of amyloid-β peptide aggregation and reduces the toxicity of its oligomers. Biomacromolecules 2020, 21, 1112–1125. [Google Scholar] [CrossRef]
- Limbocker, R.; Chia, S.; Ruggeri, F.S.; Perni, M.; Cascella, R.; Heller, G.T.; Meisl, G.; Mannini, B.; Habchi, J.; Michaels, T.C.; et al. Trodusquemine enhances Aβ 42 aggregation but suppresses its toxicity by displacing oligomers from cell membranes. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banchelli, M.; Cascella, R.; D’Andrea, C.; Cabaj, L.; Osticioli, I.; Ciofini, D.; Li, M.S.; Skupień, K.; de Angelis, M.; Siano, S.; et al. Nanoscopic insights into the surface conformation of neurotoxic amyloid β oligomers. RSC Adv. 2020, 10, 21907–21913. [Google Scholar] [CrossRef]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Aβ oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar] [CrossRef] [Green Version]
- Lacor, P.N.; Buniel, M.C.; Furlow, P.W.; Clemente, A.S.; Velasco, P.T.; Wood, M.; Viola, K.L.; Klein, W.L. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 2007, 27, 796–807. [Google Scholar] [CrossRef]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef] [Green Version]
- Cenini, G.; Cecchi, C.; Pensalfini, A.; Bonini, S.A.; Ferrari-Toninelli, G.; Liguri, G.; Memo, M.; Uberti, D. Generation of reactive oxygen species by beta amyloid fibrils and oligomers involves different intra/extracellular pathways. Amino Acids 2010, 38, 1101–1106. [Google Scholar] [CrossRef] [Green Version]
- Campioni, S.; Mannini, B.; Zampagni, M.; Pensalfini, A.; Parrini, C.; Evangelisti, E.; Relini, A.; Stefani, M.; Dobson, C.M.; Cecchi, C.; et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 2010, 6, 140–147. [Google Scholar] [CrossRef]
- Zampagni, M.; Cascella, R.; Casamenti, F.; Grossi, C.; Evangelisti, E.; Wright, D.; Becatti, M.; Liguri, G.; Mannini, B.; Campioni, S.; et al. A comparison of the biochemical modifications caused by toxic and non-toxic protein oligomers in cells. J. Cell. Mol. Med. 2011, 15, 2106–2116. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, E.; Cecchi, C.; Cascella, R.; Sgromo, C.; Becatti, M.; Dobson, C.M.; Chiti, F.; Stefani, M. Membrane lipid composition and its physicochemical properties define cell vulnerability to aberrant protein oligomers. J. Cell Sci. 2012, 125, 2416–2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascella, R.; Conti, S.; Tatini, F.; Evangelisti, E.; Scartabelli, T.; Casamenti, F.; Wilson, M.R.; Chiti, F.; Cecchi, C. Extracellular chaperones prevent Aβ42-induced toxicity in rat brains. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 1217–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, M.; Shughrue, P.; Wolfe, A.; McCampbell, A. Method for Detection of Amyloid Beta Oligomers in a Fluid Sample and Uses Thereof. U.S. Patent Application No. 13/544,554, 28 2013. [Google Scholar]
- Resenberger, U.K.; Harmeier, A.; Woerner, A.C.; Goodman, J.L.; Müller, V.; Krishnan, R.; Vabulas, R.M.; Kretzschmar, H.A.; Lindquist, S.; Hartl, F.U.; et al. The cellular prion protein mediates neurotoxic signalling of β-sheet-rich conformers independent of prion replication. EMBO J. 2011, 30, 2057–2070. [Google Scholar] [CrossRef]
- Picotti, P.; De Franceschi, G.; Frare, E.; Spolaore, B.; Zambonin, M.; Chiti, F.; de Laureto, P.P.; Fontana, A. Amyloid fibril formation and disaggregation of fragment 1-29 of apomyoglobin: Insights into the effect of pH on protein fibrillogenesis. J. Mol. Biol. 2007, 367, 1237–1245. [Google Scholar] [CrossRef]
- De Simone, A.; Dhulesia, A.; Soldi, G.; Vendruscolo, M.; Hsu, S.T.D.; Chiti, F.; Dobson, C.M. Experimental free energy surfaces reveal the mechanisms of maintenance of protein solubility. Proc. Natl. Acad. Sci. USA 2011, 108, 21057–21062. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Jiang, L.; Zhang, H.; Shimoda, L.A.; DeBerardinis, R.J.; Semenza, G.L. Analysis of hypoxia-induced metabolic reprogramming. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2014; Volume 542, pp. 425–455. [Google Scholar]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rezvani Boroujeni, E.; Hosseini, S.M.; Fani, G.; Cecchi, C.; Chiti, F. Soluble Prion Peptide 107–120 Protects Neuroblastoma SH-SY5Y Cells against Oligomers Associated with Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 7273. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197273
Rezvani Boroujeni E, Hosseini SM, Fani G, Cecchi C, Chiti F. Soluble Prion Peptide 107–120 Protects Neuroblastoma SH-SY5Y Cells against Oligomers Associated with Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(19):7273. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197273
Chicago/Turabian StyleRezvani Boroujeni, Elham, Seyed Masoud Hosseini, Giulia Fani, Cristina Cecchi, and Fabrizio Chiti. 2020. "Soluble Prion Peptide 107–120 Protects Neuroblastoma SH-SY5Y Cells against Oligomers Associated with Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 19: 7273. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197273