Neurotrophic Factor BDNF, Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
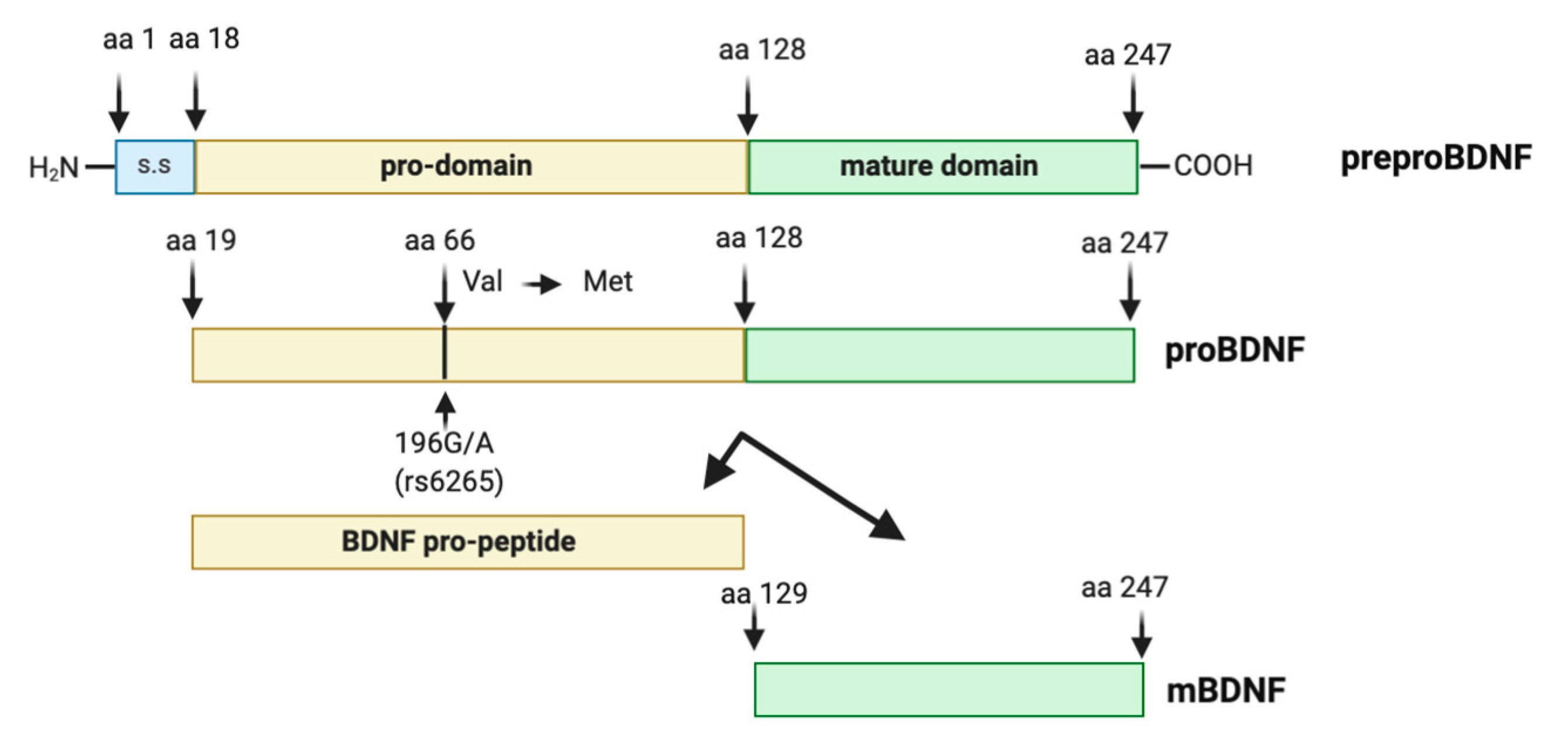
2. The Human BDNF Gene: Transcripts and Variants
2.1. BDNF Transcripts
2.2. miRNAs and BDNF
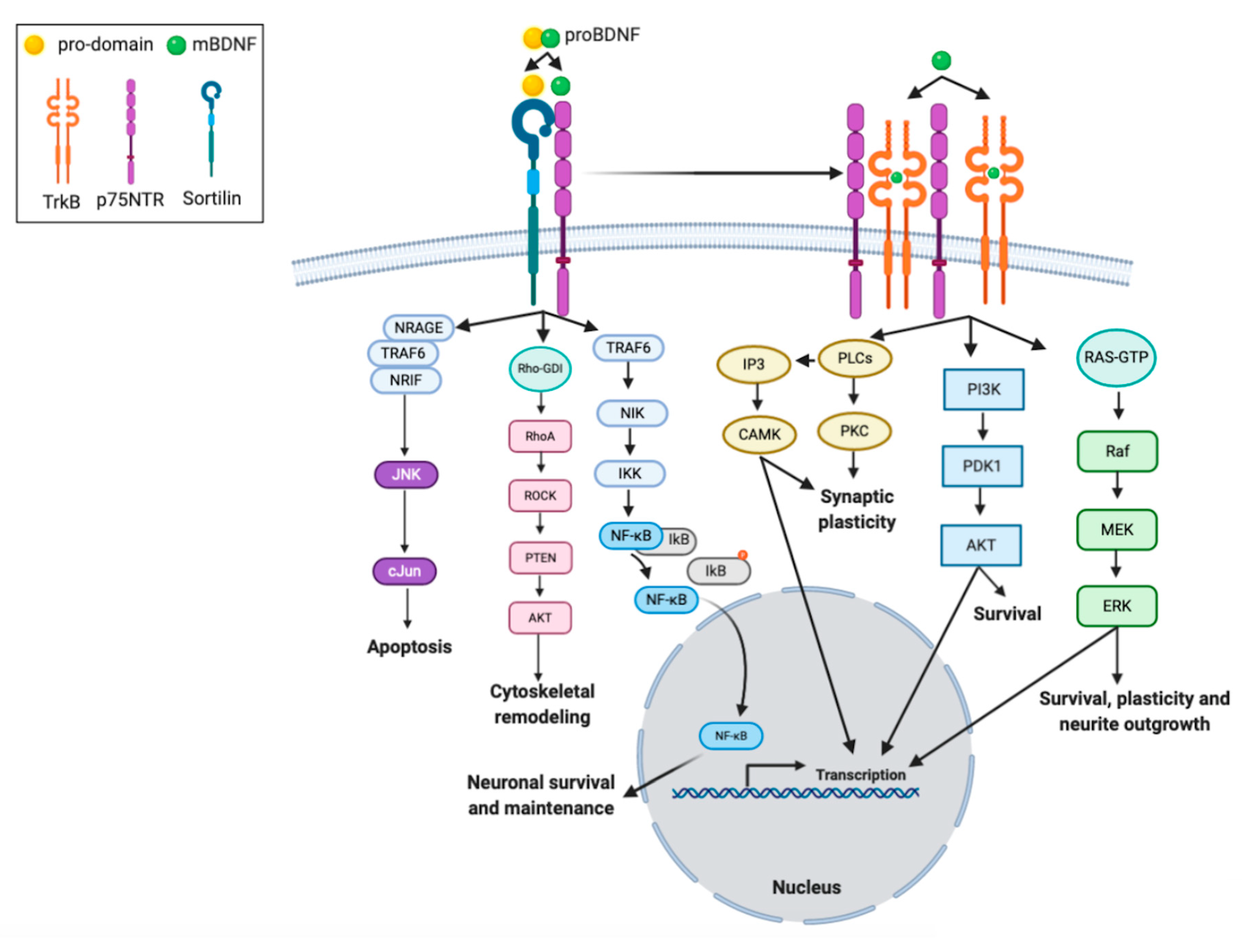
2.3. Biology of BDNF
2.4. The Human BDNF Variant Val66Met
3. Neuroplasticity in MDD: The Effects of Antidepressant Therapies
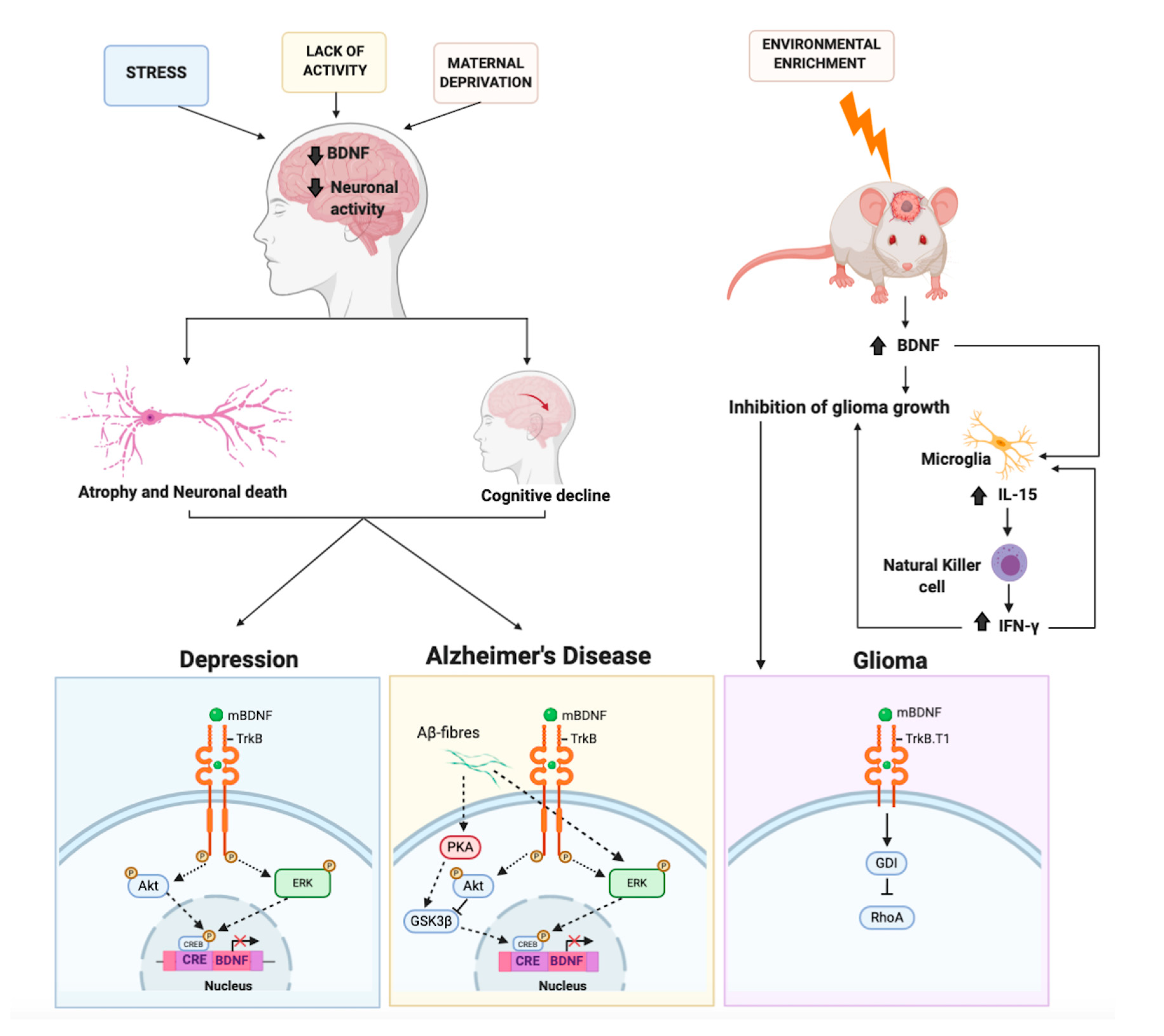
3.1. Major Depressive Disorder
3.2. BDNF and Neuronal Plasticity
3.3. BDNF and Synaptic Plasticity
3.4. BDNF in Depressed Patients
3.5. Effect of Antidepressant Therapies on Plasticity BDNF-Mediated
3.5.1. BDNF and Antidepressant Treatments
3.5.2. Beneficial Effects of Exercise on Plasticity: The Role of BDNF
4. The Protective Role of BDNF on Neurodegeneration
4.1. The Protective Role of BDNF on Alzheimer’s Disease
4.2. BDNF and Ras-ERK-CREB Signaling in Alzheimer’s Disease
5. BDNF and Brain Cancer: An Unexpected Role. An Oncogene or a Tumor Suppressor?
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| AP1 | Activator Protein 1 |
| BCL2 | B-cell lymphoma 2 |
| BDNF | Brain-Derived Neurotrophic Factor |
| BHLHB2 | Helix-loop-helix protein |
| CaMK | Ca2+/calmodulin-dependent protein kinase |
| cAMP/CRE | Cyclic adenosine monophosphate calcium response element |
| CaREs | Calcium-responsive elements |
| CREB | CREB |
| Ctsb | Myokine cathepsin B |
| CUMS | Chronic Unpredicted Mild Stress model |
| DAG | 1,2-diacylglycerol |
| ERK | Extracellular signal-regulated kinase |
| GTP-ases | guanosine triphosphate hydrolases |
| JNK | c-Jun amino terminal kinase |
| IKK | Multi-subunit IκB kinase |
| IkB | Inhibitor of kB |
| L-VGCC | L-type voltage gated calcium channel |
| LTP | Long-Term Potentiation |
| MAPK | Mitogen-activated protein kinase |
| MDD | Major depressive disorder |
| Met | Methionine |
| miRNAs | microRNAs |
| MMP | Matrix Metalloproteases |
| MPP | 1-methyl-4-phenylpyridinium |
| NF-kB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| NMDA | N-Methyl-D-Aspartate |
| NRAGE | Neurotrophin receptor-interacting MAGE homologue |
| NRIF | Neurotrophin receptor interacting factor |
| PD | Parkinson’s disease |
| p75NTR | p75 neurotrophin receptor |
| PI3K | Phosphoinositide 3-kinases |
| PKC | Protein kinase C |
| PLC | Phospholipase C |
| Rho | Ras homolog |
| ROCK | Rho-associated protein kinase |
| SIRT1 | Sirtuin 1 |
| SNPs | Polymorphism |
| SSRIs | Serotonin reuptake inhibitor |
| TRAF6 | Tumor necrosis factor receptor associated factor 6 |
| TrkB | Tropomycin receptor kinase B |
| Val | Valine |
| VDCCs | L-type voltage-dependent Ca2+ channels |
| Vps10p | Vacuolar protein sorting 10 protein |
References
- Andreska, T.; Aufmkolk, S.; Sauer, M.; Blum, R. High abundance of BDNF within glutamatergic presynapses of cultured hippocampal neurons. Front. Cell Neurosci. 2014, 8, 107. [Google Scholar] [CrossRef] [Green Version]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Münch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef] [Green Version]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R.; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [Green Version]
- Bartkowska, K.; Paquin, A.; Gauthier, A.S.; Kaplan, D.R.; Miller, F.D. Trk signaling regulates neural precursor cell proliferation and differentiation during cortical development. Development 2007, 134, 4369–4380. [Google Scholar] [CrossRef] [Green Version]
- Vilar, M.; Mira, H. Regulation of Neurogenesis by Neurotrophins during Adulthood: Expected and Unexpected Roles. Front. Neurosci. 2016, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P.; Moehl, K.; Ghena, N.; Schmaedick, M.; Cheng, A. Intermittent metabolic switching, neuroplasticity and brain health. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef]
- Numakawa, T.; Odaka, H.; Adachi, N. Actions of Brain-Derived Neurotrophin Factor in the Neurogenesis and Neuronal Function, and Its Involvement in the Pathophysiology of Brain Diseases. Int. J. Mol. Sci. 2018, 19, 3650. [Google Scholar] [CrossRef] [Green Version]
- Loprinzi, P.D.; Day, S.; Deming, R. Acute Exercise Intensity and Memory Function: Evaluation of the Transient Hypofrontality Hypothesis. Medicina (Kaunas) 2019, 55, 445. [Google Scholar] [CrossRef] [Green Version]
- Fabel, K.; Wolf, S.A.; Ehninger, D.; Babu, H.; Leal-Galicia, P.; Kempermann, G. Additive effects of physical exercise and environmental enrichment on adult hippocampal neurogenesis in mice. Front. Neurosci. 2009, 3, 50. [Google Scholar] [CrossRef] [Green Version]
- Knaepen, K.; Goekint, M.; Heyman, E.M.; Meeusen, R. Neuroplasticity-exercise-induced response of peripheral brain-derived neurotrophic factor: A systematic review of experimental studies in human subjects. Sports Med. 2010, 40, 765–801. [Google Scholar] [CrossRef]
- Szuhany, K.L.; Bugatti, M.; Otto, M.W. A meta-analytic review of the effects of exercise on brain-derived neurotrophic factor. J. Psychiatr. Res. 2015, 60, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Praag, H.; Shubert, T.; Zhao, C.; Gage, F.H. Exercise enhances learning and hippocampal neurogenesis in aged mice. J. Neurosci. 2005, 25, 8680–8685. [Google Scholar] [CrossRef] [PubMed]
- Tuon, T.; Souza, P.S.; Santos, M.F.; Pereira, F.T.; Pedroso, G.S.; Luciano, T.F.; De Souza, C.T.; Dutra, R.C.; Silveira, P.C.; Pinho, R.A. Physical Training Regulates Mitochondrial Parameters and Neuroinflammatory Mechanisms in an Experimental Model of Parkinson’s Disease. Oxid. Med. Cell. Longev. 2015, 2015, 261809. [Google Scholar] [CrossRef] [PubMed]
- Vilela, T.C.; Muller, A.P.; Damiani, A.P.; Macan, T.P.; da Silva, S.; Canteiro, P.B.; de Sena Casagrande, A.; Pedroso, G.D.S.; Nesi, R.T.; de Andrade, V.M.; et al. Strength and Aerobic Exercises Improve Spatial Memory in Aging Rats Through Stimulating Distinct Neuroplasticity Mechanisms. Mol. Neurobiol. 2017, 54, 7928–7937. [Google Scholar] [CrossRef]
- Itami, C.; Kimura, F.; Kohno, T.; Matsuoka, M.; Ichikawa, M.; Tsumoto, T.; Nakamura, S. Brain-derived neurotrophic factor-dependent unmasking of "silent" synapses in the developing mouse barrel cortex. Proc. Natl. Acad. Sci. USA 2003, 100, 13069–13074. [Google Scholar] [CrossRef] [Green Version]
- Edelmann, E.; Lessmann, V.; Brigadski, T. Pre-and postsynaptic twists in BDNF secretion and action in synaptic plasticity. Neuropharmacology 2014, 76 Pt C, 610–627. [Google Scholar] [CrossRef]
- Nakahashi, T.; Fujimura, H.; Altar, C.A.; Li, J.; Kambayashi, J.; Tandon, N.N.; Sun, B. Vascular endothelial cells synthesize and secrete brain-derived neurotrophic factor. FEBS Lett. 2000, 470, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Ward, N.; Boswell, M.; Katz, D.M. Secretion of brain-derived neurotrophic factor from brain microvascular endothelial cells. Eur. J. Neurosci. 2006, 23, 1665–1670. [Google Scholar] [CrossRef]
- Pius-Sadowska, E.; Machaliński, B. BDNF-A key player in cardiovascular system. J. Mol. Cell. Cardiol. 2017, 110, 54–60. [Google Scholar] [CrossRef]
- Anders, Q.S.; Ferreira, L.V.B.; Rodrigues, L.C.M.; Nakamura-Palacios, E.M. BDNF mRNA Expression in Leukocytes and Frontal Cortex Function in Drug Use Disorder. Front. Psychiatry 2020, 11, 469. [Google Scholar] [CrossRef]
- Chacón-Fernández, P.; Säuberli, K.; Colzani, M.; Moreau, T.; Ghevaert, C.; Barde, Y.A. Brain-derived Neurotrophic Factor in Megakaryocytes. J. Biol. Chem. 2016, 291, 9872–9881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amadio, P.; Sandrini, L.; Ieraci, A.; Tremoli, E.; Barbieri, S.S. Effect of Clotting Duration and Temperature on BDNF Measurement in Human Serum. Int. J. Mol. Sci. 2017, 18, 1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meek, T.H.; Wisse, B.E.; Thaler, J.P.; Guyenet, S.J.; Matsen, M.E.; Fischer, J.D.; Taborsky, G.J.; Schwartz, M.W.; Morton, G.J. BDNF action in the brain attenuates diabetic hyperglycemia via insulin-independent inhibition of hepatic glucose production. Diabetes 2013, 62, 1512–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marosi, K.; Mattson, M.P. BDNF mediates adaptive brain and body responses to energetic challenges. Trends Endocrinol. Metab. 2014, 25, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delezie, J.; Weihrauch, M.; Maier, G.; Tejero, R.; Ham, D.J.; Gill, J.F.; Karrer-Cardel, B.; Rüegg, M.A.; Tabares, L.; Handschin, C. BDNF is a mediator of glycolytic fiber-type specification in mouse skeletal muscle. Proc. Natl. Acad. Sci. USA 2019, 116, 16111–16120. [Google Scholar] [CrossRef] [Green Version]
- Tettamanti, G.; Cattaneo, A.G.; Gornati, R.; de Eguileor, M.; Bernardini, G.; Binelli, G. Phylogenesis of brain-derived neurotrophic factor (BDNF) in vertebrates. Gene 2010, 450, 85–93. [Google Scholar] [CrossRef]
- Metsis, M.; Timmusk, T.; Arenas, E.; Persson, H. Differential usage of multiple brain-derived neurotrophic factor promoters in the rat brain following neuronal activation. Proc. Natl. Acad. Sci. USA 1993, 90, 8802–8806. [Google Scholar] [CrossRef] [Green Version]
- Timmusk, T.; Lendahl, U.; Funakoshi, H.; Arenas, E.; Persson, H.; Metsis, M. Identification of brain-derived neurotrophic factor promoter regions mediating tissue-specific, axotomy-, and neuronal activity-induced expression in transgenic mice. J. Cell Biol. 1995, 128, 185–199. [Google Scholar] [CrossRef]
- Timmusk, T.; Palm, K.; Metsis, M.; Reintam, T.; Paalme, V.; Saarma, M.; Persson, H. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron 1993, 10, 475–489. [Google Scholar] [CrossRef]
- Vashishta, A.; Habas, A.; Pruunsild, P.; Zheng, J.J.; Timmusk, T.; Hetman, M. Nuclear factor of activated T-cells isoform c4 (NFATc4/NFAT3) as a mediator of antiapoptotic transcription in NMDA receptor-stimulated cortical neurons. J. Neurosci. 2009, 29, 15331–15340. [Google Scholar] [CrossRef]
- Volpicelli, F.; Caiazzo, M.; Greco, D.; Consales, C.; Leone, L.; Perrone-Capano, C.; Colucci D’Amato, L.; di Porzio, U. Bdnf gene is a downstream target of Nurr1 transcription factor in rat midbrain neurons in vitro. J. Neurochem. 2007, 102, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Aid, T.; Kazantseva, A.; Piirsoo, M.; Palm, K.; Timmusk, T. Mouse and rat BDNF gene structure and expression revisited. J. Neurosci. Res. 2007, 85, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Pruunsild, P.; Kazantseva, A.; Aid, T.; Palm, K.; Timmusk, T. Dissecting the human BDNF locus: Bidirectional transcription, complex splicing, and multiple promoters. Genomics 2007, 90, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Simms, B.A.; Zamponi, G.W. Neuronal voltage-gated calcium channels: Structure, function, and dysfunction. Neuron 2014, 82, 24–45. [Google Scholar] [CrossRef] [Green Version]
- Shieh, P.B.; Hu, S.C.; Bobb, K.; Timmusk, T.; Ghosh, A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron 1998, 20, 727–740. [Google Scholar] [CrossRef] [Green Version]
- Tao, X.; Finkbeiner, S.; Arnold, D.B.; Shaywitz, A.J.; Greenberg, M.E. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 1998, 20, 709–726. [Google Scholar] [CrossRef] [Green Version]
- Tabuchi, A.; Sakaya, H.; Kisukeda, T.; Fushiki, H.; Tsuda, M. Involvement of an upstream stimulatory factor as well as cAMP-responsive element-binding protein in the activation of brain-derived neurotrophic factor gene promoter I. J. Biol. Chem. 2002, 277, 35920–35931. [Google Scholar] [CrossRef] [Green Version]
- Pruunsild, P.; Sepp, M.; Orav, E.; Koppel, I.; Timmusk, T. Identification of cis-elements and transcription factors regulating neuronal activity-dependent transcription of human BDNF gene. J. Neurosci. 2011, 31, 3295–3308. [Google Scholar] [CrossRef] [Green Version]
- Tao, X.; West, A.E.; Chen, W.G.; Corfas, G.; Greenberg, M.E. A calcium-responsive transcription factor, CaRF, that regulates neuronal activity-dependent expression of BDNF. Neuron 2002, 33, 383–395. [Google Scholar] [CrossRef] [Green Version]
- Zheng, F.; Wang, H. NMDA-mediated and self-induced bdnf exon IV transcriptions are differentially regulated in cultured cortical neurons. Neurochem. Int. 2009, 54, 385–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipsky, R.H.; Xu, K.; Zhu, D.; Kelly, C.; Terhakopian, A.; Novelli, A.; Marini, A.M. Nuclear factor kappaB is a critical determinant in N-methyl-D-aspartate receptor-mediated neuroprotection. J. Neurochem. 2001, 78, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Zhou, X.; Moon, C.; Wang, H. Regulation of brain-derived neurotrophic factor expression in neurons. Int. J. Physiol. Pathophysiol. Pharmacol. 2012, 4, 188–200. [Google Scholar] [PubMed]
- Jiang, X.; Tian, F.; Du, Y.; Copeland, N.G.; Jenkins, N.A.; Tessarollo, L.; Wu, X.; Pan, H.; Hu, X.Z.; Xu, K.; et al. BHLHB2 controls Bdnf promoter 4 activity and neuronal excitability. J. Neurosci. 2008, 28, 1118–1130. [Google Scholar] [CrossRef]
- Sakata, K.; Woo, N.H.; Martinowich, K.; Greene, J.S.; Schloesser, R.J.; Shen, L.; Lu, B. Critical role of promoter IV-driven BDNF transcription in GABAergic transmission and synaptic plasticity in the prefrontal cortex. Proc. Natl. Acad. Sci. USA 2009, 106, 5942–5947. [Google Scholar] [CrossRef] [Green Version]
- Keller, S.; Sarchiapone, M.; Zarrilli, F.; Videtic, A.; Ferraro, A.; Carli, V.; Sacchetti, S.; Lembo, F.; Angiolillo, A.; Jovanovic, N.; et al. Increased BDNF promoter methylation in the Wernicke area of suicide subjects. Arch. Gen. Psychiatry 2010, 67, 258–267. [Google Scholar] [CrossRef]
- Tadić, A.; Müller-Engling, L.; Schlicht, K.F.; Kotsiari, A.; Dreimüller, N.; Kleimann, A.; Bleich, S.; Lieb, K.; Frieling, H. Methylation of the promoter of brain-derived neurotrophic factor exon IV and antidepressant response in major depression. Mol. Psychiatry 2014, 19, 281–283. [Google Scholar] [CrossRef]
- Lieb, K.; Dreimüller, N.; Wagner, S.; Schlicht, K.; Falter, T.; Neyazi, A.; Müller-Engling, L.; Bleich, S.; Tadić, A.; Frieling, H. BDNF Plasma Levels and BDNF Exon IV Promoter Methylation as Predictors for Antidepressant Treatment Response. Front. Psychiatry 2018, 9, 511. [Google Scholar] [CrossRef] [Green Version]
- Sakata, K.; Duke, S.M. Lack of BDNF expression through promoter IV disturbs expression of monoamine genes in the frontal cortex and hippocampus. Neuroscience 2014, 260, 265–275. [Google Scholar] [CrossRef]
- Hill, J.L.; Hardy, N.F.; Jimenez, D.V.; Maynard, K.R.; Kardian, A.S.; Pollock, C.J.; Schloesser, R.J.; Martinowich, K. Loss of promoter IV-driven BDNF expression impacts oscillatory activity during sleep, sensory information processing and fear regulation. Transl. Psychiatry 2016, 6, e873. [Google Scholar] [CrossRef]
- Volpicelli, F.; Speranza, L.; Pulcrano, S.; De Gregorio, R.; Crispino, M.; De Sanctis, C.; Leopoldo, M.; Lacivita, E.; di Porzio, U.; Bellenchi, G.C.; et al. The microRNA-29a Modulates Serotonin 5-HT7 Receptor Expression and Its Effects on Hippocampal Neuronal Morphology. Mol. Neurobiol. 2019, 56, 8617–8627. [Google Scholar] [CrossRef] [PubMed]
- De Gregorio, R.; Pulcrano, S.; De Sanctis, C.; Volpicelli, F.; Guatteo, E.; von Oerthel, L.; Latagliata, E.C.; Esposito, R.; Piscitelli, R.M.; Perrone-Capano, C.; et al. miR-34b/c Regulates Wnt1 and Enhances Mesencephalic Dopaminergic Neuron Differentiation. Stem Cell Rep. 2018, 10, 1237–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNeill, E.; Van Vactor, D. MicroRNAs shape the neuronal landscape. Neuron 2012, 75, 363–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keifer, J.; Zheng, Z.; Ambigapathy, G. A MicroRNA-BDNF Negative Feedback Signaling Loop in Brain: Implications for Alzheimer′s Disease. Microrna 2015, 4, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Khani-Habibabadi, F.; Askari, S.; Zahiri, J.; Javan, M.; Behmanesh, M. Novel BDNF-regulatory microRNAs in neurodegenerative disorders pathogenesis: An in silico study. Comput. Biol. Chem. 2019, 83, 107153. [Google Scholar] [CrossRef] [PubMed]
- Schratt, G.M.; Tuebing, F.; Nigh, E.A.; Kane, C.G.; Sabatini, M.E.; Kiebler, M.; Greenberg, M.E. A brain-specific microRNA regulates dendritic spine development. Nature 2006, 439, 283–289. [Google Scholar] [CrossRef]
- Baby, N.; Alagappan, N.; Dheen, S.T.; Sajikumar, S. MicroRNA-134-5p inhibition rescues long-term plasticity and synaptic tagging/capture in an Aβ(1-42)-induced model of Alzheimer’s disease. Aging Cell 2020, 19, e13046. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Wang, W.Y.; Mao, Y.W.; Gräff, J.; Guan, J.S.; Pan, L.; Mak, G.; Kim, D.; Su, S.C.; Tsai, L.H. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 2010, 466, 1105–1109. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Xu, L.; Qu, C.; Sun, H.; Zhang, J. Resveratrol prevents cognitive deficits induced by chronic unpredictable mild stress: Sirt1/miR-134 signalling pathway regulates CREB/BDNF expression in hippocampus in vivo and in vitro. Behav. Brain Res. 2018, 349, 1–7. [Google Scholar] [CrossRef]
- Xin, C.; Xia, J.; Liu, Y.; Zhang, Y. MicroRNA-202-3p Targets Brain-Derived Neurotrophic Factor and Is Involved in Depression-Like Behaviors. Neuropsychiatr. Dis. Treat. 2020, 16, 1073–1083. [Google Scholar] [CrossRef]
- Yang, C.R.; Zhang, X.Y.; Liu, Y.; Du, J.Y.; Liang, R.; Yu, M.; Zhang, F.Q.; Mu, X.F.; Li, F.; Zhou, L.; et al. Antidepressant Drugs Correct the Imbalance Between proBDNF/p75NTR/Sortilin and Mature BDNF/TrkB in the Brain of Mice with Chronic Stress. Neurotox. Res. 2020, 37, 171–182. [Google Scholar] [CrossRef] [PubMed]
- You, Y.H.; Qin, Z.Q.; Zhang, H.L.; Yuan, Z.H.; Yu, X. MicroRNA-153 promotes brain-derived neurotrophic factor and hippocampal neuron proliferation to alleviate autism symptoms through inhibition of JAK-STAT pathway by LEPR. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Rostamian Delavar, M.; Baghi, M.; Safaeinejad, Z.; Kiani-Esfahani, A.; Ghaedi, K.; Nasr-Esfahani, M.H. Differential expression of miR-34a, miR-141, and miR-9 in MPP+-treated differentiated PC12 cells as a model of Parkinson′s disease. Gene 2018, 662, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Shu, F.; Wang, X.; Wang, F.; Wu, L.; Li, L.; Lv, H. Inhibition of microRNA-375 ameliorated ketamine-induced neurotoxicity in human embryonic stem cell derived neurons. Eur. J. Pharmacol. 2019, 844, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.D.; Zheng, X.C.; Huang, F.Y.; Gao, F.; You, M.Z.; Zheng, T. MicroRNA-107 regulates anesthesia-induced neural injury in embryonic stem cell derived neurons. IUBMB Life 2019, 71, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barde, Y.A.; Edgar, D.; Thoenen, H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982, 1, 549–553. [Google Scholar] [CrossRef]
- Foltran, R.B.; Diaz, S.L. BDNF isoforms: A round trip ticket between neurogenesis and serotonin? J. Neurochem. 2016, 138, 204–221. [Google Scholar] [CrossRef]
- Mizui, T.; Ishikawa, Y.; Kumanogoh, H.; Kojima, M. Neurobiological actions by three distinct subtypes of brain-derived neurotrophic factor: Multi-ligand model of growth factor signaling. Pharmacol. Res. 2016, 105, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Wu, Y.; Sousa, N.; Almeida, O.F. SMAD pathway mediation of BDNF and TGF beta 2 regulation of proliferation and differentiation of hippocampal granule neurons. Development 2005, 132, 3231–3242. [Google Scholar] [CrossRef] [Green Version]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef]
- Pang, P.T.; Teng, H.K.; Zaitsev, E.; Woo, N.T.; Sakata, K.; Zhen, S.; Teng, K.K.; Yung, W.H.; Hempstead, B.L.; Lu, B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science 2004, 306, 487–491. [Google Scholar] [CrossRef]
- Mizoguchi, H.; Nakade, J.; Tachibana, M.; Ibi, D.; Someya, E.; Koike, H.; Kamei, H.; Nabeshima, T.; Itohara, S.; Takuma, K.; et al. Matrix metalloproteinase-9 contributes to kindled seizure development in pentylenetetrazole-treated mice by converting pro-BDNF to mature BDNF in the hippocampus. J. Neurosci. 2011, 31, 12963–12971. [Google Scholar] [CrossRef]
- Vafadari, B.; Salamian, A.; Kaczmarek, L. MMP-9 in translation: From molecule to brain physiology, pathology, and therapy. J. Neurochem. 2016, 139 (Suppl. S2), 91–114. [Google Scholar] [CrossRef] [Green Version]
- Mowla, S.J.; Farhadi, H.F.; Pareek, S.; Atwal, J.K.; Morris, S.J.; Seidah, N.G.; Murphy, R.A. Biosynthesis and post-translational processing of the precursor to brain-derived neurotrophic factor. J. Biol. Chem. 2001, 276, 12660–12666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conner, J.M.; Lauterborn, J.C.; Yan, Q.; Gall, C.M.; Varon, S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: Evidence for anterograde axonal transport. J. Neurosci. 1997, 17, 2295–2313. [Google Scholar] [CrossRef] [Green Version]
- Dieni, S.; Matsumoto, T.; Dekkers, M.; Rauskolb, S.; Ionescu, M.S.; Deogracias, R.; Gundelfinger, E.D.; Kojima, M.; Nestel, S.; Frotscher, M.; et al. BDNF and its pro-peptide are stored in presynaptic dense core vesicles in brain neurons. J. Cell Biol. 2012, 196, 775–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Siao, C.J.; Nagappan, G.; Marinic, T.; Jing, D.; McGrath, K.; Chen, Z.Y.; Mark, W.; Tessarollo, L.; Lee, F.S.; et al. Neuronal release of proBDNF. Nat. Neurosci. 2009, 12, 113–115. [Google Scholar] [CrossRef]
- Yang, J.L.; Lin, Y.T.; Chuang, P.C.; Bohr, V.A.; Mattson, M.P. BDNF and exercise enhance neuronal DNA repair by stimulating CREB-mediated production of apurinic/apyrimidinic endonuclease 1. Neuromolecular Med. 2014, 16, 161–174. [Google Scholar] [CrossRef]
- Du, J.; Feng, L.; Yang, F.; Lu, B. Activity- and Ca(2+)-dependent modulation of surface expression of brain-derived neurotrophic factor receptors in hippocampal neurons. J. Cell Biol. 2000, 150, 1423–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer-Franke, A.; Wilkinson, G.A.; Kruttgen, A.; Hu, M.; Munro, E.; Hanson, M.G.; Reichardt, L.F.; Barres, B.A. Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron 1998, 21, 681–693. [Google Scholar] [CrossRef] [Green Version]
- Deinhardt, K.; Chao, M.V. Shaping neurons: Long and short range effects of mature and proBDNF signalling upon neuronal structure. Neuropharmacology 2014, 76 Pt C, 603–609. [Google Scholar] [CrossRef] [Green Version]
- Anastasia, A.; Deinhardt, K.; Chao, M.V.; Will, N.E.; Irmady, K.; Lee, F.S.; Hempstead, B.L.; Bracken, C. Val66Met polymorphism of BDNF alters prodomain structure to induce neuronal growth cone retraction. Nat. Commun. 2013, 4, 2490. [Google Scholar] [CrossRef] [Green Version]
- Teng, H.K.; Teng, K.K.; Lee, R.; Wright, S.; Tevar, S.; Almeida, R.D.; Kermani, P.; Torkin, R.; Chen, Z.Y.; Lee, F.S.; et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J. Neurosci. 2005, 25, 5455–5463. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Colicos, M.A.; Barker, P.A.; Kennedy, T.E. p75 neurotrophin receptor expression is induced in apoptotic neurons after seizure. J. Neurosci. 1999, 19, 6887–6896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichardt, L.F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 1545–1564. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, D.R.; Miller, F.D. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 2000, 10, 381–391. [Google Scholar] [CrossRef]
- Gonzalez, A.; Moya-Alvarado, G.; Gonzalez-Billaut, C.; Bronfman, F.C. Cellular and molecular mechanisms regulating neuronal growth by brain-derived neurotrophic factor. Cytoskeleton (Hoboken) 2016, 73, 612–628. [Google Scholar] [CrossRef] [Green Version]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [Green Version]
- Minichiello, L. TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci. 2009, 10, 850–860. [Google Scholar] [CrossRef]
- Patel, A.V.; Krimm, R.F. BDNF is required for the survival of differentiated geniculate ganglion neurons. Dev. Biol. 2010, 340, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Shehadah, A.; Pal, A.; Zacharek, A.; Cui, X.; Cui, Y.; Roberts, C.; Lu, M.; Zeitlin, A.; Hariri, R.; et al. Neuroprotective effect of human placenta-derived cell treatment of stroke in rats. Cell Transplant. 2013, 22, 871–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Wang, R.; Wang, Y.; Gao, J.; Feng, L.; Yang, Z. Distinct Impacts of Fullerene on Cognitive Functions of Dementia vs. Non-dementia Mice. Neurotox. Res. 2019, 36, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Gorski, J.A.; Zeiler, S.R.; Tamowski, S.; Jones, K.R. Brain-derived neurotrophic factor is required for the maintenance of cortical dendrites. J. Neurosci. 2003, 23, 6856–6865. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; Fernández, J.R.; Zegarek, G.F.; Lo, S.B.; Firestein, B.L. BDNF-promoted increases in proximal dendrites occur via CREB-dependent transcriptional regulation of cypin. J. Neurosci. 2011, 31, 9735–9745. [Google Scholar] [CrossRef] [Green Version]
- Orefice, L.L.; Waterhouse, E.G.; Partridge, J.G.; Lalchandani, R.R.; Vicini, S.; Xu, B. Distinct roles for somatically and dendritically synthesized brain-derived neurotrophic factor in morphogenesis of dendritic spines. J. Neurosci. 2013, 33, 11618–11632. [Google Scholar] [CrossRef] [Green Version]
- Zagrebelsky, M.; Tacke, C.; Korte, M. BDNF signaling during the lifetime of dendritic spines. Cell Tissue Res. 2020. [Google Scholar] [CrossRef]
- Yoshii, A.; Constantine-Paton, M. Postsynaptic localization of PSD-95 is regulated by all three pathways downstream of TrkB signaling. Front. Synaptic Neurosci. 2014, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.D.; Wu, C.L.; Hwang, W.C.; Yang, D.I. More Insight into BDNF against Neurodegeneration: Anti-Apoptosis, Anti-Oxidation, and Suppression of Autophagy. Int. J. Mol. Sci. 2017, 18, 545. [Google Scholar] [CrossRef] [Green Version]
- Opazo, P.; Watabe, A.M.; Grant, S.G.; O’Dell, T.J. Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. J. Neurosci. 2003, 23, 3679–3688. [Google Scholar] [CrossRef] [Green Version]
- Leal, G.; Bramham, C.R.; Duarte, C.B. BDNF and Hippocampal Synaptic Plasticity. Vitam. Horm. 2017, 104, 153–195. [Google Scholar] [CrossRef]
- Baydyuk, M.; Xu, B. BDNF signaling and survival of striatal neurons. Front. Cell. Neurosci. 2014, 8, 254. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, E.G.; An, J.J.; Orefice, L.L.; Baydyuk, M.; Liao, G.Y.; Zheng, K.; Lu, B.; Xu, B. BDNF promotes differentiation and maturation of adult-born neurons through GABAergic transmission. J. Neurosci. 2012, 32, 14318–14330. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Poo, M.M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef]
- Jaworski, J.; Spangler, S.; Seeburg, D.P.; Hoogenraad, C.C.; Sheng, M. Control of dendritic arborization by the phosphoinositide-3’-kinase-Akt-mammalian target of rapamycin pathway. J. Neurosci. 2005, 25, 11300–11312. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Zhang, M.X.; Swank, M.W.; Kunz, J.; Wu, G.Y. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J. Neurosci. 2005, 25, 11288–11299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkbeiner, S.; Tavazoie, S.F.; Maloratsky, A.; Jacobs, K.M.; Harris, K.M.; Greenberg, M.E. CREB: A major mediator of neuronal neurotrophin responses. Neuron 1997, 19, 1031–1047. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Kornhauser, J.M.; Xia, Z.; Thiele, E.A.; Greenberg, M.E. Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate CREB serine 133 phosphorylation. Mol. Cell. Biol. 1998, 18, 1946–1955. [Google Scholar] [CrossRef] [Green Version]
- Segal, M.; Kreher, U.; Greenberger, V.; Braun, K. Is fragile X mental retardation protein involved in activity-induced plasticity of dendritic spines? Brain Res. 2003, 972, 9–15. [Google Scholar] [CrossRef]
- Petryshen, T.L.; Sabeti, P.C.; Aldinger, K.A.; Fry, B.; Fan, J.B.; Schaffner, S.F.; Waggoner, S.G.; Tahl, A.R.; Sklar, P. Population genetic study of the brain-derived neurotrophic factor (BDNF) gene. Mol. Psychiatry 2010, 15, 810–815. [Google Scholar] [CrossRef] [Green Version]
- Momose, Y.; Murata, M.; Kobayashi, K.; Tachikawa, M.; Nakabayashi, Y.; Kanazawa, I.; Toda, T. Association studies of multiple candidate genes for Parkinson’s disease using single nucleotide polymorphisms. Ann. Neurol. 2002, 51, 133–136. [Google Scholar] [CrossRef]
- Ventriglia, M.; Bocchio Chiavetto, L.; Benussi, L.; Binetti, G.; Zanetti, O.; Riva, M.A.; Gennarelli, M. Association between the BDNF 196 A/G polymorphism and sporadic Alzheimer’s disease. Mol. Psychiatry 2002, 7, 136–137. [Google Scholar] [CrossRef] [Green Version]
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003, 112, 257–269. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.Y.; Patel, P.D.; Sant, G.; Meng, C.X.; Teng, K.K.; Hempstead, B.L.; Lee, F.S. Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. J. Neurosci. 2004, 24, 4401–4411. [Google Scholar] [CrossRef] [PubMed]
- Chiaruttini, C.; Vicario, A.; Li, Z.; Baj, G.; Braiuca, P.; Wu, Y.; Lee, F.S.; Gardossi, L.; Baraban, J.M.; Tongiorgi, E. Dendritic trafficking of BDNF mRNA is mediated by translin and blocked by the G196A (Val66Met) mutation. Proc. Natl. Acad. Sci. USA 2009, 106, 16481–16486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.J. Salivary gland low-intensity pulsed ultrasound (LIPUS) stimulation as a potential treatment for various BDNF-implicated neuropsychiatric disorders. Med. Hypotheses 2020, 137, 109560. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Huang, T.L. Brain-derived neurotrophic factor and mental disorders. Biomed. J. 2020, 43, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Di Lazzaro, V.; Pellegrino, G.; Di Pino, G.; Corbetto, M.; Ranieri, F.; Brunelli, N.; Paolucci, M.; Bucossi, S.; Ventriglia, M.C.; Brown, P.; et al. Val66Met BDNF gene polymorphism influences human motor cortex plasticity in acute stroke. Brain Stimul. 2015, 8, 92–96. [Google Scholar] [CrossRef] [Green Version]
- Morin-Moncet, O.; Latulipe-Loiselle, A.; Therrien-Blanchet, J.M.; Theoret, H. BDNF Val66Met polymorphism is associated with altered activity-dependent modulation of short-interval intracortical inhibition in bilateral M1. PLoS ONE 2018, 13, e0197505. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T.; Fukui, K.; Takeuchi, H.; Yokota, S.; Kikuchi, Y.; Tomita, H.; Taki, Y.; Kawashima, R. Effects of the BDNF Val66Met Polymorphism on Gray Matter Volume in Typically Developing Children and Adolescents. Cereb. Cortex 2016, 26, 1795–1803. [Google Scholar] [CrossRef] [Green Version]
- Meng, W.D.; Sun, S.J.; Yang, J.; Chu, R.X.; Tu, W.; Liu, Q. Elevated Serum Brain-Derived Neurotrophic Factor (BDNF) but not BDNF Gene Val66Met Polymorphism Is Associated with Autism Spectrum Disorders. Mol. Neurobiol. 2017, 54, 1167–1172. [Google Scholar] [CrossRef]
- Montag, C.; Basten, U.; Stelzel, C.; Fiebach, C.J.; Reuter, M. The BDNF Val66Met polymorphism and anxiety: Support for animal knock-in studies from a genetic association study in humans. Psychiatry Res. 2010, 179, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Kim, J.; Namgung, E.; Lee, D.W.; Kim, G.H.; Kim, M.; Kim, N.; Kim, T.D.; Kim, S.; Lyoo, I.K.; et al. The BDNF Val66Met Polymorphism Affects the Vulnerability of the Brain Structural Network. Front. Hum. Neurosci. 2017, 11, 400. [Google Scholar] [CrossRef] [PubMed]
- Hariri, A.R.; Goldberg, T.E.; Mattay, V.S.; Kolachana, B.S.; Callicott, J.H.; Egan, M.F.; Weinberger, D.R. Brain-derived neurotrophic factor val66met polymorphism affects human memory-related hippocampal activity and predicts memory performance. J. Neurosci. 2003, 23, 6690–6694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezawas, L.; Verchinski, B.A.; Mattay, V.S.; Callicott, J.H.; Kolachana, B.S.; Straub, R.E.; Egan, M.F.; Meyer-Lindenberg, A.; Weinberger, D.R. The brain-derived neurotrophic factor val66met polymorphism and variation in human cortical morphology. J. Neurosci. 2004, 24, 10099–10102. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.C.; Milev, P.; O’Leary, D.S.; Librant, A.; Andreasen, N.C.; Wassink, T.H. Cognitive and magnetic resonance imaging brain morphometric correlates of brain-derived neurotrophic factor Val66Met gene polymorphism in patients with schizophrenia and healthy volunteers. Arch. Gen. Psychiatry 2006, 63, 731–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montag, C.; Weber, B.; Fliessbach, K.; Elger, C.; Reuter, M. The BDNF Val66Met polymorphism impacts parahippocampal and amygdala volume in healthy humans: Incremental support for a genetic risk factor for depression. Psychol. Med. 2009, 39, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Du, X.; Yin, G.; Zhang, Y.; Li, X.; Cai, J.; Huang, X.; Ning, Y.; Soares, J.C.; Wu, F.; et al. Effects of smoking on cognition and BDNF levels in a male Chinese population: Relationship with BDNF Val66Met polymorphism. Sci. Rep. 2019, 9, 217. [Google Scholar] [CrossRef] [Green Version]
- Altmann, V.; Schumacher-Schuh, A.F.; Rieck, M.; Callegari-Jacques, S.M.; Rieder, C.R.; Hutz, M.H. Val66Met BDNF polymorphism is associated with Parkinson’s disease cognitive impairment. Neurosci. Lett. 2016, 615, 88–91. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, J.; Guo, Y.; Dong, G.; Zou, W.; Chen, Z. Association between BDNF G196A (Val66Met) polymorphism and cognitive impairment in patients with Parkinson’s disease: A meta-analysis. Braz. J. Med. Biol. Res. 2019, 52, e8443. [Google Scholar] [CrossRef] [Green Version]
- Franzmeier, N.; Ren, J.; Damm, A.; Monté-Rubio, G.; Boada, M.; Ruiz, A.; Ramirez, A.; Jessen, F.; Düzel, E.; Rodríguez Gómez, O.; et al. The BDNF. Mol. Psychiatry 2019. [Google Scholar] [CrossRef]
- Yin, Y.; Su, X.; Pan, L.; Li, C. BDNF Val66Met polymorphism and cognitive impairment in Parkinson’s disease-a meta-analysis. Neurol. Sci. 2019, 40, 1901–1907. [Google Scholar] [CrossRef] [PubMed]
- Dalby, R.B.; Elfving, B.; Poulsen, P.H.; Foldager, L.; Frandsen, J.; Videbech, P.; Rosenberg, R. Plasma brain-derived neurotrophic factor and prefrontal white matter integrity in late-onset depression and normal aging. Acta Psychiatr. Scand. 2013, 128, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Han, K.M.; Won, E.; Yoon, B.J.; Lee, M.S.; Ham, B.J. Association of brain-derived neurotrophic factor DNA methylation and reduced white matter integrity in the anterior corona radiata in major depression. J. Affect. Disord. 2015, 172, 74–80. [Google Scholar] [CrossRef]
- Youssef, M.M.; Underwood, M.D.; Huang, Y.Y.; Hsiung, S.C.; Liu, Y.; Simpson, N.R.; Bakalian, M.J.; Rosoklija, G.B.; Dwork, A.J.; Arango, V.; et al. Association of BDNF Val66Met Polymorphism and Brain BDNF Levels with Major Depression and Suicide. Int. J. Neuropsychopharmacol. 2018, 21, 528–538. [Google Scholar] [CrossRef]
- Caldieraro, M.A.; McKee, M.; Leistner-Segal, S.; Vares, E.A.; Kubaski, F.; Spanemberg, L.; Brusius-Facchin, A.C.; Fleck, M.P.; Mischoulon, D. Val66Met polymorphism association with serum BDNF and inflammatory biomarkers in major depression. World J. Biol. Psychiatry 2018, 19, 402–409. [Google Scholar] [CrossRef]
- Rong, C.; Park, C.; Rosenblat, J.D.; Subramaniapillai, M.; Zuckerman, H.; Fus, D.; Lee, Y.L.; Pan, Z.; Brietzke, E.; Mansur, R.B.; et al. Predictors of Response to Ketamine in Treatment Resistant Major Depressive Disorder and Bipolar Disorder. Int. J. Environ. Res. Public Health 2018, 15, 771. [Google Scholar] [CrossRef] [Green Version]
- Mandolini, G.M.; Lazzaretti, M.; Pigoni, A.; Delvecchio, G.; Soares, J.C.; Brambilla, P. The impact of BDNF Val66Met polymorphism on cognition in Bipolar Disorder: A review: Special Section on ″Translational and Neuroscience Studies in Affective Disorders″Section Editor, Maria Nobile MD, PhD. This Section of JAD focuses on the relevance of translational and neuroscience studies in providing a better understanding of the neural basis of affective disorders. The main aim is to briefly summaries relevant research findings in clinical neuroscience with particular regards to specific innovative topics in mood and anxiety disorders. J. Affect. Disord. 2019, 243, 552–558. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.C.; Chuang, Y.C.; Huang, C.W.; Lui, C.C.; Lee, C.C.; Hsu, S.W.; Lin, P.H.; Lu, Y.T.; Chang, Y.T.; Hsu, C.W.; et al. Interictal serum brain-derived neurotrophic factor level reflects white matter integrity, epilepsy severity, and cognitive dysfunction in chronic temporal lobe epilepsy. Epilepsy Behav. 2016, 59, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, M.K.; Thompson, P.J.; Wandschneider, B.; Foulkes, A.; de Tisi, J.; Stretton, J.; Perona, M.; Thom, M.; Bonelli, S.B.; Burdett, J.; et al. The impact of brain-derived neurotrophic factor Val66Met polymorphism on cognition and functional brain networks in patients with intractable partial epilepsy. CNS Neurosci. Ther. 2019, 25, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Doherty, C.; Hogue, O.; Floden, D.P.; Altemus, J.B.; Najm, I.M.; Eng, C.; Busch, R.M. BDNF and COMT, but not APOE, alleles are associated with psychiatric symptoms in refractory epilepsy. Epilepsy Behav. 2019, 94, 131–136. [Google Scholar] [CrossRef]
- Kheirollahi, M.; Kazemi, E.; Ashouri, S. Brain-Derived Neurotrophic Factor Gene Val66Met Polymorphism and Risk of Schizophrenia: A Meta-analysis of Case-Control Studies. Cell. Mol. Neurobiol. 2016, 36, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Zhang, G.; Du, X.; Zhang, Y.; Yin, G.; Dai, J.; He, M.X.; Soares, J.C.; Li, X.; Zhang, X.Y. Suicide attempt, clinical correlates, and BDNF Val66Met polymorphism in chronic patients with schizophrenia. Neuropsychology 2018, 32, 199–205. [Google Scholar] [CrossRef]
- Huang, E.; Hettige, N.C.; Zai, G.; Tomasi, J.; Huang, J.; Zai, C.C.; Pivac, N.; Nikolac Perkovic, M.; Tiwari, A.K.; Kennedy, J.L. BDNF Val66Met polymorphism and clinical response to antipsychotic treatment in schizophrenia and schizoaffective disorder patients: A meta-analysis. Pharmacogenomics J. 2019, 19, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Deng, C. BDNF as a pharmacogenetic target for antipsychotic treatment of schizophrenia. Neurosci. Lett. 2020, 726, 133870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.; Machan, J.T.; Hayes, L.; Zervas, M. Molecular organization and timing of Wnt1 expression define cohorts of midbrain dopamine neuron progenitors in vivo. J. Comp. Neurol. 2011, 519, 2978–3000. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Cejudo, J.; Gutiérrez-Fernández, M.; Otero-Ortega, L.; Rodríguez-Frutos, B.; Fuentes, B.; Vallejo-Cremades, M.T.; Hernanz, T.N.; Cerdán, S.; Díez-Tejedor, E. Brain-derived neurotrophic factor administration mediated oligodendrocyte differentiation and myelin formation in subcortical ischemic stroke. Stroke 2015, 46, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Balkaya, M.; Cho, S. Genetics of stroke recovery: BDNF val66met polymorphism in stroke recovery and its interaction with aging. Neurobiol. Dis. 2019, 126, 36–46. [Google Scholar] [CrossRef]
- Lohia, R.; Salari, R.; Brannigan, G. Sequence specificity despite intrinsic disorder: How a disease-associated Val/Met polymorphism rearranges tertiary interactions in a long disordered protein. PLoS Comput. Biol. 2019, 15, e1007390. [Google Scholar] [CrossRef]
- Rock, P.L.; Roiser, J.P.; Riedel, W.J.; Blackwell, A.D. Cognitive impairment in depression: A systematic review and meta-analysis. Psychol. Med. 2014, 44, 2029–2040. [Google Scholar] [CrossRef] [Green Version]
- Severino, V.; Farina, A.; Colucci-D’Amato, L.; Reccia, M.G.; Volpicelli, F.; Parente, A.; Chambery, A. Secretome profiling of differentiated neural mes-c-myc A1 cell line endowed with stem cell properties. Biochim. Biophys. Acta 2013, 1834, 2385–2395. [Google Scholar] [CrossRef]
- Colucci-D’Amato, L.; Farina, A.; Vissers, J.P.; Chambery, A. Quantitative neuroproteomics: Classical and novel tools for studying neural differentiation and function. Stem Cell Rev. Rep. 2011, 7, 77–93. [Google Scholar] [CrossRef]
- Di Lieto, A.; Leo, D.; Volpicelli, F.; di Porzio, U.; Colucci-D’Amato, L. FLUOXETINE modifies the expression of serotonergic markers in a differentiation-dependent fashion in the mesencephalic neural cell line A1 mes c-myc. Brain Res. 2007, 1143, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nawa, Y.; Kaneko, H.; Oda, M.; Tsubonoya, M.; Hiroi, T.; Gentile, M.T.; Colucci-D’Amato, L.; Takahashi, R.; Matsui, H. Functional characterization of the neuron-restrictive silencer element in the human tryptophan hydroxylase 2 gene expression. J. Neurochem. 2017, 142, 827–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentile, M.T.; Nawa, Y.; Lunardi, G.; Florio, T.; Matsui, H.; Colucci-D’Amato, L. Tryptophan hydroxylase 2 (TPH2) in a neuronal cell line: Modulation by cell differentiation and NRSF/rest activity. J. Neurochem. 2012, 123, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Heninger, G.R.; Nestler, E.J. A molecular and cellular theory of depression. Arch. Gen. Psychiatry 1997, 54, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Colucci-D’Amato, L.; di Porzio, U. Neurogenesis in adult CNS: From denial to opportunities and challenges for therapy. Bioessays 2008, 30, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Colucci-D’Amato, L.; Bonavita, V.; di Porzio, U. The end of the central dogma of neurobiology: Stem cells and neurogenesis in adult CNS. Neurol. Sci. 2006, 27, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Cramer, S.C.; Sur, M.; Dobkin, B.H.; O’Brien, C.; Sanger, T.D.; Trojanowski, J.Q.; Rumsey, J.M.; Hicks, R.; Cameron, J.; Chen, D.; et al. Harnessing neuroplasticity for clinical applications. Brain 2011, 134, 1591–1609. [Google Scholar] [CrossRef]
- Castrén, E. Neuronal network plasticity and recovery from depression. JAMA Psychiatry 2013, 70, 983–989. [Google Scholar] [CrossRef]
- Snapyan, M.; Lemasson, M.; Brill, M.S.; Blais, M.; Massouh, M.; Ninkovic, J.; Gravel, C.; Berthod, F.; Götz, M.; Barker, P.A.; et al. Vasculature guides migrating neuronal precursors in the adult mammalian forebrain via brain-derived neurotrophic factor signaling. J. Neurosci. 2009, 29, 4172–4188. [Google Scholar] [CrossRef] [Green Version]
- Bergami, M.; Rimondini, R.; Santi, S.; Blum, R.; Götz, M.; Canossa, M. Deletion of TrkB in adult progenitors alters newborn neuron integration into hippocampal circuits and increases anxiety-like behavior. Proc. Natl. Acad. Sci. USA 2008, 105, 15570–15575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.L.; Tong, K.Y.; Yip, S.P. Relationship of serum brain-derived neurotrophic factor (BDNF) and health-related lifestyle in healthy human subjects. Neurosci. Lett. 2008, 447, 124–128. [Google Scholar] [CrossRef]
- Zafra, F.; Lindholm, D.; Castrén, E.; Hartikka, J.; Thoenen, H. Regulation of brain-derived neurotrophic factor and nerve growth factor mRNA in primary cultures of hippocampal neurons and astrocytes. J. Neurosci. 1992, 12, 4793–4799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castrén, E.; Antila, H. Neuronal plasticity and neurotrophic factors in drug responses. Mol. Psychiatry 2017, 22, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Castrén, E.; Zafra, F.; Thoenen, H.; Lindholm, D. Light regulates expression of brain-derived neurotrophic factor mRNA in rat visual cortex. Proc. Natl. Acad. Sci. USA 1992, 89, 9444–9448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianfranceschi, L.; Siciliano, R.; Walls, J.; Morales, B.; Kirkwood, A.; Huang, Z.J.; Tonegawa, S.; Maffei, L. Visual cortex is rescued from the effects of dark rearing by overexpression of BDNF. Proc. Natl. Acad. Sci. USA 2003, 100, 12486–12491. [Google Scholar] [CrossRef] [Green Version]
- Hensch, T.K.; Bilimoria, P.M. Re-opening Windows: Manipulating Critical Periods for Brain Development. Cerebrum 2012, 2012, 11. [Google Scholar]
- Hong, E.J.; McCord, A.E.; Greenberg, M.E. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron 2008, 60, 610–624. [Google Scholar] [CrossRef] [Green Version]
- Volpicelli, F.; Speranza, L.; di Porzio, U.; Crispino, M.; Perrone-Capano, C. The serotonin receptor 7 and the structural plasticity of brain circuits. Front. Behav. Neurosci. 2014, 8, 318. [Google Scholar] [CrossRef] [Green Version]
- Cazorla, M.; Arrang, J.M.; Prémont, J. Pharmacological characterization of six trkB antibodies reveals a novel class of functional agents for the study of the BDNF receptor. Br. J. Pharmacol. 2011, 162, 947–960. [Google Scholar] [CrossRef] [Green Version]
- Park, S.W.; Nhu, l.H.; Cho, H.Y.; Seo, M.K.; Lee, C.H.; Ly, N.N.; Choi, C.M.; Lee, B.J.; Kim, G.M.; Seol, W.; et al. p11 mediates the BDNF-protective effects in dendritic outgrowth and spine formation in B27-deprived primary hippocampal cells. J. Affect. Disord. 2016, 196, 1–10. [Google Scholar] [CrossRef]
- Katoh-Semba, R.; Asano, T.; Ueda, H.; Morishita, R.; Takeuchi, I.K.; Inaguma, Y.; Kato, K. Riluzole enhances expression of brain-derived neurotrophic factor with consequent proliferation of granule precursor cells in the rat hippocampus. FASEB J. 2002, 16, 1328–1330. [Google Scholar] [CrossRef]
- Cavanaugh, J.E.; Ham, J.; Hetman, M.; Poser, S.; Yan, C.; Xia, Z. Differential regulation of mitogen-activated protein kinases ERK1/2 and ERK5 by neurotrophins, neuronal activity, and cAMP in neurons. J. Neurosci. 2001, 21, 434–443. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.; Cavanaugh, J.E.; Leak, R.K.; Perez, R.G.; Zigmond, M.J. Rapid activation of ERK by 6-hydroxydopamine promotes survival of dopaminergic cells. J. Neurosci. Res. 2008, 86, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Rauti, R.; Cellot, G.; D’Andrea, P.; Colliva, A.; Scaini, D.; Tongiorgi, E.; Ballerini, L. BDNF impact on synaptic dynamics: Extra or intracellular long-term release differently regulates cultured hippocampal synapses. Mol. Brain 2020, 13, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernfors, P.; Lee, K.F.; Jaenisch, R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature 1994, 368, 147–150. [Google Scholar] [CrossRef]
- Gao, X.; Smith, G.M.; Chen, J. Impaired dendritic development and synaptic formation of postnatal-born dentate gyrus granular neurons in the absence of brain-derived neurotrophic factor signaling. Exp. Neurol. 2009, 215, 178–190. [Google Scholar] [CrossRef]
- Mariga, A.; Zavadil, J.; Ginsberg, S.D.; Chao, M.V. Withdrawal of BDNF from hippocampal cultures leads to changes in genes involved in synaptic function. Dev. Neurobiol. 2015, 75, 173–192. [Google Scholar] [CrossRef] [Green Version]
- Aarse, J.; Herlitze, S.; Manahan-Vaughan, D. The requirement of BDNF for hippocampal synaptic plasticity is experience-dependent. Hippocampus 2016, 26, 739–751. [Google Scholar] [CrossRef] [Green Version]
- Magariños, A.M.; Li, C.J.; Gal Toth, J.; Bath, K.G.; Jing, D.; Lee, F.S.; McEwen, B.S. Effect of brain-derived neurotrophic factor haploinsufficiency on stress-induced remodeling of hippocampal neurons. Hippocampus 2011, 21, 253–264. [Google Scholar] [CrossRef] [Green Version]
- von Bohlen Und Halbach, O.; von Bohlen Und Halbach, V. BDNF effects on dendritic spine morphology and hippocampal function. Cell Tissue Res. 2018, 373, 729–741. [Google Scholar] [CrossRef]
- von Bohlen und Halbach, O.; Minichiello, L.; Unsicker, K. TrkB but not trkC receptors are necessary for postnatal maintenance of hippocampal spines. Neurobiol. Aging 2008, 29, 1247–1255. [Google Scholar] [CrossRef]
- Dokter, M.; Busch, R.; Poser, R.; Vogt, M.A.; von Bohlen Und Halbach, V.; Gass, P.; Unsicker, K.; von Bohlen Und Halbach, O. Implications of p75NTR for dentate gyrus morphology and hippocampus-related behavior revisited. Brain Struct. Funct. 2015, 220, 1449–1462. [Google Scholar] [CrossRef]
- Lin, P.Y.; Kavalali, E.T.; Monteggia, L.M. Genetic Dissection of Presynaptic and Postsynaptic BDNF-TrkB Signaling in Synaptic Efficacy of CA3-CA1 Synapses. Cell Rep. 2018, 24, 1550–1561. [Google Scholar] [CrossRef] [Green Version]
- Eyman, M.; Cefaliello, C.; Mandile, P.; Piscopo, S.; Crispino, M.; Giuditta, A. Training old rats selectively modulates synaptosomal protein synthesis. J. Neurosci. Res. 2013, 91, 20–29. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Sidiropoulou, K.; Kallergi, E.; Dalezios, Y.; Tavernarakis, N. Modulation of Autophagy by BDNF Underlies Synaptic Plasticity. Cell Metab. 2017, 26, 230–242. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, H.; Altar, C.A.; Chen, R.; Nakamura, T.; Nakahashi, T.; Kambayashi, J.; Sun, B.; Tandon, N.N. Brain-derived neurotrophic factor is stored in human platelets and released by agonist stimulation. Thromb. Haemost. 2002, 87, 728–734. [Google Scholar] [CrossRef] [Green Version]
- Klein, A.B.; Williamson, R.; Santini, M.A.; Clemmensen, C.; Ettrup, A.; Rios, M.; Knudsen, G.M.; Aznar, S. Blood BDNF concentrations reflect brain-tissue BDNF levels across species. Int. J. Neuropsychopharmacol. 2011, 14, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Mondal, A.C.; Fatima, M. Direct and indirect evidences of BDNF and NGF as key modulators in depression: Role of antidepressants treatment. Int. J. Neurosci. 2019, 129, 283–296. [Google Scholar] [CrossRef]
- Molendijk, M.L.; Spinhoven, P.; Polak, M.; Bus, B.A.; Penninx, B.W.; Elzinga, B.M. Serum BDNF concentrations as peripheral manifestations of depression: Evidence from a systematic review and meta-analyses on 179 associations (N=9484). Mol. Psychiatry 2014, 19, 791–800. [Google Scholar] [CrossRef]
- Polyakova, M.; Stuke, K.; Schuemberg, K.; Mueller, K.; Schoenknecht, P.; Schroeter, M.L. BDNF as a biomarker for successful treatment of mood disorders: A systematic & quantitative meta-analysis. J. Affect. Disord. 2015, 174, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T.; Yoshimura, R.; Ikuta, T.; Iwata, N. Brain-Derived Neurotrophic Factor and Major Depressive Disorder: Evidence from Meta-Analyses. Front. Psychiatry 2017, 8, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra-Millàs, M. Are the changes in the peripheral brain-derived neurotrophic factor levels due to platelet activation? World J. Psychiatry 2016, 6, 84–101. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.P.; Naughton, M.; Dowling, J.; Walsh, A.; Ismail, F.; Shorten, G.; Scott, L.; McLoughlin, D.M.; Cryan, J.F.; Dinan, T.G.; et al. Serum BDNF as a peripheral biomarker of treatment-resistant depression and the rapid antidepressant response: A comparison of ketamine and ECT. J. Affect. Disord. 2015, 186, 306–311. [Google Scholar] [CrossRef]
- Lee, B.H.; Kim, Y.K. Reduced platelet BDNF level in patients with major depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 849–853. [Google Scholar] [CrossRef]
- Pandey, G.N.; Dwivedi, Y.; Rizavi, H.S.; Ren, X.; Zhang, H.; Pavuluri, M.N. Brain-derived neurotrophic factor gene and protein expression in pediatric and adult depressed subjects. Prog. Neuropsychopharmacol. Biol. Psychiatry 2010, 34, 645–651. [Google Scholar] [CrossRef]
- Serra-Millàs, M.; López-Vílchez, I.; Navarro, V.; Galán, A.M.; Escolar, G.; Penadés, R.; Catalán, R.; Fañanás, L.; Arias, B.; Gastó, C. Changes in plasma and platelet BDNF levels induced by S-citalopram in major depression. Psychopharmacology 2011, 216, 1–8. [Google Scholar] [CrossRef]
- Dwivedi, Y. The Neurobiological Basis of Suicide; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Pandey, G.N.; Ren, X.; Rizavi, H.S.; Conley, R.R.; Roberts, R.C.; Dwivedi, Y. Brain-derived neurotrophic factor and tyrosine kinase B receptor signalling in post-mortem brain of teenage suicide victims. Int. J. Neuropsychopharmacol. 2008, 11, 1047–1061. [Google Scholar] [CrossRef] [Green Version]
- Hosang, G.M.; Shiles, C.; Tansey, K.E.; McGuffin, P.; Uher, R. Interaction between stress and the BDNF Val66Met polymorphism in depression: A systematic review and meta-analysis. BMC Med. 2014, 12, 7. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Du, W.; Shao, F.; Wang, W. Cognitive dysfunction and epigenetic alterations of the BDNF gene are induced by social isolation during early adolescence. Behav. Brain Res. 2016, 313, 177–183. [Google Scholar] [CrossRef]
- Yan, T.; Wang, L.; Kuang, W.; Xu, J.; Li, S.; Chen, J.; Yang, Y. Brain-derived neurotrophic factor Val66Met polymorphism association with antidepressant efficacy: A systematic review and meta-analysis. Asia Pac. Psychiatry 2014, 6, 241–251. [Google Scholar] [CrossRef]
- Castrén, E.; Rantamäki, T. The role of BDNF and its receptors in depression and antidepressant drug action: Reactivation of developmental plasticity. Dev. Neurobiol. 2010, 70, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Crispino, M.; Volpicelli, F.; Perrone-Capano, C. Role of the Serotonin Receptor 7 in Brain Plasticity: From Development to Disease. Int. J. Mol. Sci. 2020, 21, 505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alenina, N.; Klempin, F. The role of serotonin in adult hippocampal neurogenesis. Behav. Brain Res. 2015, 277, 49–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malberg, J.E.; Eisch, A.J.; Nestler, E.J.; Duman, R.S. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 2000, 20, 9104–9110. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, L.; Saxe, M.; Gross, C.; Surget, A.; Battaglia, F.; Dulawa, S.; Weisstaub, N.; Lee, J.; Duman, R.; Arancio, O.; et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003, 301, 805–809. [Google Scholar] [CrossRef] [Green Version]
- Rantamäki, T.; Castrén, E. Targeting TrkB neurotrophin receptor to treat depression. Expert Opin. Ther. Targets 2008, 12, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Speranza, L.; Labus, J.; Volpicelli, F.; Guseva, D.; Lacivita, E.; Leopoldo, M.; Bellenchi, G.C.; di Porzio, U.; Bijata, M.; Perrone-Capano, C.; et al. Serotonin 5-HT7 receptor increases the density of dendritic spines and facilitates synaptogenesis in forebrain neurons. J. Neurochem. 2017, 141, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Speranza, L.; Giuliano, T.; Volpicelli, F.; De Stefano, M.E.; Lombardi, L.; Chambery, A.; Lacivita, E.; Leopoldo, M.; Bellenchi, G.C.; di Porzio, U.; et al. Activation of 5-HT7 receptor stimulates neurite elongation through mTOR, Cdc42 and actin filaments dynamics. Front. Behav. Neurosci. 2015, 9, 62. [Google Scholar] [CrossRef] [Green Version]
- Speranza, L.; Chambery, A.; Di Domenico, M.; Crispino, M.; Severino, V.; Volpicelli, F.; Leopoldo, M.; Bellenchi, G.C.; di Porzio, U.; Perrone-Capano, C. The serotonin receptor 7 promotes neurite outgrowth via ERK and Cdk5 signaling pathways. Neuropharmacology 2013, 67, 155–167. [Google Scholar] [CrossRef]
- Popova, N.K.; Naumenko, V.S. Neuronal and behavioral plasticity: The role of serotonin and BDNF systems tandem. Expert Opin. Ther. Targets 2019, 23, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Castrén, E. Neurotrophins and psychiatric disorders. Handb. Exp. Pharmacol. 2014, 220, 461–479. [Google Scholar] [CrossRef] [PubMed]
- Castrén, E.; Kojima, M. Brain-derived neurotrophic factor in mood disorders and antidepressant treatments. Neurobiol. Dis. 2017, 97, 119–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castrén, E.; Rantamäki, T. Role of brain-derived neurotrophic factor in the aetiology of depression: Implications for pharmacological treatment. CNS Drugs 2010, 24, 1–7. [Google Scholar] [CrossRef]
- Lepack, A.E.; Fuchikami, M.; Dwyer, J.M.; Banasr, M.; Duman, R.S. BDNF release is required for the behavioral actions of ketamine. Int. J. Neuropsychopharmacol. 2014, 18. [Google Scholar] [CrossRef] [Green Version]
- Abdallah, C.G.; Sanacora, G.; Duman, R.S.; Krystal, J.H. Ketamine and rapid-acting antidepressants: A window into a new neurobiology for mood disorder therapeutics. Annu. Rev. Med. 2015, 66, 509–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteggia, L.M.; Zarate, C. Antidepressant actions of ketamine: From molecular mechanisms to clinical practice. Curr. Opin. Neurobiol. 2015, 30, 139–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björkholm, C.; Monteggia, L.M. BDNF-a key transducer of antidepressant effects. Neuropharmacology 2016, 102, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshaw, B.A.; Malberg, J.E.; Lucki, I. Central administration of IGF-I and BDNF leads to long-lasting antidepressant-like effects. Brain Res. 2005, 1037, 204–208. [Google Scholar] [CrossRef]
- Eisch, A.J.; Bolaños, C.A.; de Wit, J.; Simonak, R.D.; Pudiak, C.M.; Barrot, M.; Verhaagen, J.; Nestler, E.J. Brain-derived neurotrophic factor in the ventral midbrain-nucleus accumbens pathway: A role in depression. Biol. Psychiatry 2003, 54, 994–1005. [Google Scholar] [CrossRef]
- Monteggia, L.M.; Luikart, B.; Barrot, M.; Theobold, D.; Malkovska, I.; Nef, S.; Parada, L.F.; Nestler, E.J. Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol. Psychiatry 2007, 61, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Monteggia, L.M.; Barrot, M.; Powell, C.M.; Berton, O.; Galanis, V.; Gemelli, T.; Meuth, S.; Nagy, A.; Greene, R.W.; Nestler, E.J. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc. Natl. Acad. Sci. USA 2004, 101, 10827–10832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saarelainen, T.; Hendolin, P.; Lucas, G.; Koponen, E.; Sairanen, M.; MacDonald, E.; Agerman, K.; Haapasalo, A.; Nawa, H.; Aloyz, R.; et al. Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J. Neurosci. 2003, 23, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Luikart, B.W.; Birnbaum, S.; Chen, J.; Kwon, C.H.; Kernie, S.G.; Bassel-Duby, R.; Parada, L.F. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 2008, 59, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.J.; Lee, F.S.; Li, X.Y.; Bambico, F.; Duman, R.S.; Aghajanian, G.K. Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol. Psychiatry 2012, 71, 996–1005. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Fogaça, M.V.; Deyama, S.; Li, X.Y.; Fukumoto, K.; Duman, R.S. BDNF release and signaling are required for the antidepressant actions of GLYX-13. Mol. Psychiatry 2018, 23, 2007–2017. [Google Scholar] [CrossRef]
- Herring, M.P.; Puetz, T.W.; O’Connor, P.J.; Dishman, R.K. Effect of exercise training on depressive symptoms among patients with a chronic illness: A systematic review and meta-analysis of randomized controlled trials. Arch. Intern. Med. 2012, 172, 101–111. [Google Scholar] [CrossRef]
- Ota, K.T.; Duman, R.S. Environmental and pharmacological modulations of cellular plasticity: Role in the pathophysiology and treatment of depression. Neurobiol. Dis. 2013, 57, 28–37. [Google Scholar] [CrossRef] [Green Version]
- Schuch, F.B.; Vancampfort, D.; Richards, J.; Rosenbaum, S.; Ward, P.B.; Stubbs, B. Exercise as a treatment for depression: A meta-analysis adjusting for publication bias. J. Psychiatr. Res. 2016, 77, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, P.; Brassard, P.; Adser, H.; Pedersen, M.V.; Leick, L.; Hart, E.; Secher, N.H.; Pedersen, B.K.; Pilegaard, H. Evidence for a release of brain-derived neurotrophic factor from the brain during exercise. Exp. Physiol. 2009, 94, 1062–1069. [Google Scholar] [CrossRef]
- Seifert, T.; Brassard, P.; Wissenberg, M.; Rasmussen, P.; Nordby, P.; Stallknecht, B.; Adser, H.; Jakobsen, A.H.; Pilegaard, H.; Nielsen, H.B.; et al. Endurance training enhances BDNF release from the human brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R372–R377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Zheng, Y.L.; Yin, X.; Xu, S.J.; Tian, D.; Zhang, C.Y.; Wang, S.; Ma, J.Z. Excessive Treadmill Training Enhances Brain-Specific MicroRNA-34a in the Mouse Hippocampus. Front. Mol. Neurosci. 2020, 13, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Assis, G.G.; de Almondes, K.M. Exercise-dependent BDNF as a Modulatory Factor for the Executive Processing of Individuals in Course of Cognitive Decline. A Systematic Review. Front. Psychol. 2017, 8, 584. [Google Scholar] [CrossRef] [PubMed]
- Pajonk, F.G.; Wobrock, T.; Gruber, O.; Scherk, H.; Berner, D.; Kaizl, I.; Kierer, A.; Müller, S.; Oest, M.; Meyer, T.; et al. Hippocampal plasticity in response to exercise in schizophrenia. Arch. Gen. Psychiatry 2010, 67, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, H.Y.; Becke, A.; Berron, D.; Becker, B.; Sah, N.; Benoni, G.; Janke, E.; Lubejko, S.T.; Greig, N.H.; Mattison, J.A.; et al. Running-Induced Systemic Cathepsin B Secretion Is Associated with Memory Function. Cell Metab. 2016, 24, 332–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Laznik-Bogoslavski, D.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise induces hippocampal BDNF through a PGC-1α/FNDC5 pathway. Cell Metab. 2013, 18, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Müller, P.; Duderstadt, Y.; Lessmann, V.; Müller, N.G. Lactate and BDNF: Key Mediators of Exercise Induced Neuroplasticity? J. Clin. Med. 2020, 9, 1136. [Google Scholar] [CrossRef]
- Piotrowicz, Z.; Chalimoniuk, M.; Płoszczyca, K.; Czuba, M.; Langfort, J. Exercise-Induced Elevated BDNF Level Does Not Prevent Cognitive Impairment Due to Acute Exposure to Moderate Hypoxia in Well-Trained Athletes. Int. J. Mol. Sci. 2020, 21, 5569. [Google Scholar] [CrossRef]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Hock, C.; Heese, K.; Hulette, C.; Rosenberg, C.; Otten, U. Region-specific neurotrophin imbalances in Alzheimer disease: Decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch. Neurol. 2000, 57, 846–851. [Google Scholar] [CrossRef] [Green Version]
- Zuccato, C.; Marullo, M.; Conforti, P.; MacDonald, M.E.; Tartari, M.; Cattaneo, E. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington′s disease. Brain Pathol. 2008, 18, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Narisawa-Saito, M.; Wakabayashi, K.; Tsuji, S.; Takahashi, H.; Nawa, H. Regional specificity of alterations in NGF, BDNF and NT-3 levels in Alzheimer′s disease. Neuroreport 1996, 7, 2925–2928. [Google Scholar] [CrossRef]
- Hoxha, E.; Lippiello, P.; Zurlo, F.; Balbo, I.; Santamaria, R.; Tempia, F.; Miniaci, M.C. The Emerging Role of Altered Cerebellar Synaptic Processing in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Bylykbashi, E.; Chatila, Z.K.; Lee, S.W.; Pulli, B.; Clemenson, G.D.; Kim, E.; Rompala, A.; Oram, M.K.; Asselin, C.; et al. Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science 2018, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleiman, S.F.; Henry, J.; Al-Haddad, R.; El Hayek, L.; Abou Haidar, E.; Stringer, T.; Ulja, D.; Karuppagounder, S.S.; Holson, E.B.; Ratan, R.R.; et al. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body β-hydroxybutyrate. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Colucci-D’Amato, L.; Perrone-Capano, C.; di Porzio, U. Chronic activation of ERK and neurodegenerative diseases. Bioessays 2003, 25, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Kirouac, L.; Rajic, A.J.; Cribbs, D.H.; Padmanabhan, J. Activation of Ras-ERK Signaling and GSK-3 by Amyloid Precursor Protein and Amyloid Beta Facilitates Neurodegeneration in Alzheimer′s Disease. eNeuro 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Nizzari, M.; Barbieri, F.; Gentile, M.T.; Passarella, D.; Caorsi, C.; Diaspro, A.; Taglialatela, M.; Pagano, A.; Colucci-D’Amato, L.; Florio, T.; et al. Amyloid-β protein precursor regulates phosphorylation and cellular compartmentalization of microtubule associated protein tau. J. Alzheimers Dis. 2012, 29, 211–227. [Google Scholar] [CrossRef]
- Bartolotti, N.; Segura, L.; Lazarov, O. Diminished CRE-Induced Plasticity is Linked to Memory Deficits in Familial Alzheimer’s Disease Mice. J. Alzheimers Dis. 2016, 50, 477–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, E.; Fahnestock, M. CREB expression mediates amyloid β-induced basal BDNF downregulation. Neurobiol. Aging 2015, 36, 2406–2413. [Google Scholar] [CrossRef] [PubMed]
- Thiele, C.J.; Li, Z.; McKee, A.E. On Trk--the TrkB signal transduction pathway is an increasingly important target in cancer biology. Clin. Cancer Res. 2009, 15, 5962–5967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.; Zhou, L.I.; Lim, Y.; Yang, M.; Zhu, Y.H.; Li, Z.W.; Fu, D.L.; Zhou, X.F. Mature brain-derived neurotrophic factor and its receptor TrkB are upregulated in human glioma tissues. Oncol. Lett. 2015, 10, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Colucci-D’Amato, G.L.; D’Alessio, A.; Califano, D.; Cali, G.; Rizzo, C.; Nitsch, L.; Santelli, G.; de Franciscis, V. Abrogation of nerve growth factor-induced terminal differentiation by ret oncogene involves perturbation of nuclear translocation of ERK. J. Biol. Chem. 2000, 275, 19306–19314. [Google Scholar] [CrossRef] [Green Version]
- Radin, D.P.; Patel, P. BDNF: An Oncogene or Tumor Suppressor? Anticancer. Res. 2017, 37, 3983–3990. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Prager, B.C.; Wu, Q.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Yang, K.; Morton, A.R.; Zhou, W.; Zhu, Z.; et al. Reciprocal Signaling between Glioblastoma Stem Cells and Differentiated Tumor Cells Promotes Malignant Progression. Cell Stem Cell 2018, 22, 514–528. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, S.; D’Alessandro, G.; Chece, G.; Brau, F.; Maggi, L.; Rosa, A.; Porzia, A.; Mainiero, F.; Esposito, V.; Lauro, C.; et al. Enriched environment reduces glioma growth through immune and non-immune mechanisms in mice. Nat. Commun. 2015, 6, 6623. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, S.; Porzia, A.; Mainiero, F.; Di Angelantonio, S.; Cortese, B.; Basilico, B.; Pagani, F.; Cignitti, G.; Chece, G.; Maggio, R.; et al. Environmental stimuli shape microglial plasticity in glioma. Elife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Colucci-D’Amato, L.; Cimaglia, G. As a potential source of neuroactive compounds to promote and restore neural functions. J. Tradit. Complement. Med. 2020, 10, 309–314. [Google Scholar] [CrossRef]
- Gentile, M.T.; Ciniglia, C.; Reccia, M.G.; Volpicelli, F.; Gatti, M.; Thellung, S.; Florio, T.; Melone, M.A.; Colucci-D’Amato, L. Ruta graveolens L. induces death of glioblastoma cells and neural progenitors, but not of neurons, via ERK 1/2 and AKT activation. PLoS ONE 2015, 10, e0118864. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colucci-D’Amato, L.; Speranza, L.; Volpicelli, F. Neurotrophic Factor BDNF, Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer. Int. J. Mol. Sci. 2020, 21, 7777. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207777
Colucci-D’Amato L, Speranza L, Volpicelli F. Neurotrophic Factor BDNF, Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer. International Journal of Molecular Sciences. 2020; 21(20):7777. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207777
Chicago/Turabian StyleColucci-D’Amato, Luca, Luisa Speranza, and Floriana Volpicelli. 2020. "Neurotrophic Factor BDNF, Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer" International Journal of Molecular Sciences 21, no. 20: 7777. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207777