Molecular Modeling Study of c-KIT/PDGFRα Dual Inhibitors for the Treatment of Gastrointestinal Stromal Tumors



Abstract
:
1. Introduction
2. Results
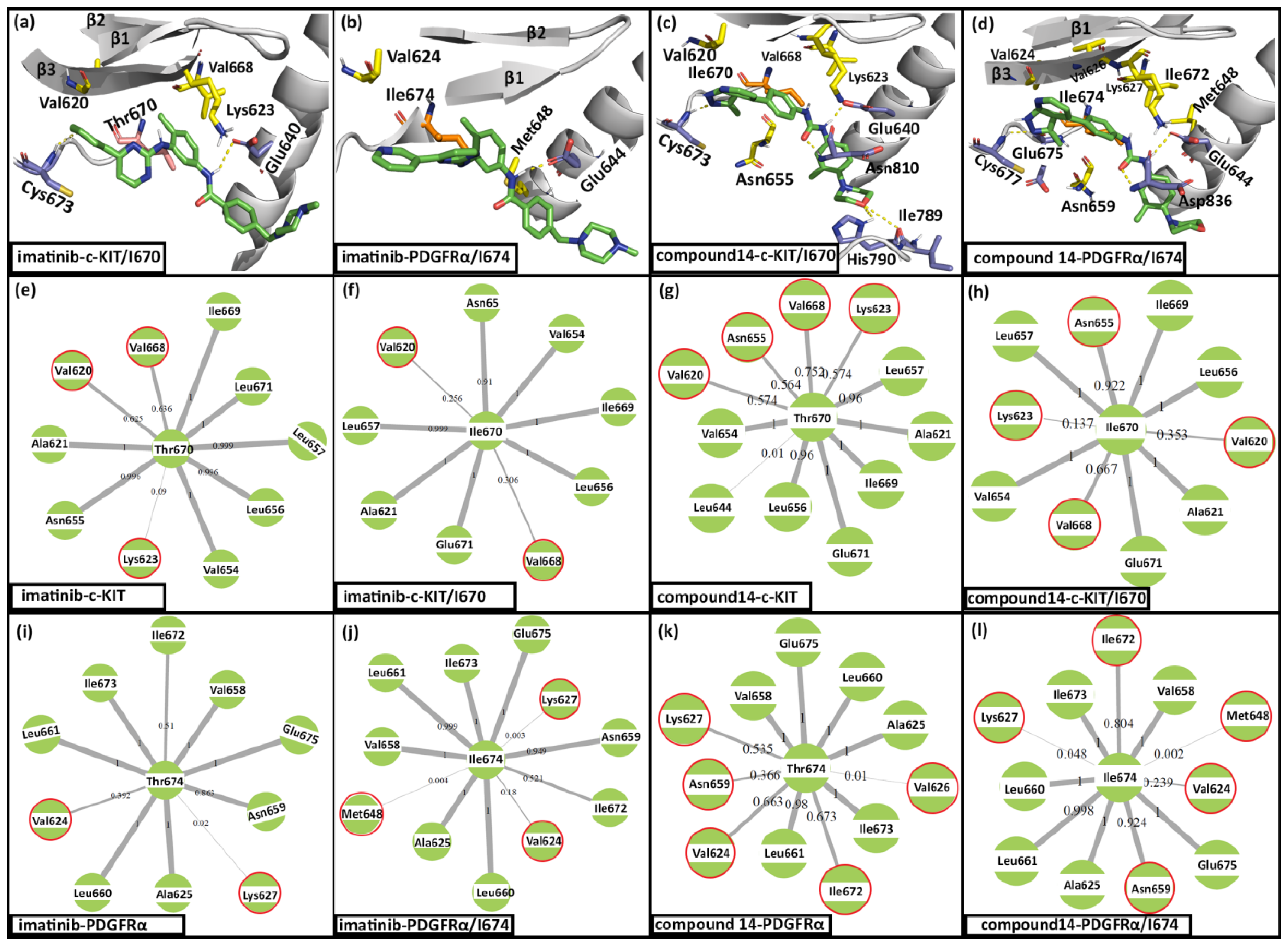
2.1. Molecular Docking
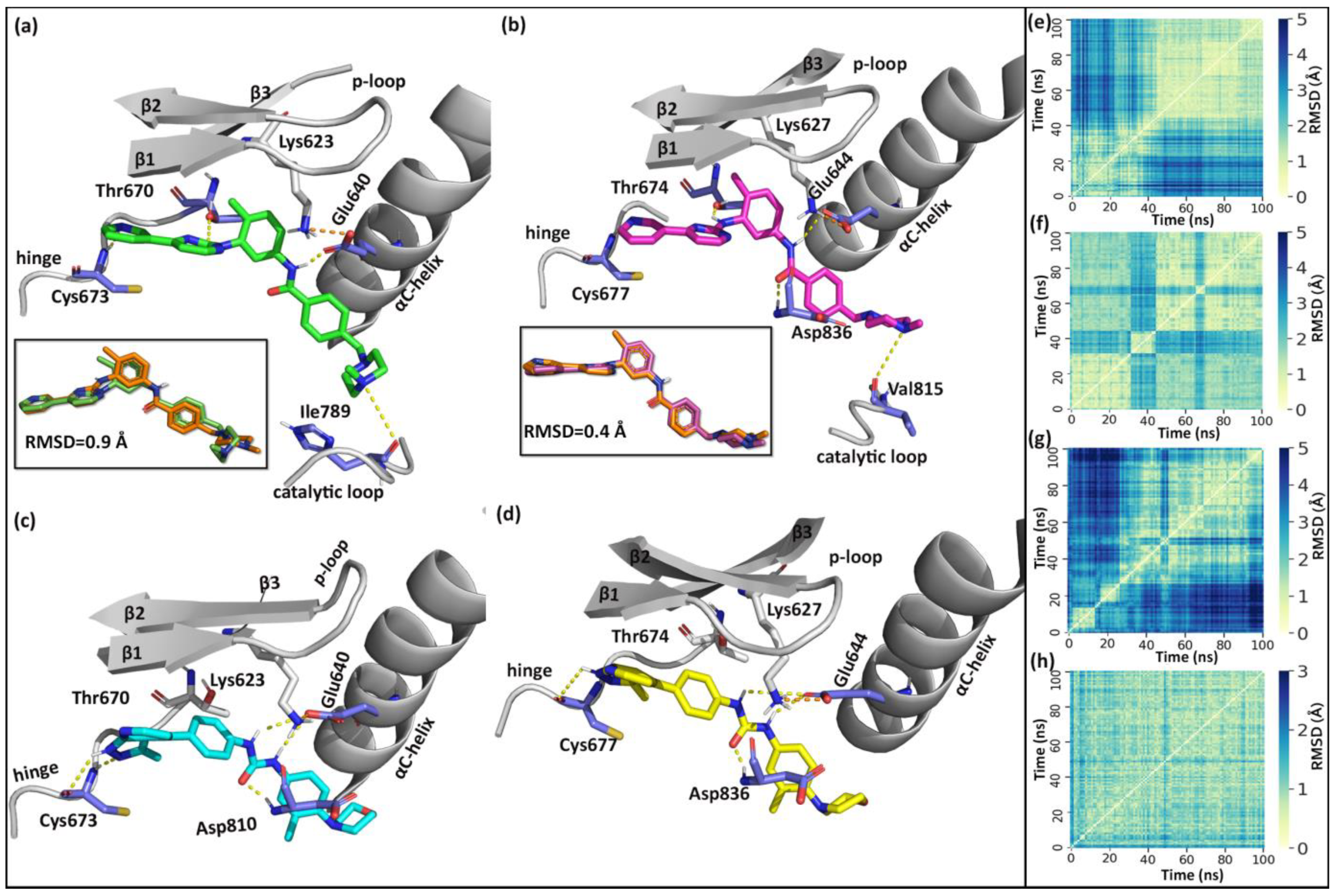
2.2. Molecular Dynamics Simulation
2.3. Evaluation of Binding Energy
2.4. D-QSAR
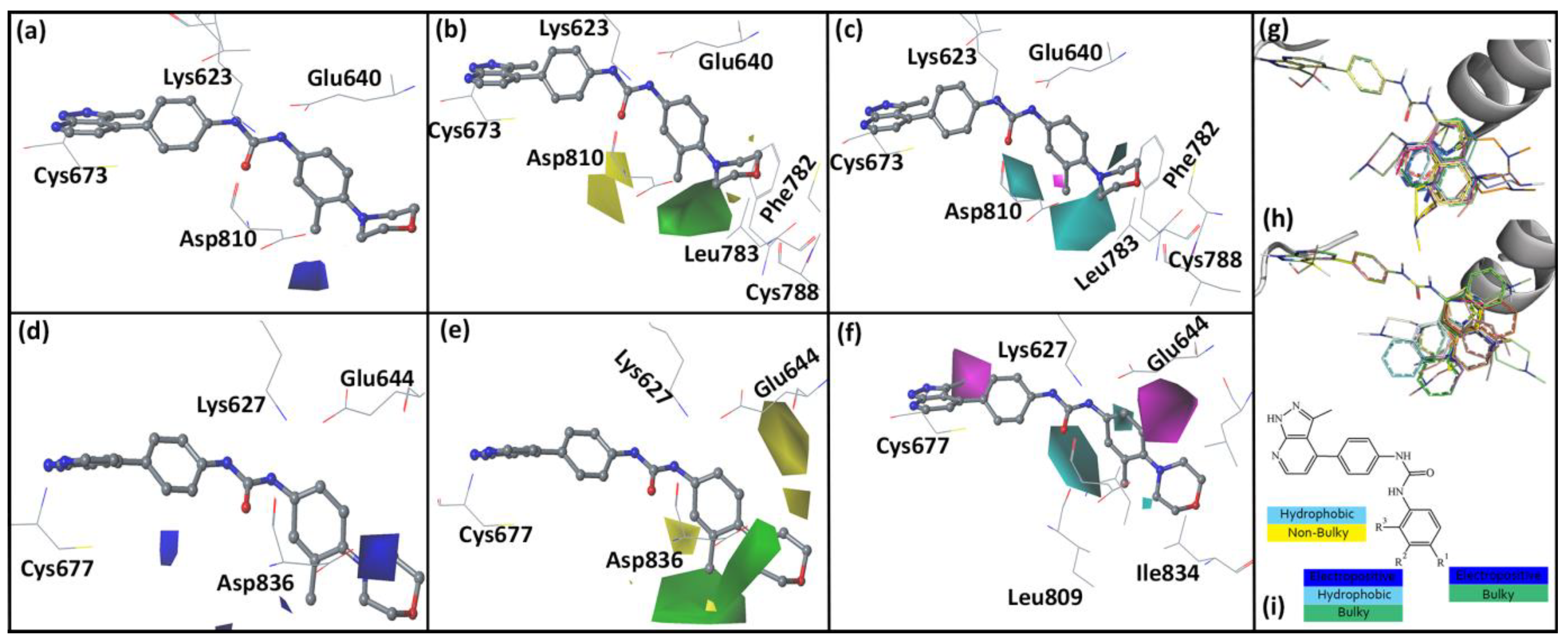
2.5. Analysis of Contour Map
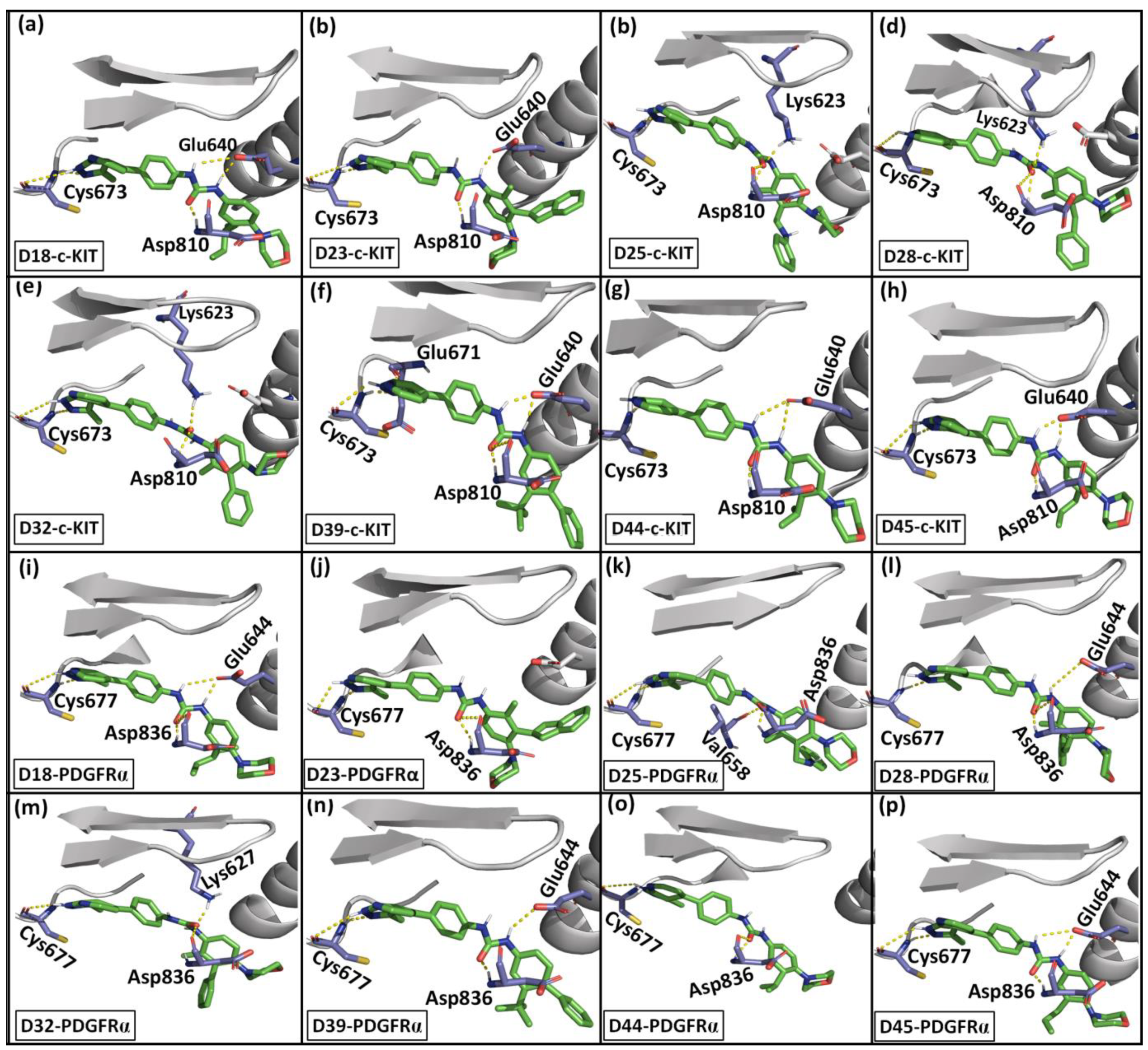
2.6. Designed Compounds
3. Discussion
4. Methodology
4.1. Data Preparation
4.2. Molecular Docking
4.3. Molecular Dynamics Simulation
4.4. Evaluation of Binding Energy
4.5. 3D-QSAR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| S | Steric |
| E | Electrostatic |
| H | Hydrophobic |
| ADMET | Absorption, distribution, metabolism, excretion and toxicity |
| BE | Binding energy |
| BS SD | Bootstrap standard deviation |
| CADD | Computer-aided drug discovery |
| c-KIT | Stem cell factor receptor |
| CoMFA | Comparative molecular field analysis |
| CoMSIA | Comparative molecular similarity indices analysis |
| FDA | Food and Drug Administration |
| FGFR | Fibroblast growth factor receptor |
| FLT3 | Fms like tyrosine kinase 3 |
| GISTs | Gastrointestinal stromal tumors |
| ICC | Interstitial cells of Cajal |
| MD | Molecular dynamics |
| MM/PBSA | Molecular mechanics energies combined with the Poisson–Boltzmann and surface area continuum solvation |
| ONC | Optimal number of components |
| PDB | Protein data bank |
| PDGFRa | Platelet derived growth factor receptor alpha |
| RAF1 | Rapidly accelerated fibrosarcoma 1 |
| RET | Rearranged during transfection |
| RMSD | Root mean square deviation |
| SASA | Solvent accessible surface area |
| SEE | Standard error of estimation |
| VEGFR | Vascular endothelial growth factor receptor |
| 3D-QSAR | Three-dimensional quantitative structure–activity relationship |
References
- Yang, Z.; Wang, F.; Liu, S.; Guan, W. Comparative clinical features and short-term outcomes of gastric and small intestinal gastrointestinal stromal tumours: A retrospective study. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Kindblom, L.-G.; Remotti, H.E.; Aldenborg, F.; Meis-Kindblom, J.M. Gastrointestinal pacemaker cell tumor (GIPACT): Gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am. J. Pathol. 1998, 152, 1259. [Google Scholar]
- Miettinen, M.; Lasota, J. Gastrointestinal stromal tumors–definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Arch. 2001, 438, 1–12. [Google Scholar] [CrossRef]
- Nilsson, B.; Bümming, P.; Meis-Kindblom, J.M.; Odén, A.; Dortok, A.; Gustavsson, B.; Sablinska, K.; Kindblom, L.G. Gastrointestinal stromal tumors: The incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era: A population-based study in western Sweden. Cancer 2005, 103, 821–829. [Google Scholar] [CrossRef]
- Mol, C.D.; Dougan, D.R.; Schneider, T.R.; Skene, R.J.; Kraus, M.L.; Scheibe, D.N.; Snell, G.P.; Zou, H.; Sang, B.-C.; Wilson, K.P. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J. Biol. Chem. 2004, 279, 31655–31663. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Yan, X.-E.; Yin, Y.; Yun, C.-H. Structural and biochemical studies of the PDGFRA kinase domain. Biochem. Biophys. Res. Commun. 2016, 477, 667–672. [Google Scholar] [CrossRef] [Green Version]
- Rosnet, O.; Birnbaum, D. Hematopoietic receptors of class III receptor-type tyrosine kinases. Crit. Rev. Oncog. 1993, 4, 595. [Google Scholar]
- Heldin, C.-H. Dimerization of cell surface receptors in signal transduction. Cell 1995, 80, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, S.R.; Mohammadi, M.; Schlessinger, J. Autoregulatory mechanisms in protein-tyrosine kinases. J. Biol. Chem. 1998, 273, 11987–11990. [Google Scholar] [CrossRef] [Green Version]
- Rosenkranz, S.; Ikuno, Y.; Leong, F.L.; Klinghoffer, R.A.; Miyake, S.; Band, H.; Kazlauskas, A. Src family kinases negatively regulate platelet-derived growth factor α receptor-dependent signaling and disease progression. J. Biol. Chem. 2000, 275, 9620–9627. [Google Scholar] [CrossRef] [Green Version]
- Kelly, J.D.; Haldeman, B.; Grant, F.J.; Murray, M.J.; Seifert, R.; Bowen-Pope, D.F.; Cooper, J.; Kazlauskas, A. Platelet-derived growth factor (PDGF) stimulates PDGF receptor subunit dimerization and intersubunit trans-phosphorylation. J. Biol. Chem. 1991, 266, 8987–8992. [Google Scholar] [PubMed]
- Yin, Y.-Q.; Liu, C.-J.; Zhang, B.; Wen, Y.; Yin, Y. Association between CT imaging features and KIT mutations in small intestinal gastrointestinal stromal tumors. Sci. Rep. 2019, 9, 1–7. [Google Scholar] [CrossRef]
- Xu, Z.; Huo, X.; Tang, C.; Ye, H.; Nandakumar, V.; Lou, F.; Zhang, D.; Jiang, S.; Sun, H.; Dong, H. Frequent KIT mutations in human gastrointestinal stromal tumors. Sci. Rep. 2014, 4, 5907. [Google Scholar] [CrossRef] [Green Version]
- Rubin, B.P.; Heinrich, M.C.; Corless, C.L. Gastrointestinal stromal tumour. Lancet 2007, 369, 1731–1741. [Google Scholar] [CrossRef]
- Rammohan, A.; Sathyanesan, J.; Rajendran, K.; Pitchaimuthu, A.; Perumal, S.-K.; Srinivasan, U.; Ramasamy, R.; Palaniappan, R.; Govindan., M. A gist of gastrointestinal stromal tumors: A review. World J. Gastrointest. Oncol. 2013, 5, 102. [Google Scholar] [CrossRef]
- Belinsky, M.G.; Cai, K.Q.; Zhou, Y.; Luo, B.; Pei, J.; Rink, L.; von Mehren, M. Succinate dehydrogenase deficiency in a PDGFRA mutated GIST. BMC Cancer 2017, 17, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Call, J.W.; Wang, Y.; Montoya, D.; Scherzer, N.J.; Heinrich, M.C. Survival in advanced GIST has improved over time and correlates with increased access to post-imatinib tyrosine kinase inhibitors: Results from Life Raft Group Registry. Clin. Sarcoma Res. 2019, 9, 4. [Google Scholar] [CrossRef]
- Zhao, R.; Wang, Y.; Huang, Y.; Cui, Y.; Xiaoting, W.; Chen, Y.; Zhuang, W.; Zhou, Y.; Wu, X. Adjuvant imatinib for patients with high-risk gastrointestinal stromal tumors: A retrospective cohort study. Sci. Rep. 2017, 7, 16834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corless, C.L.; Schroeder, A.; Griffith, D.; Town, A.; McGreevey, L.; Harrell, P.; Shiraga, S.; Bainbridge, T.; Morich, J.; Heinrich, M.C. PDGFRA Mutations in Gastrointestinal Stromal Tumors: Frequency, Spectrum and In Vitro Sensitivity to Imatinib. J. Clin. Oncol. 2005, 23, 5357–5364. [Google Scholar] [CrossRef] [PubMed]
- Janeway, K.A.; Albritton, K.H.; Abbeele, A.D.V.D.; D’Amato, G.Z.; Pedrazzoli, P.; Siena, S.; Picus, J.; Butrynski, J.E.; Schlemmer, M.; Heinrich, M.C.; et al. Sunitinib treatment in pediatric patients with advanced GIST following failure of imatinib. Pediatr. Blood Cancer 2009, 52, 767–771. [Google Scholar] [CrossRef]
- Waddell, T.; Cunningham, D. Evaluation of regorafenib in colorectal cancer and GIST. Lancet 2012, 381, 273–275. [Google Scholar] [CrossRef]
- Serrano, C.; Mariño-Enríquez, A.; Tao, D.L.; Ketzer, J.; Eilers, G.; Zhu, M.; Yu, C.; Mannan, A.M.; Rubin, B.P.; Demetri, G.D.; et al. Complementary activity of tyrosine kinase inhibitors against secondary kit mutations in imatinib-resistant gastrointestinal stromal tumours. Br. J. Cancer 2019, 120, 612–620. [Google Scholar] [CrossRef] [Green Version]
- Son, M.K.; Ryu, M.-H.; Park, J.O.; Im, S.-A.; Kim, T.-Y.; Lee, S.J.; Ryoo, B.-Y.; Park, S.R.; Kang, Y.-K. Efficacy and Safety of Regorafenib in Korean Patients with Advanced Gastrointestinal Stromal Tumor after Failure of Imatinib and Sunitinib: A Multicenter Study Based on the Management Access Program. Cancer Res. Treat. 2017, 49, 350–357. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.-K.; Yoo, C.; Ryoo, B.-Y.; Lee, J.J.; Tan, E.; Park, I.; Park, J.H.; Choi, Y.J.; Jo, J.; Ryu, J.-S.; et al. Phase II study of dovitinib in patients with metastatic and/or unresectable gastrointestinal stromal tumours after failure of imatinib and sunitinib. Br. J. Cancer 2013, 109, 2309–2315. [Google Scholar] [CrossRef]
- Le Cesne, A.; Blay, J.-Y.; Bui, B.N.; Bouché, O.; Adenis, A.; Domont, J.; Cioffi, A.; Ray-Coquard, I.; Lassau, N.; Bonvalot, S.; et al. Phase II study of oral masitinib mesilate in imatinib-naïve patients with locally advanced or metastatic gastro-intestinal stromal tumour (GIST). Eur. J. Cancer 2010, 46, 1344–1351. [Google Scholar] [CrossRef]
- Von Mehren, M.; Tetzlaff, E.D.; Macaraeg, M.; Davis, J.; Agarwal, V.; Ramachandran, A.; Heinrich, M.C. Dose escalating study of crenolanib besylate in advanced GIST patients with PDGFRA D842V activating mutations. J. Clin. Oncol. 2016, 34, 11010. [Google Scholar] [CrossRef]
- Smith, B.D.; Kaufman, M.D.; Lu, W.-P.; Gupta, A.; Leary, C.B.; Wise, S.C.; Rutkoski, T.J.; Ahn, Y.M.; Al-Ani, G.; Bulfer, S.L.; et al. Ripretinib (DCC-2618) Is a Switch Control Kinase Inhibitor of a Broad Spectrum of Oncogenic and Drug-Resistant KIT and PDGFRA Variants. Cancer Cell 2019, 35, 738–751.e9. [Google Scholar] [CrossRef]
- Ravegnini, G.; Nannini, M.; Sammarini, G.; Astolfi, A.; Biasco, G.; Pantaleo, M.A.; Hrelia, P.; Angelini, S. Personalized Medicine in Gastrointestinal Stromal Tumor (GIST): Clinical Implications of the Somatic and Germline DNA Analysis. Int. J. Mol. Sci. 2015, 16, 15592–15608. [Google Scholar] [CrossRef] [Green Version]
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharmacal Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Keretsu, S.; Balasubramanian, P.K.; Bhujbal, S.P.; Cho, S.J. Receptor-guided 3D-Quantitative Structure-Activity Relationship and Docking Studies of 6-Substituted 2-Arylaminopurines as CDK2 Kinase Inhibitors. Bull. Korean Chem. Soc. 2017, 38, 1275–1284. [Google Scholar] [CrossRef]
- Pasha, F.A.; Cho, S.J.; Beg, Y.; Tripathi, Y.B. Quantum chemical QSAR study of flavones and their radical-scavenging activity. Med. Chem. Res. 2007, 16, 408–417. [Google Scholar] [CrossRef]
- Juan, A.A.S.; Cho, S.J. 3D-QSAR study of microsomal prostaglandin E2 synthase(mPGES-1) inhibitors. J. Mol. Model. 2007, 13, 601–610. [Google Scholar] [CrossRef]
- Keretsu, S.; Bhujbal, S.P.; Cho, S.J. Computational study of paroxetine-like inhibitors reveals new molecular insight to inhibit GRK2 with selectivity over ROCK1. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Keretsu, S.; Bhujbal, S.P.; Cho, S.J. Molecular modeling studies of pyrrolo[2,3-d]pyrimidin-4-amine derivatives as JAK1 inhibitors based on 3D-QSAR, molecular docking, molecular dynamics (MD) and MM-PBSA calculations. J. Biomol. Struct. Dyn. 2020, 2020, 1–13. [Google Scholar] [CrossRef]
- Keretsu, S.; Bhujbal, S.P.; Cho, S.J. Docking and 3D-QSAR Studies of Hydrazone and Triazole Derivatives for Selective Inhibition of GRK2 over ROCK2. Lett. Drug Des. Discov. 2020, 17, 618–632. [Google Scholar] [CrossRef]
- Keretsu, S.; Bhujbal, S.P.; Cho, S.J. Computational Study of Pyrimidin-2-Aminopyrazol-Hydroxamate-based JAK2 Inhibitors for the Treatment of Myeloproliferative Neoplasms. Bull. Korean Chem. Soc. 2020, 41, 542–551. [Google Scholar] [CrossRef]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef]
- Klebe, G. Comparative molecular similarity indices analysis: CoMSIA. Perspect. Drug Discov. Des. 1998, 87–104. [Google Scholar] [CrossRef]
- Lu, Y.; Mao, F.; Li, X.; Zheng, X.; Wang, M.; Xu, Q.; Zhu, J.; Li, J. Discovery of Potent, Selective Stem Cell Factor Receptor/Platelet Derived Growth Factor Receptor Alpha (c-KIT/PDGFRα) Dual Inhibitor for the Treatment of Imatinib-Resistant Gastrointestinal Stromal Tumors (GISTs). J. Med. Chem. 2017, 60, 5099–5119. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Ross, C.; Nizami, B.; Glenister, M.; Amamuddy, O.S.; Atilgan, A.R.; Atilgan, C.; Bishop, Ö.T. MODE-TASK: Large-scale protein motion tools. Bioinformatics 2018, 34, 3759–3763. [Google Scholar] [CrossRef] [Green Version]
- Tamborini, E.; Pricl, S.; Negri, T.; Lagonigro, M.S.; Miselli, F.; Greco, A.; Gronchi, A.; Casali, P.G.; Ferrone, M.; Fermeglia, M.; et al. Functional analyses and molecular modeling of two c-Kit mutations responsible for imatinib secondary resistance in GIST patients. Oncogene 2006, 25, 6140–6146. [Google Scholar] [CrossRef] [Green Version]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, H.; Van Der Spoel, D.; Van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; Van Der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, J.A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2009, 31, 671–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, R.; Kumar, R.; Lynn, A. Open Source Drug Discovery Consortium g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Bang, S.J.; Cho, S.J. Comparative molecular field analysis (CoMFA) and comparative molecular similarity index analysis (CoMSIA) study of mutagen X. Bull. Korean Chem. Soc. 2004, 25, 1525–1530. [Google Scholar]
- Gadhe, C.G.; Madhavan, T.; Kothandan, G.; Cho, S.J. In Silico Quantitative Structure-Activity Relationship Studies on P-gp Modulators of Tetrahydroisoquinoline-Ethyl-Phenylamine Series. BMC Struct. Biol. 2011, 11, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadhe, C.G.; Kothandan, G.; Cho, S.J. Large variation in electrostatic contours upon addition of steric parameters and the effect of charge calculation schemes in CoMFA on mutagenicity of MX analogues. Mol. Simul. 2012, 38, 861–871. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Structure A | ||||
| Compounds | Structures | R1 | c-KIT (pIC50) | PDGFRα (pIC50) |
| 1 |  | 8.62 | 7.06 | |
| 2 |  | 8.43 | 7.49 | |
| 3 |  | >4.3 | >4.3 | |
| 4 |  | 5.2 | >4.3 | |
| 5 | A |  | 8.41 | 7.66 |
| 6 | A |  | 4.96 | 6.52 |
| 7 | A |  | 5.74 | 7.19 |
| 8 | A |  | 5.32 | 6.58 |
| 9 | A |  | 5.62 | 6.74 |
| 10 | A |  | 6.70 | 4.67 |
| 11 | A |  | 7.72 | 6.67 |
| 12 | A |  | 5.71 | 5.75 |
| 13 | A |  | 8.14 | 6.90 |
| 14 | A |  | 8.62 | 8.14 |
| 15 | A |  | 5.65 | 5.96 |
| 16 | A |  | 5.34 | 6.03 |
| 17 | A |  | 6.40 | 4.72 |
| 18 | A |  | 5.19 | 5.39 |
| 19 | A |  | 7.92 | 6.87 |
| 20 | A |  | 7.92 | 6.75 |
| 21 | A |  | 8.59 | 7.08 |
| 22 | A |  | 8.03 | 6.51 |
| 23 | A |  | 4.66 | 5.34 |
| 24 | A |  | 4.4 | >4.3 |
| 25 | A |  | 4.7 | >4.3 |
| 26 | A |  | 4.69 | 4.36 |
| 27 | A |  | 8.46 | 6.79 |
| 28 | A |  | 8.85 | 7.57 |
| 29 | A |  | 6.49 | 5.95 |
| 30 | A |  | 5.85 | 5.83 |
| 31 | A |  | 6.26 | 5.95 |
| 32 | A |  | 6.50 | 6.14 |
| 33 | A |  | 6.84 | 6.24 |
| 34 | A |  | >4.3 | >4.3 |
| 35 | A |  | 7.29 | 6.41 |
| 36 | A |  | 7.59 | 6.80 |
| 37 | A |  | 7.11 | 6.33 |
| 38 | A |  | >4.3 | >4.3 |
| 39 | A |  | 8.43 | 7.09 |
| 40 | A |  | 8.77 | 7.62 |
| 41 | A |  | 8.21 | 6.87 |
| 42 | A |  | 8.06 | 7.08 |
| 43 | A |  | 6.92 | 6.39 |
| 44 | A |  | 7.70 | 6.40 |
| 45 | A |  | 7.85 | 6.50 |
| 46 | A |  | 8.72 | 7.66 |
| 47 | A |  | 7.70 | 6.91 |
| 48 | A |  | 8.44 | 7.01 |
| Complexes | Van der Waals (kJ/Mol) | Electrostatics (kJ/Mol) | Polar Solvation (kJ/Mol) | SASA (kJ/Mol) | Total Binding Energy (kJ/Mol) |
|---|---|---|---|---|---|
| Imatinib–c-KIT | −260 | −74 | 257 | −28 | −105 |
| Imatinib–PDGFRα | −244 | −58 | 225 | −27 | −104 |
| Compound 14–c-KIT | −257 | −57 | 219 | −25 | −120 |
| Compound 14–PDGFRα | −251 | −55 | 213 | −25 | −118 |
| Compound 31–c-KIT | −183 | −48 | 185 | −19 | −65 |
| Compound 31–PDGFRα | −180 | −50 | 163 | −18 | −85 |
| Imatinib–c-KIT/I670 | −227 | −48 | 228 | −26 | −73 |
| Imatinib–PDGFRα/I674 | −248 | −18 | 227 | −28 | −67 |
| Compound 14–c-KIT/I670 | −250 | −61 | 205 | −25 | −131 |
| Compound 14–PDGFRα/I674 | −265 | −39 | 203 | −25 | −126 |
| c-KIT Residues | Compound 14–c-KIT (kJ/mol) | Imatinib–c-KIT (kJ/mol) | PDGFRα Residues | Compound 14–PDGFRα (kJ/mol) | Imatinib–PDGFRα (kJ/mol) |
|---|---|---|---|---|---|
| Asp572 | −0.9 | −0.7 | Glu587 | −0.66 | −0.42 |
| Leu595 | −2.6 | −3.4 | Leu599 | −2.48 | −2.87 |
| Val603 | −5.4 | −3.2 | Gly600 | −0.91 | −0.82 |
| Ala621 | −2.7 | −3.1 | Val607 | −5.23 | −5.10 |
| Val620 | −0.7 | −0.8 | Val608 | −1.10 | −0.69 |
| Val622 | −1.0 | −1.7 | Glu609 | −1.31 | −2.33 |
| Glu635 | −0.8 | −0.7 | Val624 | −0.76 | −1.10 |
| Val643 | −3.1 | −3.6 | Ala625 | −2.20 | −1.87 |
| Leu644 | −6.7 | −6.6 | Val626 | −1.15 | −1.84 |
| Leu647 | −2.1 | −0.5 | Glu637 | −0.83 | −0.96 |
| Ile653 | −3.0 | −0.9 | Ile647 | −4.19 | −4.35 |
| Val654 | −5.2 | −7.6 | Met648 | −8.93 | −7.98 |
| Tyr672 | −3.9 | −5.6 | Leu651 | −2.11 | −0.96 |
| Cys673 | −2.1 | −2.5 | Ile657 | −2.52 | −0.76 |
| Gly676 | −0.8 | −0.4 | Val658 | −5.46 | −5.28 |
| Leu783 | −2.9 | −0.6 | Ile672 | −0.83 | −2.95 |
| Cys788 | −1.3 | −1.8 | Tyr676 | −3.39 | −4.99 |
| His790 | −3.6 | −1.9 | Cys677 | −2.48 | −1.82 |
| Asp792 | −1.0 | −0.2 | Gly680 | −0.92 | −0.76 |
| Leu799 | −4.9 | −4.3 | Leu809 | −2.58 | −1.14 |
| Lys807 | −1.1 | 2.2 | Cys814 | −2.27 | −1.81 |
| Ile808 | −1.0 | 0.4 | Leu825 | −5.59 | −5.59 |
| Cys809 | −6.4 | −6.2 | Ile834 | −1.21 | −0.40 |
| Asp810 | −2.1 | 9.3 | Cys835 | −6.27 | −5.11 |
| Phe811 | −6.8 | −4.0 | Asp836 | −5.11 | 1.89 |
| Asp851 | −1.5 | −0.5 | Phe837 | −4.87 | −7.16 |
| Parameters | CoMFA (c-KIT) | CoMSIA (c-KIT) | CoMFA (PDGFRα) | CoMSIA (PDGFRα) |
|---|---|---|---|---|
| q2 | 0.63 | 0.6 | 0.61 | 0.62 |
| ONC | 6 | 5 | 6 | 3 |
| r2 | 0.98 | 0.9 | 0.98 | 0.81 |
| SEE | 0.2 | 0.46 | 0.12 | 0.39 |
| F value | 204 | 43 | 232 | 46 |
| BS r2 | 0.98 | 0.94 | 0.98 | 0.97 |
| BS SD | 0.15 | 0.32 | 0.1 | 0.14 |
| r2pred | 0.59 | 0.58 | 0.56 | 0.59 |
| Influence of different fields (%) | ||||
| S | 59 | 50 | 67 | 42 |
| E | 41 | - | 33 | - |
| H | - | 50 | - | 58 |
 | |||||
|---|---|---|---|---|---|
| Compounds | R1 | R2 | R3 | Predicted Activity (pIC50) | |
| c-KIT | PDGFRα | ||||
| Compound D18 |  |  | H | 10.4 | 8.3 |
| Compound D23 |  |  | CH3 | 10.1 | 8.2 |
| Compound D25 |  |  | CH3 | 10.5 | 8.1 |
| Compound D28 |  |  | CH3 | 9.6 | 8.4 |
| Compound D32 |  |  | CH3 | 9.1 | 8.3 |
| Compound D39 |  |  | H | 10.3 | 8.1 |
| Compound D44 |  |  | H | 10.2 | 8.3 |
| Compound D45 |  |  | H | 9.3 | 8.1 |
| Complexes (Designed Compounds–Receptor) | Van der Waals (kJ/Mol) | Electrostatics (kJ/Mol) | Polar Solvation (kJ/Mol) | SASA (kJ/Mol) | Total Binding Energy (kJ/Mol) |
|---|---|---|---|---|---|
| D18–c-KIT | −272 | −57 | 218 | −28 | −139 |
| D18–PDGFRα | −282 | −57 | 224 | −27 | −142 |
| D23–c-KIT | −287 | −48 | 242 | −29 | −122 |
| D23–PDGFRα | −286 | −36 | 212 | −28 | −138 |
| D28–c-KIT | −278 | −62 | 242 | −27 | −126 |
| D28–PDGFRα | −289 | −38 | 227 | −29 | −129 |
| D32–c-KIT | −271 | −53 | 229 | −27 | −122 |
| D32–PDGFRα | −283 | −44 | 224 | −28 | −130 |
| D39–c-KIT | −268 | −65 | 212 | −26 | −148 |
| D39–PDGFRα | −274 | −48 | 200 | −26 | −150 |
| D44–c-KIT | −262 | −56 | 216 | −25 | −129 |
| D44–PDGFRα | −261 | −42 | 209 | −26 | −120 |
| D45–c-KIT | −262 | −76 | 244 | −26 | −121 |
| D45–PDGFRα | −274 | −58 | 215 | −26 | −143 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keretsu, S.; Ghosh, S.; Cho, S.J. Molecular Modeling Study of c-KIT/PDGFRα Dual Inhibitors for the Treatment of Gastrointestinal Stromal Tumors. Int. J. Mol. Sci. 2020, 21, 8232. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218232
Keretsu S, Ghosh S, Cho SJ. Molecular Modeling Study of c-KIT/PDGFRα Dual Inhibitors for the Treatment of Gastrointestinal Stromal Tumors. International Journal of Molecular Sciences. 2020; 21(21):8232. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218232
Chicago/Turabian StyleKeretsu, Seketoulie, Suparna Ghosh, and Seung Joo Cho. 2020. "Molecular Modeling Study of c-KIT/PDGFRα Dual Inhibitors for the Treatment of Gastrointestinal Stromal Tumors" International Journal of Molecular Sciences 21, no. 21: 8232. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218232