Glioblastoma Exhibits Inter-Individual Heterogeneity of TSPO and LAT1 Expression in Neoplastic and Parenchymal Cells

 , ,
, ,
Abstract
:1. Introduction
2. Results
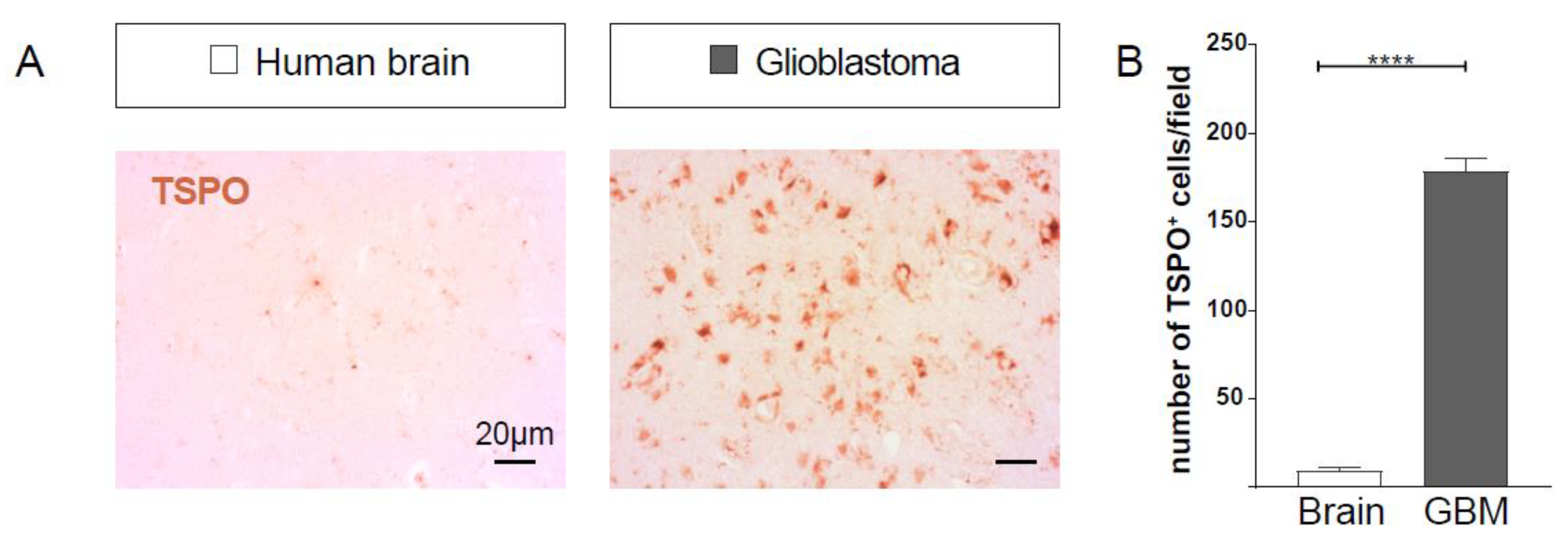
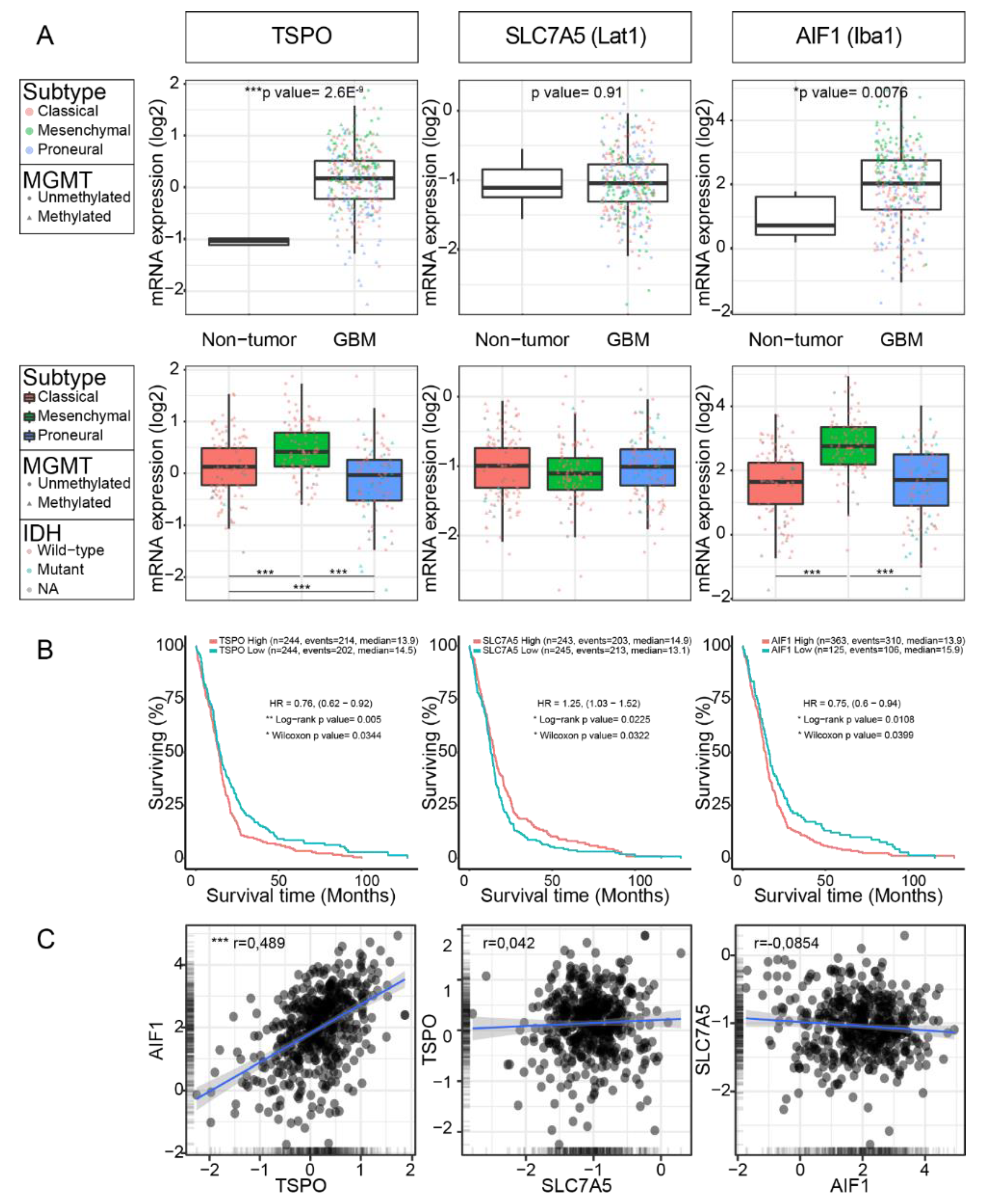
2.1. TSPO Expression is Upregulated in GBM
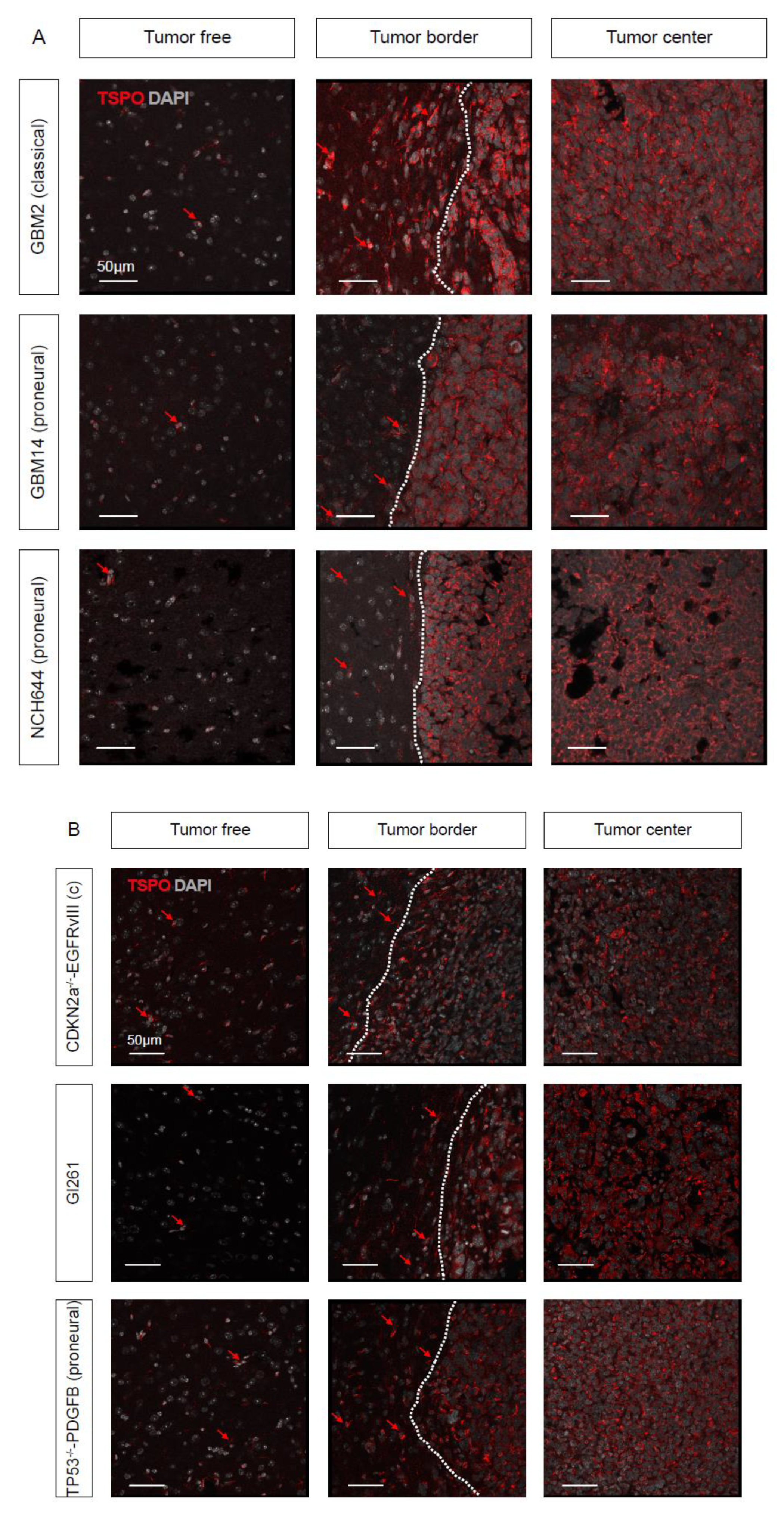
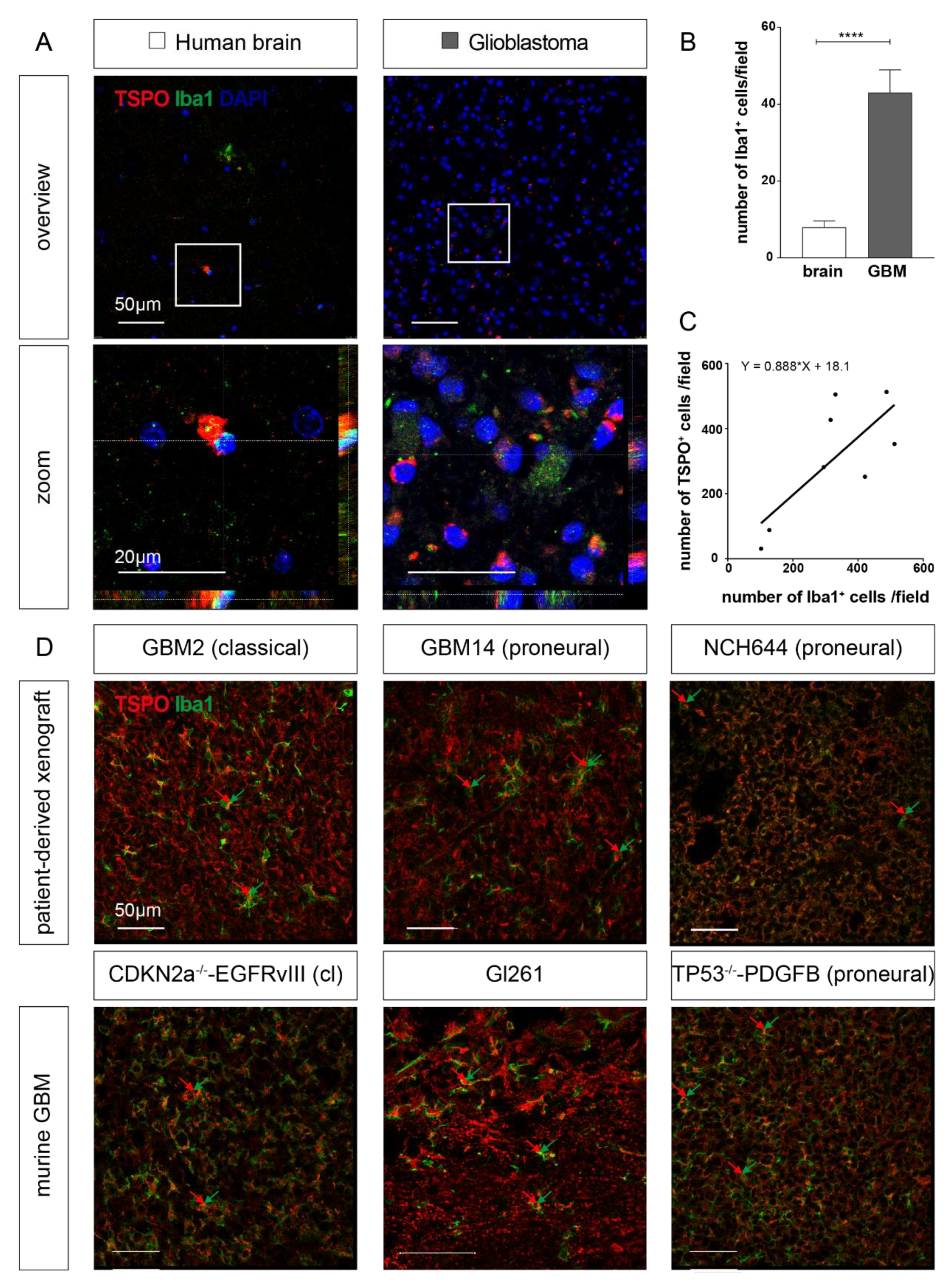
2.2. TSPO is Expressed in Parenchymal Cells of GBM
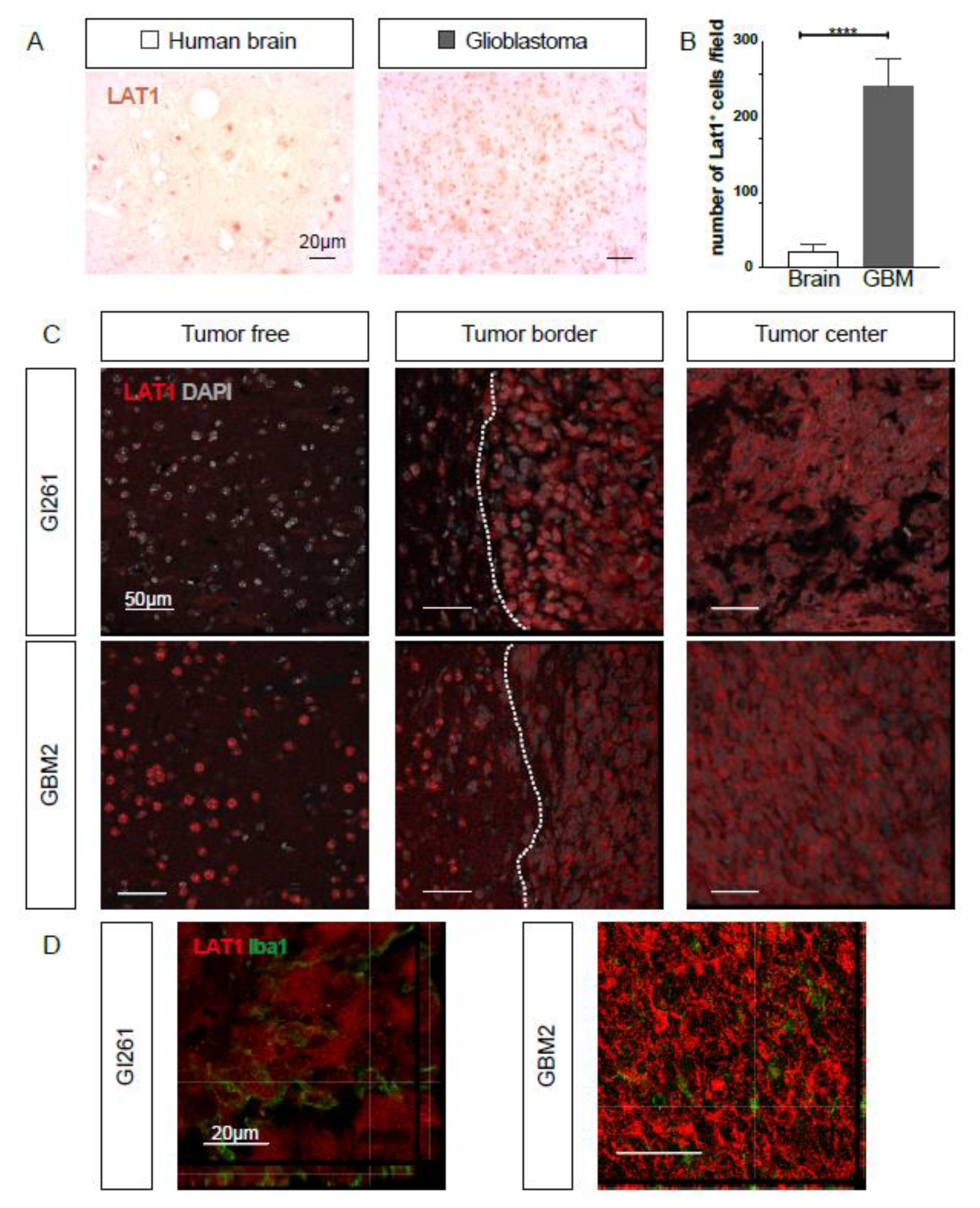
2.3. LAT1 Expression is Upregulated in GBM
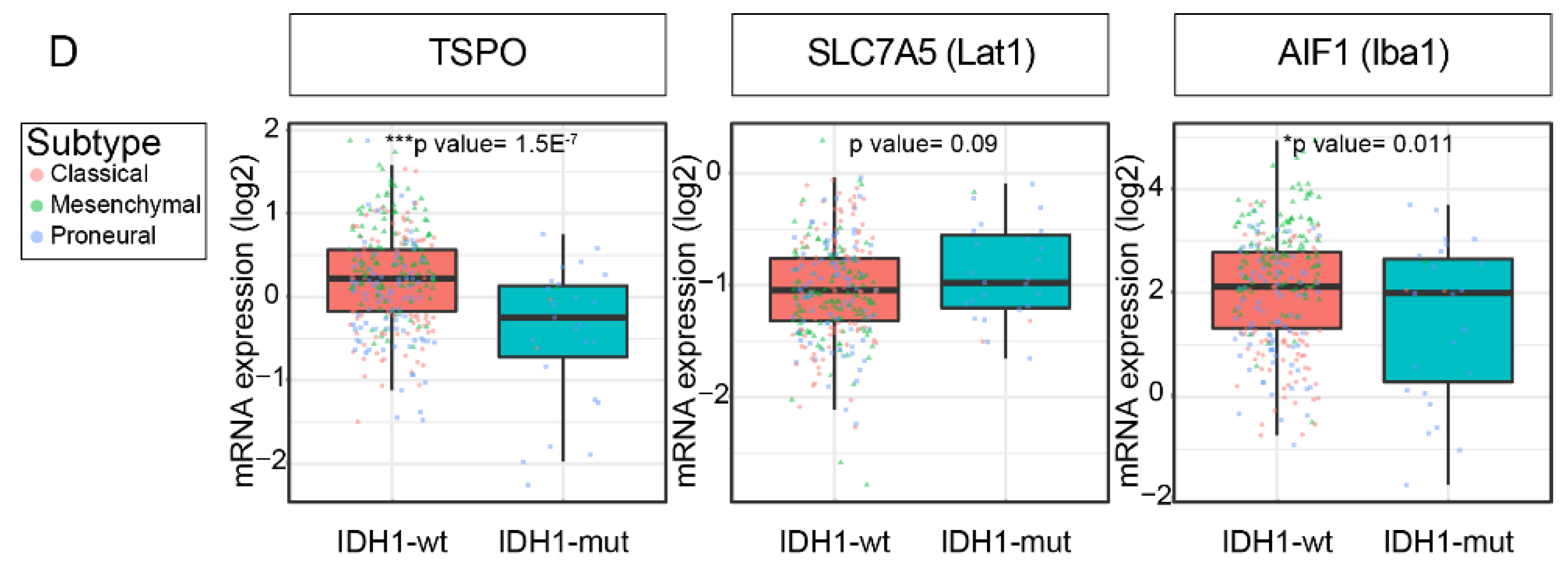
2.4. TSPO and LAT1 Expression do not Correlate in a Big Cohort of GBM Patient Samples
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Animal Experiments
4.3. Tumor Inoculation
4.4. Mouse Brain Tissue Preparation
4.5. Fluorescent Immunohistochemistry and Confocal Microscopy
4.6. Immunohistochemistry of Human Specimens
4.7. GlioVis Analysis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AIF1 | Allograft inflammatory factor-1 (also known as Iba1) |
| CX3CR1 | C-X3-C Motif Chemokine Receptor 1 (also known as fractalkine receptor) |
| GBM | Glioblastoma |
| GSC | Glioblastoma stem-like cell |
| IHC | Immunohistochemistry |
| Iba1 | Ionized calcium-binding adapter molecule 1 (encoded by the gene AIF1) |
| LAS X | Leica Application Suite X |
| LAT1 | Amino acid transporter light chain L system (encoded by the gene SLC7A5) |
| PBR | Peripheral benzodiazepine receptor (encoded by the gene TSPO) |
| PDGFRB | Platelet-derived growth factor receptor beta |
| PET | Positron emission tomography |
| SLC7A5 | Solute carrier family 7 member 5 (also known as LAT1) |
| TAM | Tumor-associated myeloid cell |
| TCGA | The Cancer Genome Atlas |
| TSPO | Translocator protein of 18 kDa (also known as PBR) |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Glioma Cell Line and GSC Cultures1 | Species | GBM Subtype |
|---|---|---|
| Gl261 glioma cell line | murine | |
| cdkn2aKOEGFRvIII GBM- | murine | classical |
| p53KOPDGFB GBM | murine | proneural |
| GBM#2 | human | classical |
| GBM#14 | human | proneural |
| NCH644 | human | proneural |
| Target | Label | Species | Catalog | Company | Concentration |
|---|---|---|---|---|---|
| anti-TSPO | rabbit | ab109497 | abcam | 1:250 | |
| anti-LAT1 | rabbit | LS-C415524 | Biozol | 1:100 | |
| anti-Iba1 | goat | ab5076 | abcam | 1:400 | |
| anti-CD31 | rat | cat. 550274 | BD Biosciences | 1:50 | |
| anti-PDGFRβ | goat | AF1042 | R&D Systems | 1:100 | |
| anti-rabbit | biotin | donkey | Cat. 711-065-152 | Jackson Immuno-Research | 1:250 |
| anti-rabbit | AF488 | donkey | Cat. A-21206 | ThermoFisher Scientific | 1:500 |
| anti-goat | biotin | donkey | B7024 | Sigma | 1:250 |
| anti-goat | AF488 | donkey | Cat. 705-545-147 | Jackson Immuno-Research | 1:500 |
| anti-goat | AF647 | donkey | Cat. 705-605-003 | Jackson Immuno-Research | 1:500 |
| anti-rabbit | AF594 | donkey | Cat. A-21207 | ThermoFisher Scientific | 1:500 |
| anti-rat | biotin | donkey | Cat. 712-065-150 | Jackson Immuno-Research | 1:250 |
| streptavidin | AF488 | Cat. 016-540-084 | Jackson Immuno-Research | 1:500 | |
| streptavidin | AF594 | Cat. 016-580-084 | Jackson Immuno-Research | 1:500 |
References
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. NeuroOncology 2018, 20, iv1–iv86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibetseder, A.; Ackerl, M.; Flechl, B.; Wohrer, A.; Widhalm, G.; Dieckmann, K.; Kreinecker, S.S.; Pichler, J.; Hainfellner, J.; Preusser, M.; et al. Outcome and molecular characteristics of adolescent and young adult patients with newly diagnosed primary glioblastoma: A study of the Society of Austrian Neurooncology (SANO). NeuroOncology 2013, 15, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karsy, M.; Guan, J.; Cohen, A.L.; Jensen, R.L.; Colman, H. New Molecular Considerations for Glioma: IDH, ATRX, BRAF, TERT, H3 K27M. Curr. Neurol. Neurosci. Rep. 2017, 17, 19. [Google Scholar] [CrossRef]
- Brito, C.; Azevedo, A.; Esteves, S.; Marques, A.R.; Martins, C.; Costa, I.; Mafra, M.; Bravo Marques, J.M.; Roque, L.; Pojo, M. Clinical insights gained by refining the 2016 WHO classification of diffuse gliomas with: EGFR amplification, TERT mutations, PTEN deletion and MGMT methylation. BMC Cancer 2019, 19, 968. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavare, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [Green Version]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [Green Version]
- Albert, N.L.; Winkelmann, I.; Suchorska, B.; Wenter, V.; Schmid-Tannwald, C.; Mille, E.; Todica, A.; Brendel, M.; Tonn, J.C.; Bartenstein, P.; et al. Early static (18)F-FET-PET scans have a higher accuracy for glioma grading than the standard 20-40 min scans. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1105–1114. [Google Scholar] [CrossRef]
- Holzgreve, A.; Brendel, M.; Gu, S.; Carlsen, J.; Mille, E.; Boning, G.; Mastrella, G.; Unterrainer, M.; Gildehaus, F.J.; Rominger, A.; et al. Monitoring of Tumor Growth with [(18)F]-FET PET in a Mouse Model of Glioblastoma: SUV Measurements and Volumetric Approaches. Front. Neurosci. 2016, 10, 260. [Google Scholar] [CrossRef]
- Suchorska, B.; Albert, N.L.; Tonn, J.C. Usefulness of PET Imaging to Guide Treatment Options in Gliomas. Curr. Treat. Options Neurol. 2016, 18, 4. [Google Scholar] [CrossRef]
- Habermeier, A.; Graf, J.; Sandhofer, B.F.; Boissel, J.P.; Roesch, F.; Closs, E.I. System L amino acid transporter LAT1 accumulates O-(2-fluoroethyl)-L-tyrosine (FET). Amino Acids 2015, 47, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Popperl, G.; Kreth, F.W.; Mehrkens, J.H.; Herms, J.; Seelos, K.; Koch, W.; Gildehaus, F.J.; Kretzschmar, H.A.; Tonn, J.C.; Tatsch, K. FET PET for the evaluation of untreated gliomas: Correlation of FET uptake and uptake kinetics with tumour grading. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Wester, H.J.; Herz, M.; Weber, W.; Heiss, P.; Senekowitsch-Schmidtke, R.; Schwaiger, M.; Stocklin, G. Synthesis and radiopharmacology of O-(2-[18F]fluoroethyl)-L-tyrosine for tumor imaging. J. Nucl. Med. 1999, 40, 205–212. [Google Scholar] [PubMed]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Glass, R.; Synowitz, M. CNS macrophages and peripheral myeloid cells in brain tumours. Acta Neuropathol. 2014, 128, 347–362. [Google Scholar] [CrossRef] [Green Version]
- Aldape, K.; Zadeh, G.; Mansouri, S.; Reifenberger, G.; von Deimling, A. Glioblastoma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 829–848. [Google Scholar] [CrossRef]
- Albert, N.L.; Unterrainer, M.; Fleischmann, D.F.; Lindner, S.; Vettermann, F.; Brunegraf, A.; Vomacka, L.; Brendel, M.; Wenter, V.; Wetzel, C.; et al. TSPO PET for glioma imaging using the novel ligand 18F-GE-180: First results in patients with glioblastoma. Eur. J. Nucl. Med. Mol. Imaging 2017. [Google Scholar] [CrossRef]
- Zinnhardt, B.; Pigeon, H.; Theze, B.; Viel, T.; Wachsmuth, L.; Fricke, I.B.; Schelhaas, S.; Honold, L.; Schwegmann, K.; Wagner, S.; et al. Combined PET Imaging of the Inflammatory Tumor Microenvironment Identifies Margins of Unique Radiotracer Uptake. Cancer Res. 2017, 77, 1831–1841. [Google Scholar] [CrossRef] [Green Version]
- Campos, B.; Wan, F.; Farhadi, M.; Ernst, A.; Zeppernick, F.; Tagscherer, K.E.; Ahmadi, R.; Lohr, J.; Dictus, C.; Gdynia, G.; et al. Differentiation therapy exerts antitumor effects on stem-like glioma cells. Clin. Cancer Res. 2010, 16, 2715–2728. [Google Scholar] [CrossRef] [Green Version]
- Drachsler, M.; Kleber, S.; Mateos, A.; Volk, K.; Mohr, N.; Chen, S.; Cirovic, B.; Tuttenberg, J.; Gieffers, C.; Sykora, J.; et al. CD95 maintains stem cell-like and non-classical EMT programs in primary human glioblastoma cells. Cell Death Dis. 2016, 7, e2209. [Google Scholar] [CrossRef] [PubMed]
- Binda, E.; Visioli, A.; Giani, F.; Trivieri, N.; Palumbo, O.; Restelli, S.; Dezi, F.; Mazza, T.; Fusilli, C.; Legnani, F.; et al. Wnt5a Drives an Invasive Phenotype in Human Glioblastoma Stem-like Cells. Cancer Res. 2017, 77, 996–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, T.S.; Holland, E.C. Animal models for glioma drug discovery. Expert Opin. Drug Discov. 2011, 6, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Mastrella, G.; Hou, M.; Li, M.; Stoecklein, V.M.; Zdouc, N.; Volmar, M.N.M.; Miletic, H.; Reinhard, S.; Herold-Mende, C.C.; Kleber, S.; et al. Targeting APLN/APLNR Improves Antiangiogenic Efficiency and Blunts Proinvasive Side Effects of VEGFA/VEGFR2 Blockade in Glioblastoma. Cancer Res. 2019, 79, 2298–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerl, K.; Nolan, K.A.; Karger, C.; Fuchs, M.; Wenger, R.H.; Stolt, C.C.; Willam, C.; Kurtz, A.; Kurt, B. Erythropoietin production by PDGFR-beta(+) cells. Pflug. Arch. 2016, 468, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Madisen, L.; Zwingman, T.A.; Sunkin, S.M.; Oh, S.W.; Zariwala, H.A.; Gu, H.; Ng, L.L.; Palmiter, R.D.; Hawrylycz, M.J.; Jones, A.R.; et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010, 13, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Aliberti, J.; Graemmel, P.; Sunshine, M.J.; Kreutzberg, G.W.; Sher, A.; Littman, D.R. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell Biol. 2000, 20, 4106–4114. [Google Scholar] [CrossRef] [Green Version]
- Wieghofer, P.; Knobeloch, K.P.; Prinz, M. Genetic targeting of microglia. Glia 2015, 63, 1–22. [Google Scholar] [CrossRef]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. NeuroOncology 2017, 19, 139–141. [Google Scholar] [CrossRef] [Green Version]
- Ghadery, C.; Best, L.A.; Pavese, N.; Tai, Y.F.; Strafella, A.P. PET Evaluation of Microglial Activation in Non-neurodegenerative Brain Diseases. Curr. Neurol. Neurosci. Rep. 2019, 19, 38. [Google Scholar] [CrossRef] [Green Version]
- Bradburn, S.; Murgatroyd, C.; Ray, N. Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A meta-analysis. Ageing Res. Rev. 2019, 50, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chaney, A.; Williams, S.R.; Boutin, H. In vivo molecular imaging of neuroinflammation in Alzheimer’s disease. J. Neurochem. 2019, 149, 438–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohno, M.; Link, J.; Dennis, L.E.; McCready, H.; Huckans, M.; Hoffman, W.F.; Loftis, J.M. Neuroinflammation in addiction: A review of neuroimaging studies and potential immunotherapies. Pharm. Biochem. Behav. 2019, 179, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R. TSPO in diverse CNS pathologies and psychiatric disease: A critical review and a way forward. Pharm. Ther. 2019, 194, 44–58. [Google Scholar] [CrossRef]
- Unterrainer, M.; Fleischmann, D.F.; Diekmann, C.; Vomacka, L.; Lindner, S.; Vettermann, F.; Brendel, M.; Wenter, V.; Ertl-Wagner, B.; Herms, J.; et al. Comparison of (18)F-GE-180 and dynamic (18)F-FET PET in high grade glioma: A double-tracer pilot study. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 580–590. [Google Scholar] [CrossRef]
- Pigeon, H.; Peres, E.A.; Truillet, C.; Jego, B.; Boumezbeur, F.; Caille, F.; Zinnhardt, B.; Jacobs, A.H.; Le Bihan, D.; Winkeler, A. TSPO-PET and diffusion-weighted MRI for imaging a mouse model of infiltrative human glioma. NeuroOncology 2019. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.; Feng, L.; Law, I.; Svarer, C.; Knudsen, G.M.; Mikkelsen, J.D.; de Nijs, R.; Larsen, V.A.; Dyssegaard, A.; Thomsen, G.; et al. TSPO Imaging in Glioblastoma Multiforme: A Direct Comparison Between 123I-CLINDE SPECT, 18F-FET PET, and Gadolinium-Enhanced MR Imaging. J. Nucl. Med. 2015, 56, 1386–1390. [Google Scholar] [CrossRef] [Green Version]
- Betlazar, C.; Harrison-Brown, M.; Middleton, R.J.; Banati, R.; Liu, G.J. Cellular Sources and Regional Variations in the Expression of the Neuroinflammatory Marker Translocator Protein (TSPO) in the Normal Brain. Int. J. Mol. Sci. 2018, 19, 2707. [Google Scholar] [CrossRef] [Green Version]
- Turkheimer, F.E.; Edison, P.; Pavese, N.; Roncaroli, F.; Anderson, A.N.; Hammers, A.; Gerhard, A.; Hinz, R.; Tai, Y.F.; Brooks, D.J. Reference and target region modeling of [11C]-(R)-PK11195 brain studies. J. Nucl. Med. 2007, 48, 158–167. [Google Scholar]
- Cosenza-Nashat, M.; Zhao, M.L.; Suh, H.S.; Morgan, J.; Natividad, R.; Morgello, S.; Lee, S.C. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol. Appl. Neurobiol. 2009, 35, 306–328. [Google Scholar] [CrossRef] [Green Version]
- Lavisse, S.; Guillermier, M.; Herard, A.S.; Petit, F.; Delahaye, M.; Van Camp, N.; Ben Haim, L.; Lebon, V.; Remy, P.; Dolle, F.; et al. Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J. Neurosci. 2012, 32, 10809–10818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckers, L.; Ory, D.; Geric, I.; Declercq, L.; Koole, M.; Kassiou, M.; Bormans, G.; Baes, M. Increased Expression of Translocator Protein (TSPO) Marks Pro-inflammatory Microglia but Does Not Predict Neurodegeneration. Mol. Imaging Biol. 2018, 20, 94–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlodavsky, E.; Soustiel, J.F. Immunohistochemical expression of peripheral benzodiazepine receptors in human astrocytomas and its correlation with grade of malignancy, proliferation, apoptosis and survival. J. NeuroOncol. 2007, 81, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Roncaroli, F.; Su, Z.; Herholz, K.; Gerhard, A.; Turkheimer, F.E. TSPO expression in brain tumours: Is TSPO a target for brain tumour imaging? Clin. Transl. Imaging 2016, 4, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, T.Y.; Jung, S.; Kim, I.Y.; Moon, K.S.; Jang, W.Y.; Park, S.J.; Kim, Y.H.; Kim, H.S.; Min, J.J.; Kim, J. Pathologic analysis of glioblastoma via multiple stereotactic biopsies of active tumor and necrosis. Oncol. Rep. 2012, 27, 707–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawashiro, H.; Otani, N.; Uozumi, Y.; Ooigawa, H.; Toyooka, T.; Suzuki, T.; Katoh, H.; Tsuzuki, N.; Ohnuki, A.; Shima, K.; et al. High expression of L-type amino acid transporter 1 in infiltrating glioma cells. Brain Tumor Pathol. 2005, 22, 89–91. [Google Scholar] [CrossRef] [PubMed]
- Youland, R.S.; Kitange, G.J.; Peterson, T.E.; Pafundi, D.H.; Ramiscal, J.A.; Pokorny, J.L.; Giannini, C.; Laack, N.N.; Parney, I.F.; Lowe, V.J.; et al. The role of LAT1 in (18)F-DOPA uptake in malignant gliomas. J. NeuroOncol. 2013, 111, 11–18. [Google Scholar] [CrossRef]
- Nedergaard, M.K.; Kristoffersen, K.; Michaelsen, S.R.; Madsen, J.; Poulsen, H.S.; Stockhausen, M.T.; Lassen, U.; Kjaer, A. The use of longitudinal 18F-FET MicroPET imaging to evaluate response to irinotecan in orthotopic human glioblastoma multiforme xenografts. PLoS ONE 2014, 9, e100009. [Google Scholar] [CrossRef]
- Nawashiro, H.; Otani, N.; Shinomiya, N.; Fukui, S.; Ooigawa, H.; Shima, K.; Matsuo, H.; Kanai, Y.; Endou, H. L-type amino acid transporter 1 as a potential molecular target in human astrocytic tumors. Int. J. Cancer 2006, 119, 484–492. [Google Scholar] [CrossRef]
- Gerl, M.; Vockl, J.; Kurt, B.; van Veen, T.A.; Kurtz, A.; Wagner, C. Inducible deletion of connexin 40 in adult mice causes hypertension and disrupts pressure control of renin secretion. Kidney Int. 2015, 87, 557–563. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Healy, S.; McMahon, J.; Owens, P.; Dockery, P.; FitzGerald, U. Threshold-based segmentation of fluorescent and chromogenic images of microglia, astrocytes and oligodendrocytes in FIJI. J. Neurosci. Methods 2018, 295, 87–103. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, L.; Kirchleitner, S.V.; Zhao, D.; Li, M.; Tonn, J.-C.; Glass, R.; Kälin, R.E. Glioblastoma Exhibits Inter-Individual Heterogeneity of TSPO and LAT1 Expression in Neoplastic and Parenchymal Cells. Int. J. Mol. Sci. 2020, 21, 612. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020612
Cai L, Kirchleitner SV, Zhao D, Li M, Tonn J-C, Glass R, Kälin RE. Glioblastoma Exhibits Inter-Individual Heterogeneity of TSPO and LAT1 Expression in Neoplastic and Parenchymal Cells. International Journal of Molecular Sciences. 2020; 21(2):612. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020612
Chicago/Turabian StyleCai, Linzhi, Sabrina V. Kirchleitner, Dongxu Zhao, Min Li, Jörg-Christian Tonn, Rainer Glass, and Roland E. Kälin. 2020. "Glioblastoma Exhibits Inter-Individual Heterogeneity of TSPO and LAT1 Expression in Neoplastic and Parenchymal Cells" International Journal of Molecular Sciences 21, no. 2: 612. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020612