Adenosine Triphosphate Accumulated Following Cerebral Ischemia Induces Neutrophil Extracellular Trap Formation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
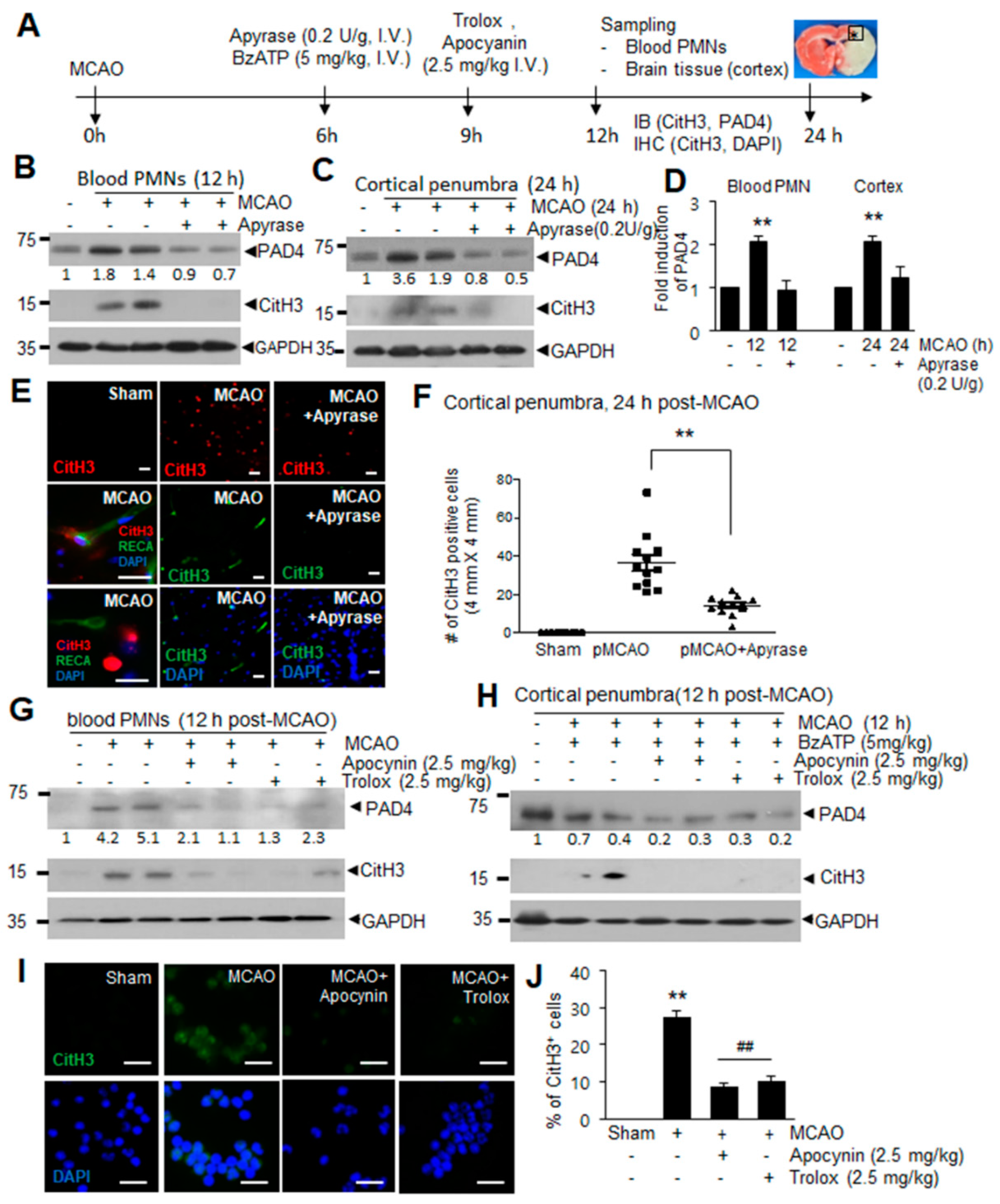
2.1. Induction of PAD4 and CitH3 in Plasma and Brain Parenchyma after MCAO and by ATP and BzATP in Normal Animal
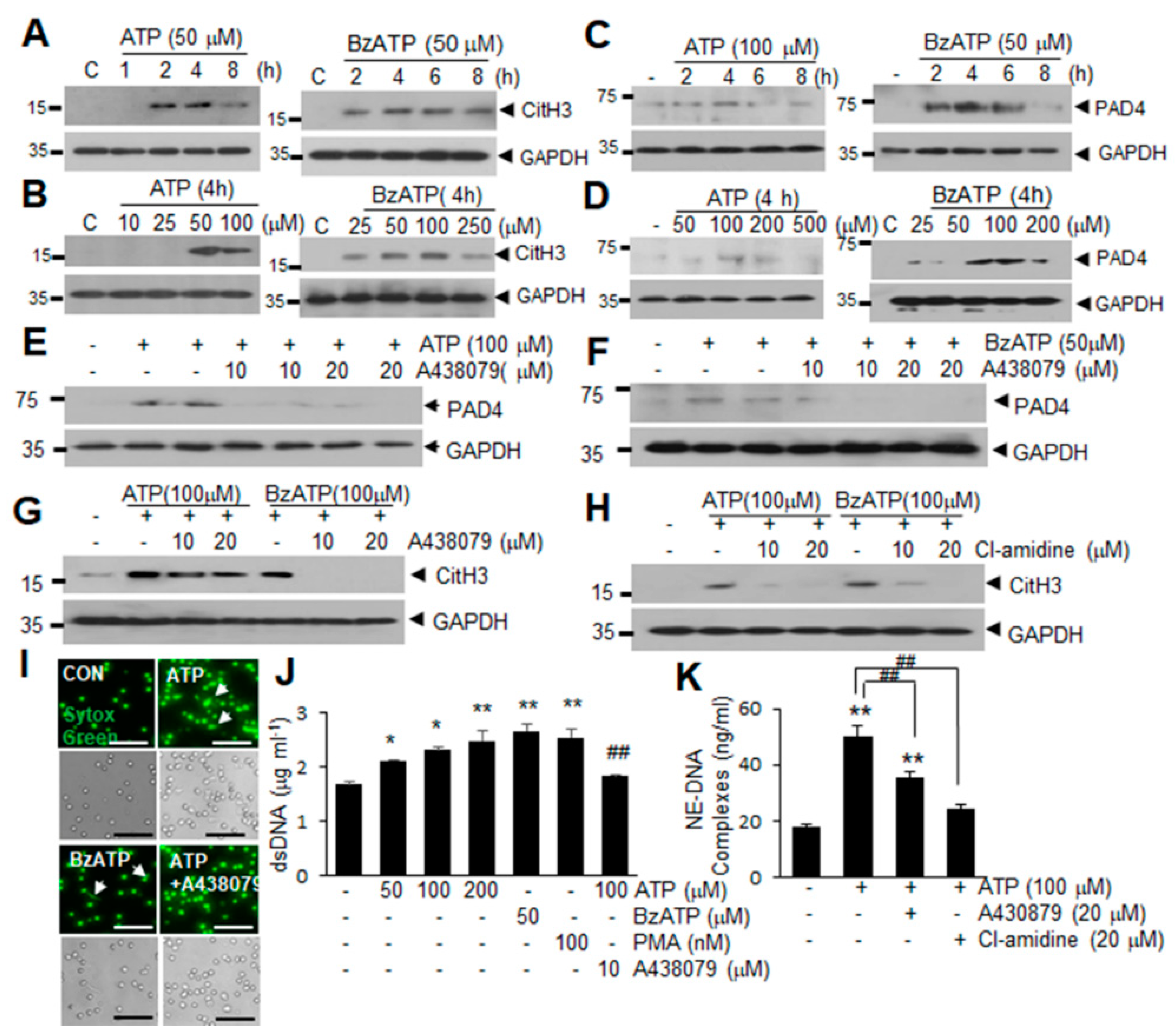
2.2. ATP Induced PAD4 and CitH3 Upregulation in Neutrophils and NETosis in a P2X7R-Dependent Manner
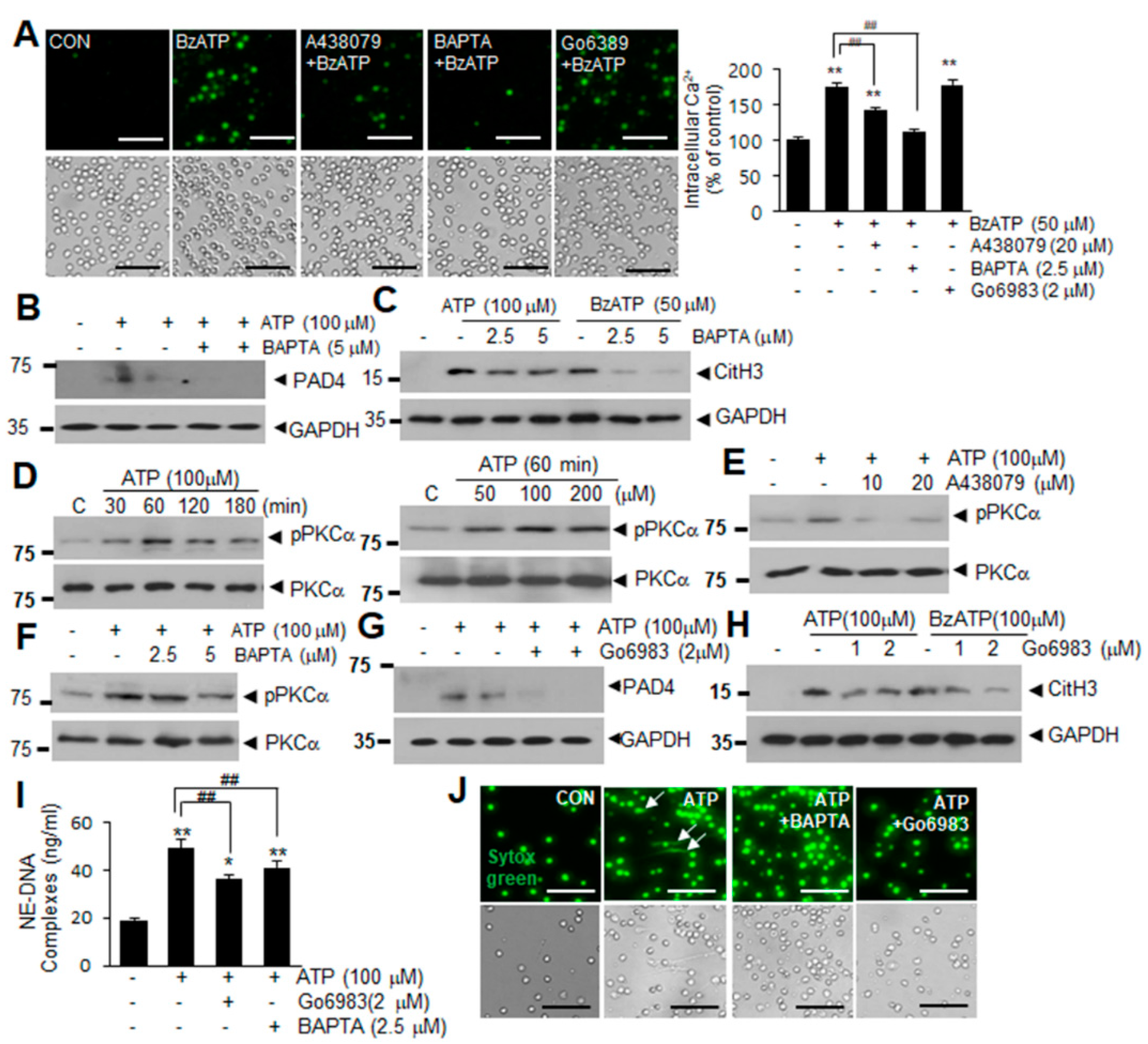
2.3. Intracellular Ca2+ Influx and PKCα Activation Are Involved in ATP or BzATP-Mediated NETosis
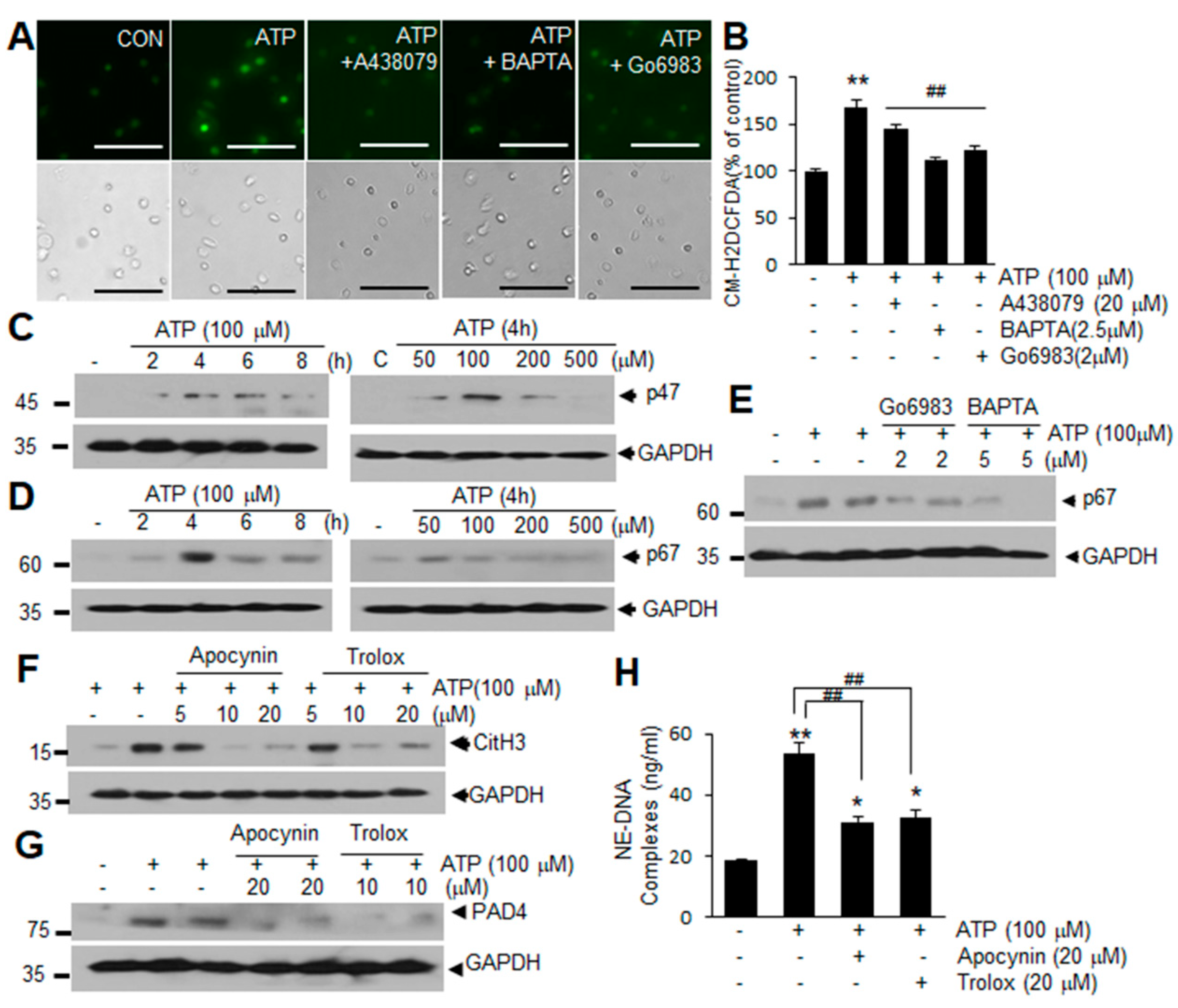
2.4. ROS Production Is Involved in ATP-Mediated Induction of NETosis
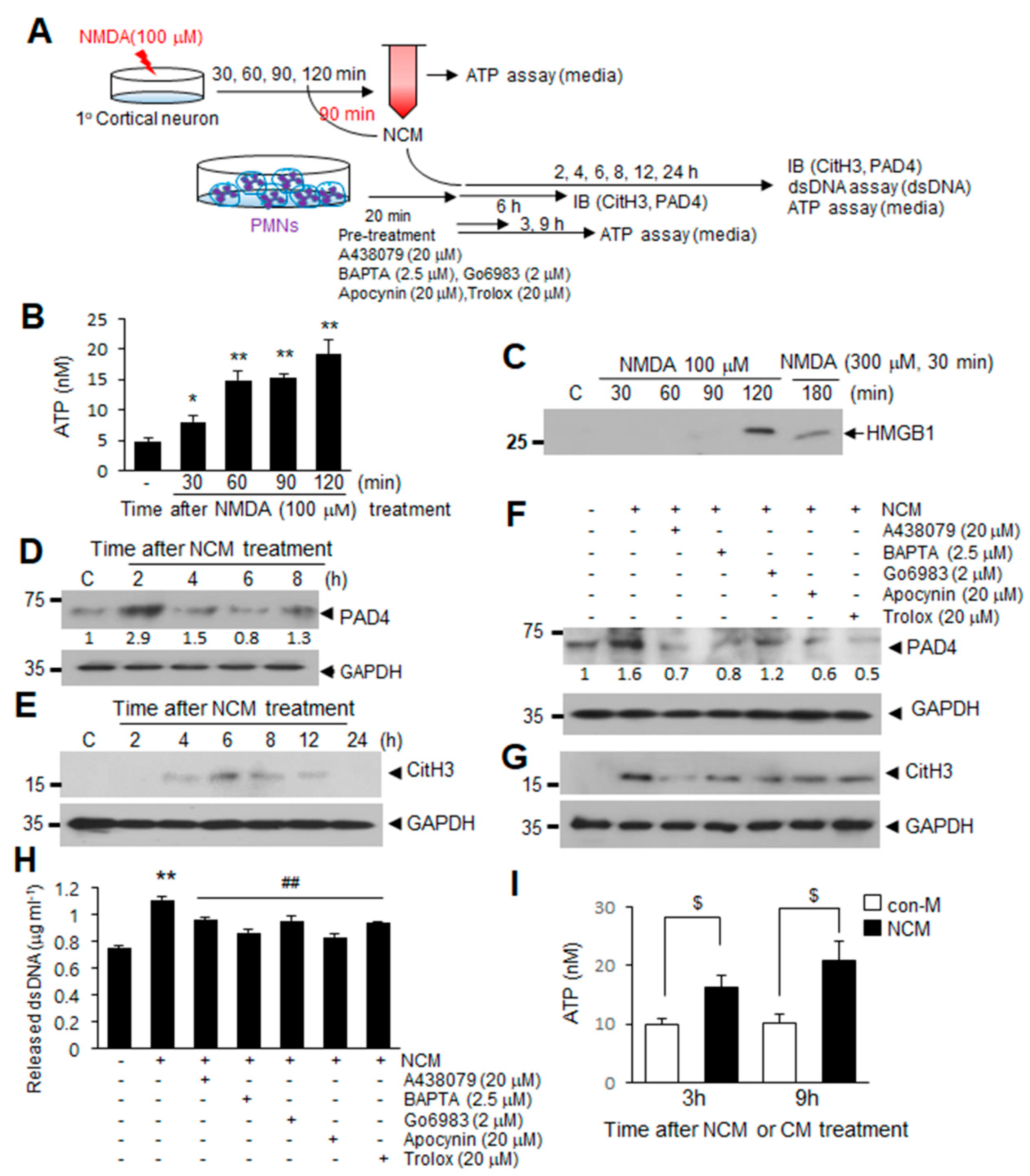
2.5. ATP Released Following NMDA-Induced Neuronal Death Induces NETosis
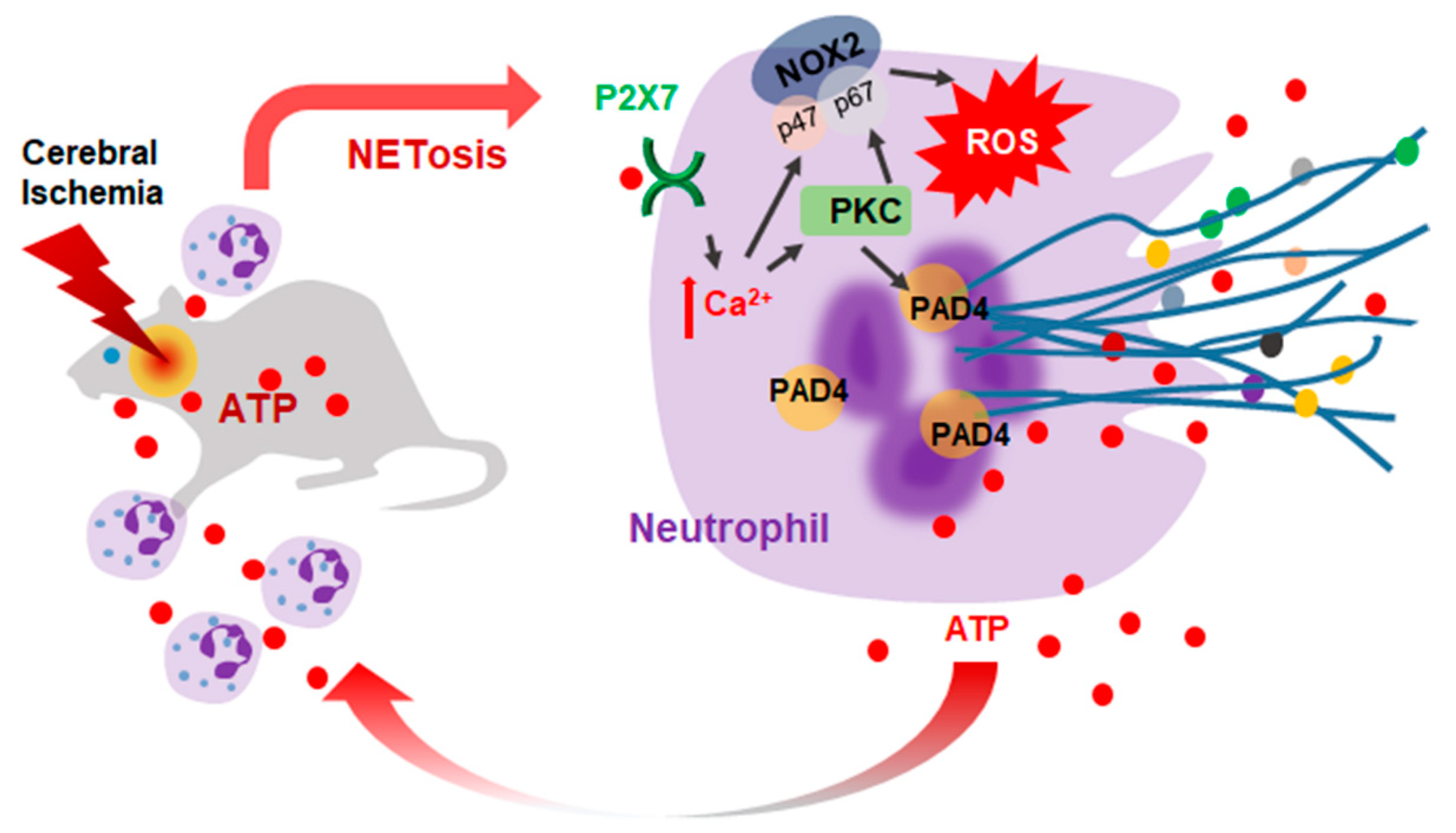
2.6. ATP-P2X7R-Mediated Induction of NETosis in the Ischemic Brain
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Surgical Procedure for Permanent MCAO
4.3. Isolation of Circulating Neutrophils
4.4. Isolation of Bone Marrow Neutrophils
4.5. Measurement of Blood ATP Levels
4.6. Immunofluorescence Staining
4.7. Immunoblotting
4.8. Preparation of Mixed Neuron-Glia Cultures
4.9. Treatment of Neutrophils with NMDA-Conditioned Media (NCM)
4.10. Quantification of Cell-Free DNA
4.11. Quantification of NETs-DNA Complexes in Cell Supernatant
4.12. Intracellular Calcium Levels
4.13. Quantification of Reactive Oxygen Species
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.A.; Phoon, M.C.; van Rooijen, N.; Chow, V.T. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Mitroulis, I.; Kambas, K.; Chrysanthopoulou, A.; Skendros, P.; Apostolidou, E.; Kourtzelis, I.; Drosos, G.I.; Boumpas, D.T.; Ritis, K. Neutrophil extracellular trap formation is associated with IL-1β and autophagy-related signaling in gout. PLoS ONE 2011, 6, e29318. [Google Scholar] [CrossRef] [PubMed]
- Arita, K.; Hashimoto, H.; Shimizu, T.; Nakashima, K.; Yamada, M.; Sato, M. Structural basis for Ca(2+)-induced activation of human PAD4. Nat. Struct. Mol. Biol. 2004, 11, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, H.O.; Chen, H.W.; Tohme, S.; Tai, S.; van der Windt, D.J.; Loughran, P.; Rosborough, B.R.; Sud, V.; Beer-Stolz, D.; Turnquist, H.R.; et al. IL-33 exacerbates liver sterile inflammation by amplifying neutrophil extracellular trap formation. J. Hepatol. 2017, 68, 130–139. [Google Scholar] [CrossRef]
- Kim, S.W.; Lee, H.; Lee, H.K.; Kim, I.D.; Lee, J.K. Neutrophil extracellular trap induced by HMGB1 exacerbates damages in the ischemic brain. Acta Neuropathol. Commun. 2019, 7, 94. [Google Scholar] [CrossRef] [Green Version]
- Allen, C.; Thornton, P.; Denes, A.; McColl, B.W.; Pierozynski, A.; Monestier, M.; Pinteaux, E.; Rothwell, N.J.; Allan, S.M. Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. J. Immunol. 2012, 189, 381–392. [Google Scholar] [CrossRef]
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef]
- Perez-de-Puig, I.; Miró-Mur, F.; Ferrer-Ferrer, M.; Gelpi, E.; Pedragosa, J.; Justicia, C.; Urra, X.; Chamorro, A.; Planas, A.M. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol. 2015, 129, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Kang, L.; Yu, H.; Yang, X.; Zhu, Y.; Bai, X.; Wang, R.; Cao, Y.; Xu, H.; Luo, H.; Lu, L.; et al. Neutrophil extracellular traps released by neutrophils impair revascularization and vascular remodeling after stroke. Nat. Commun. 2020, 11, 2488. [Google Scholar] [CrossRef]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, D.; Chiozzi, P.; Falzoni, S.; Hanau, S.; Di Virgilio, F. Purinergic modulation of interleukin-1 beta release from microglial cells stimulated with bacterial endotoxin. J. Exp. Med. 1997, 185, 79–82. [Google Scholar] [CrossRef] [Green Version]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.; Persechini, P.M.; Ojcius, D.M. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem. 2007, 82, 2871–2879. [Google Scholar] [CrossRef] [Green Version]
- Fontanils, U.; Seil, M.; Pochet, S.; El Ouaaliti, M.; Garcia-Marcos, M.; Dehaye, J.P.; Marino, A. Stimulation by P2X7 receptors of calcium-dependent production of reactive oxygen species (ROS) in rat submandibular glands. Biochim. Biophys. Acta 2010, 1800, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, M.; Katsnelson, M.A.; Dubyak, G.R.; Pearlman, E. Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1β secretion in response to ATP. Nat. Commun. 2016, 7, 10555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solini, A.; Chiozzi, P.; Morelli, A.; Fellin, R.; Di Virgilio, F. Human primary fibroblasts in vitro express a purinergic P2X7 receptor coupled to ion fluxes, microvesicle formation and IL-6 release. J. Cell Sci. 1999, 112, 297–305. [Google Scholar]
- Sluyter, R.; Dalitz, J.G.; Wiley, J.S. P2X7 receptor polymorphism impairs extracellular adenosine 5′-triphosphate-induced interleukin-18 release from human monocytes. Genes Immun. 2004, 5, 588–591. [Google Scholar] [CrossRef] [Green Version]
- Rabadi, M.; Kim, M.; Li, H.; Han, S.J.; Choi, Y.; D’Agati, V.; Lee, H.T. ATP induces PAD4 in renal proximal tubule cells via P2X7 receptor activation to exacerbate ischemic AKI. Am. J. Physiol. Renal Physiol. 2018, 314, CF293–CF305. [Google Scholar] [CrossRef]
- Sofoluwe, A.; Bacchetta, M.; Badaoui, M.; Kwak, B.R.; Chanson, M. ATP amplifies NADPH-dependent and -independent neutrophil extracellular trap formation. Sci. Rep. 2019, 9, 16556. [Google Scholar] [CrossRef]
- Volonté, C.; Amadio, S.; Cavaliere, F.; D’Ambrosi, N.; Vacca, F.; Bernardi, G. Extracellular ATP and neurodegeneration. Curr. Drug Targets CNS Neurol. Disord. 2003, 2, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Imura, Y.; Morizawa, Y.; Komatsu, R.; Shibata, K.; Shinozaki, Y.; Kasai, H.; Moriishi, K.; Moriyama, Y.; Koizumi, S. Microglia release ATP by exocytosis. Glia 2013, 61, 1320–1330. [Google Scholar] [CrossRef] [PubMed]
- Ballerini, P.; Di Iorio, P.; Ciccarelli, R.; Nargi, E.; D’Alimonte, I.; Traversa, U.; Rathbone, M.P.; Caciagli, F. Glial cells express multiple ATP binding cassette proteins which are involved in ATP release. Neuroreport 2002, 13, 1789–1792. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purine and pyrimidine receptors. Cell Mol. Life Sci. 2007, 64, 471–483. [Google Scholar] [CrossRef]
- Iyer, S.S.; Pulskens, W.P.; Sadler, J.J.; Butter, L.M.; Teske, G.J.; Ulland, T.K.; Eisenbarth, S.C.; Florquin, S.; Flavell, R.A.; Leemans, J.C.; et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc. Natl. Acad. Sci. USA 2009, 106, 20388–20393. [Google Scholar] [CrossRef] [Green Version]
- Surprenant, A.; Rassendren, F.; Kawashima, E.; North, R.A.; Buell, G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 1996, 272, 735–738. [Google Scholar] [CrossRef]
- Díaz-Vegas, A.; Campos, C.A.; Contreras-Ferrat, A.; Casas, M.; Buvinic, S.; Jaimovich, E.; Espinosa, A. ROS Production via P2Y1-PKC-NOX2 Is Triggered by Extracellular ATP after Electrical Stimulation of Skeletal Muscle Cells. PLoS ONE 2015, 10, e0129882. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, H. Extracellular metabolism of ATP and other nucleotides. Naunyn-Schmiedeberg’s Arch Pharmacol. 2000, 362, 299–309. [Google Scholar] [CrossRef]
- Melani, A.; Corti, F.; Stephan, H.; Müller, C.E.; Donati, C.; Bruni, P.; Vannucchi, M.G.; Pedata, F. Ecto-ATPase inhibition: ATP and adenosine release under physiological and ischemic in vivo conditions in the rat striatum. Exp. Neurol. 2012, 233, 193–204. [Google Scholar] [CrossRef]
- Sil, P.; Hayes, C.P.; Reaves, B.J.; Breen, P.; Quinn, S.; Sokolove, J.; Rada, B. P2Y6 Receptor Antagonist MRS2578 Inhibits Neutrophil Activation and Aggregated Neutrophil Extracellular Trap Formation Induced by Gout-Associated Monosodium Urate Crystals. J. Immunol. 2017, 98, 428–442. [Google Scholar] [CrossRef] [Green Version]
- Sil, P.; Wicklum, H.; Surell, C.; Rada, B. Macrophage-derived IL-1β enhances monosodium urate crystal-triggered NET formation. Inflamm. Res. 2017, 66, 227–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Guo, Z.; Liu, X.; Ouyang, Q.; He, C.; Burnstock, G.; Yuan, H.; Xiang, Z. Block of P2X7 receptors could partly reverse the delayed neuronal death in area CA1 of the hippocampus after transient global cerebral ischemia. Purinergic Signal. 2013, 9, 663–675. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Zhang, X.; Dai, Z.; Feng, Y.; Li, Q.; Zhang, J.H.; Liu, X.; Chen, Y.; Feng, H. P2X7 Receptor Suppression Preserves Blood-Brain Barrier through Inhibiting RhoA Activation after Experimental Intracerebral Hemorrhage in Rats. Sci. Rep. 2016, 6, 23286. [Google Scholar] [CrossRef]
- Feng, L.; Chen, Y.; Ding, R.; Fu, Z.; Yang, S.; Deng, X.; Zeng, J. P2X7R blockade prevents NLRP3 inflammasome activation and brain injury in a rat model of intracerebral hemorrhage: Involvement of peroxynitrite. J. Neuroinflamm. 2015, 12, 190. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.; Mei, B.; Li, H.; Dou, Y.; Tian, X.; Shen, M.; Chen, G. P2X7 Participates in Intracerebral Hemorrhage-Induced Secondary Brain Injury in Rats via MAPKs Signaling Pathways. Neurochem. Res. 2017, 42, 2372–2383. [Google Scholar] [CrossRef]
- Linden, J.; Koch-Nolte, F.; Dahl, G. Purine Release, Metabolism, and Signaling in the Inflammatory Response. Annu. Rev. Immunol. 2019, 37, 325–347. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, N.S.; Yu, H.C.; Tung, C.H.; Huang, K.Y.; Huang, H.B.; Lu, M.C. Increased peptidylarginine deiminases expression during the macrophage differentiation and participated inflammatory responses. Arthritis Res. Ther. 2019, 21, 108. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Castillo, Z.; Palafox-Sánchez, C.A.; Parra-Rojas, I.; Martínez-Bonilla, G.E.; del Toro-Arreola, S.; Ramírez-Dueñas, M.G.; Ocampo-Bermudes, G.; Muñoz-Valle, J.F. Comparative analysis of autoantibodies targeting peptidylarginine deiminase type 4, mutated citrullinated vimentin and cyclic citrullinated peptides in rheumatoid arthritis: Associations with cytokine profiles, clinical and genetic features. Clin. Exp. Immunol. 2015, 182, 119–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona-Rivera, C.; Purmalek, M.M.; Moore, E.; Waldman, M.; Walter, P.J.; Garraffo, H.M.; Phillips, K.A.; Preston, K.L.; Graf, J.; Kaplan, M.J. A role for muscarinic receptors in neutrophil extracellular trap formation and levamisole-induced autoimmunity. JCI Insight 2017, 2, e89780. [Google Scholar] [CrossRef]
- Meegan, J.E.; Yang, X.; Beard, R.S., Jr.; Jannaway, M.; Chatterjee, V.; Taylor-Clark, T.E.; Yuan, S.Y. Citrullinated histone 3 causes endothelial barrier dysfunction. Biochem. Biophys. Res. Commun. 2018, 503, 1498–1502. [Google Scholar] [CrossRef]
- Pieterse, E.; Rother, N.; Garsen, M.; Hofstra, J.M.; Satchell, S.C.; Hoffmann, M.; Loeven, M.A.; Knaapen, H.K.; van der Heijden, O.W.H.; Berden, J.H.M.; et al. Neutrophil Extracellular Traps Drive Endothelial-to-Mesenchymal Transition. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1371–1379. [Google Scholar] [CrossRef] [Green Version]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef]
- Gray, R.D.; Lucas, C.D.; MacKellar, A.; Li, F.; Hiersemenzel, K.; Haslett, C.; Davidson, D.J.; Rossi, A.G. Activation of conventional protein kinase C (PKC) is critical in the generation of human neutrophil extracellular traps. J. Inflamm. 2013, 10, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Rana, R.; Corbett, J.; Kadiiska, M.B.; Goldstein, J.; Mason, R.P. P2X7 receptor-NADPH oxidase axis mediates protein radical formation and Kupffer cell activation in carbon tetrachloride-mediated steatohepatitis in obese mice. Free Radic. Biol. Med. 2012, 52, 1666–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apolloni, S.; Parisi, C.; Pesaresi, M.G.; Rossi, S.; Carrì, M.T.; Cozzolino, M.; Volonté, C.; D’Ambrosi, N. The NADPH oxidase pathway is dysregulated by the P2X7 receptor in the SOD1-G93A microglia model of amyotrophic lateral sclerosis. J. Immunol. 2013, 190, 5187–5195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahl, G. ATP release through pannexon channels. Philos. Trans. R. Soc. B 2015, 370. [Google Scholar] [CrossRef]
- Jacob, F.; Pérez Novo, C.; Bachert, C.; Van Crombruggen, K. Purinergic signaling in inflammatory cells: P2 receptor expression, functional effects, and modulation of inflammatory responses. Purinergic Signal. 2013, 9, 285–306. [Google Scholar] [CrossRef] [Green Version]
- Arai, Y.; Nishinaka, Y.; Arai, T.; Morita, M.; Mizugishi, K.; Adachi, S.; Takaori-Kondo, A.; Watanabe, T.; Yamashita, K. Uric acid induces NADPH oxidase-independent neutrophil extracellular trap formation. Biochem. Biophys. Res. Commun. 2014, 443, 556–561. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.; Cuthill, I.C.; Emerson, M.; Altman, D.G.; NC3Rs Reporting Guidelines Working Group. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. J. Gene Med. 2010, 12, 561–563. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-W.; Davaanyam, D.; Seol, S.-I.; Lee, H.-K.; Lee, H.; Lee, J.-K. Adenosine Triphosphate Accumulated Following Cerebral Ischemia Induces Neutrophil Extracellular Trap Formation. Int. J. Mol. Sci. 2020, 21, 7668. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207668
Kim S-W, Davaanyam D, Seol S-I, Lee H-K, Lee H, Lee J-K. Adenosine Triphosphate Accumulated Following Cerebral Ischemia Induces Neutrophil Extracellular Trap Formation. International Journal of Molecular Sciences. 2020; 21(20):7668. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207668
Chicago/Turabian StyleKim, Seung-Woo, Dashdulam Davaanyam, Song-I Seol, Hye-Kyung Lee, Hahnbie Lee, and Ja-Kyeong Lee. 2020. "Adenosine Triphosphate Accumulated Following Cerebral Ischemia Induces Neutrophil Extracellular Trap Formation" International Journal of Molecular Sciences 21, no. 20: 7668. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207668