Exogenous Oestrogen Impacts Cell Fate Decision in the Developing Gonads: A Potential Cause of Declining Human Reproductive Health


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
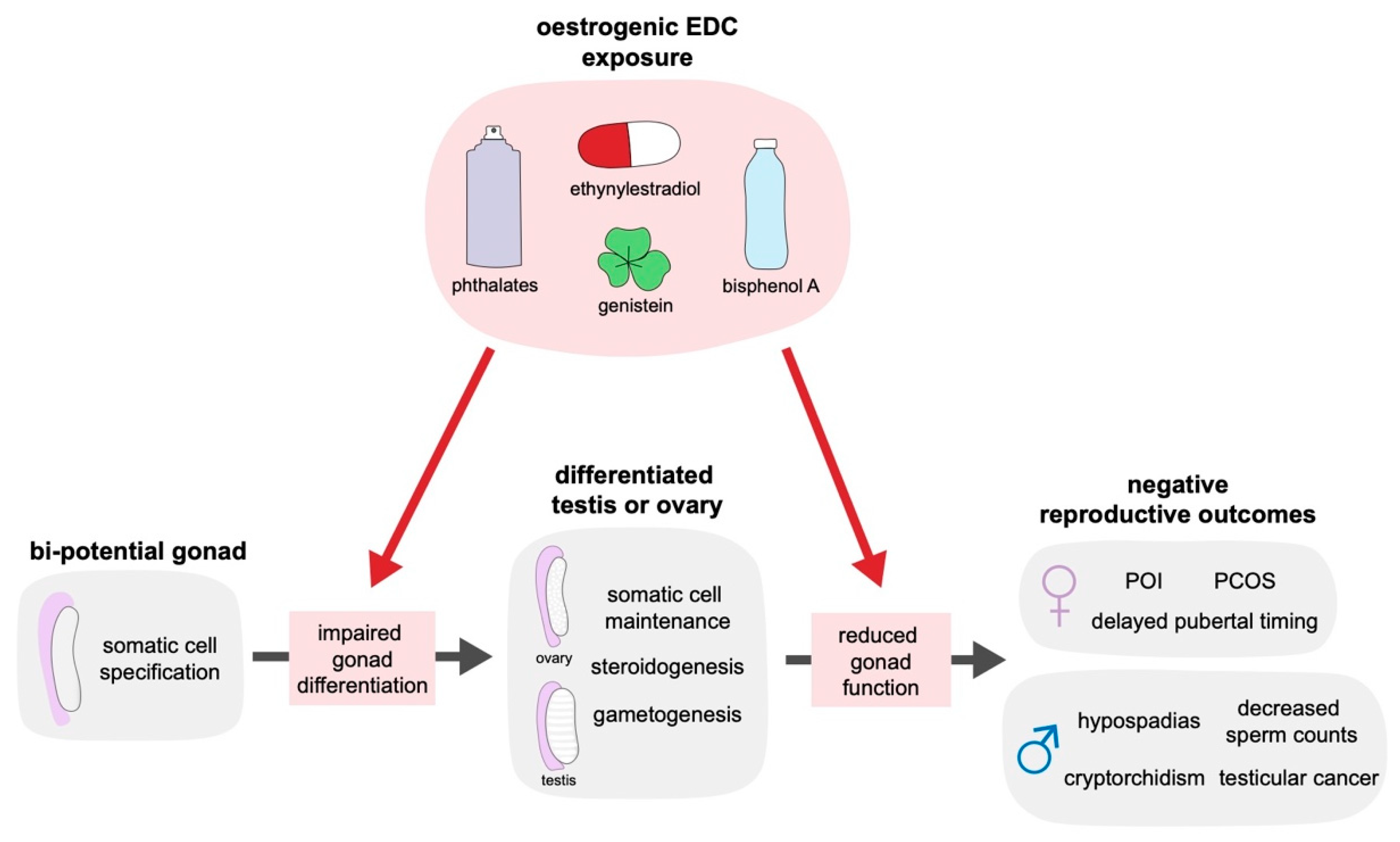
2. The Impact of Oestrogenic Endocrine Disrupting Chemicals on Reproductive Health
3. The Function of Oestrogen in the Mammalian Gonad
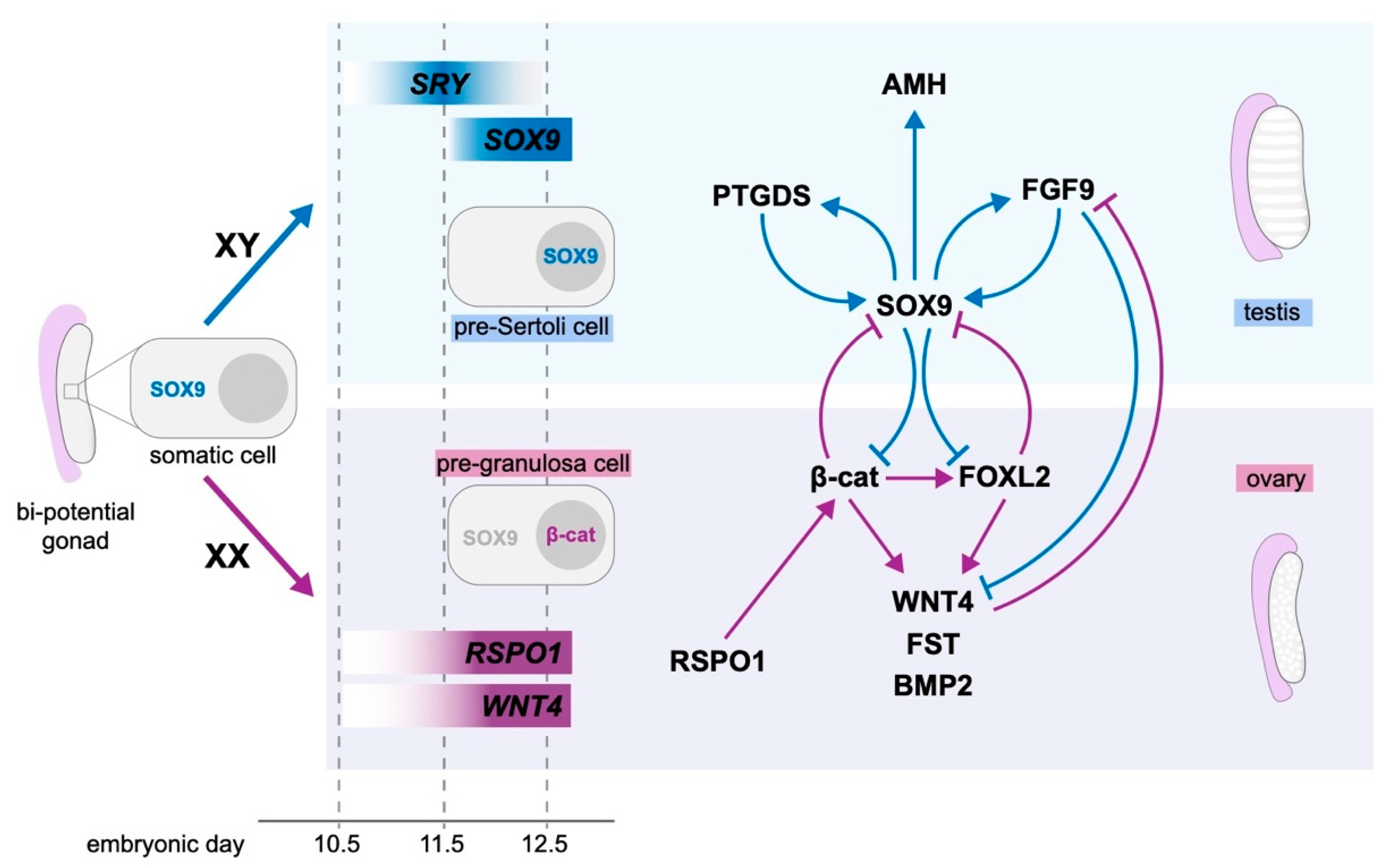
4. Molecular Control of Gonad Differentiation
4.1. Testis Development
4.2. Ovarian Development
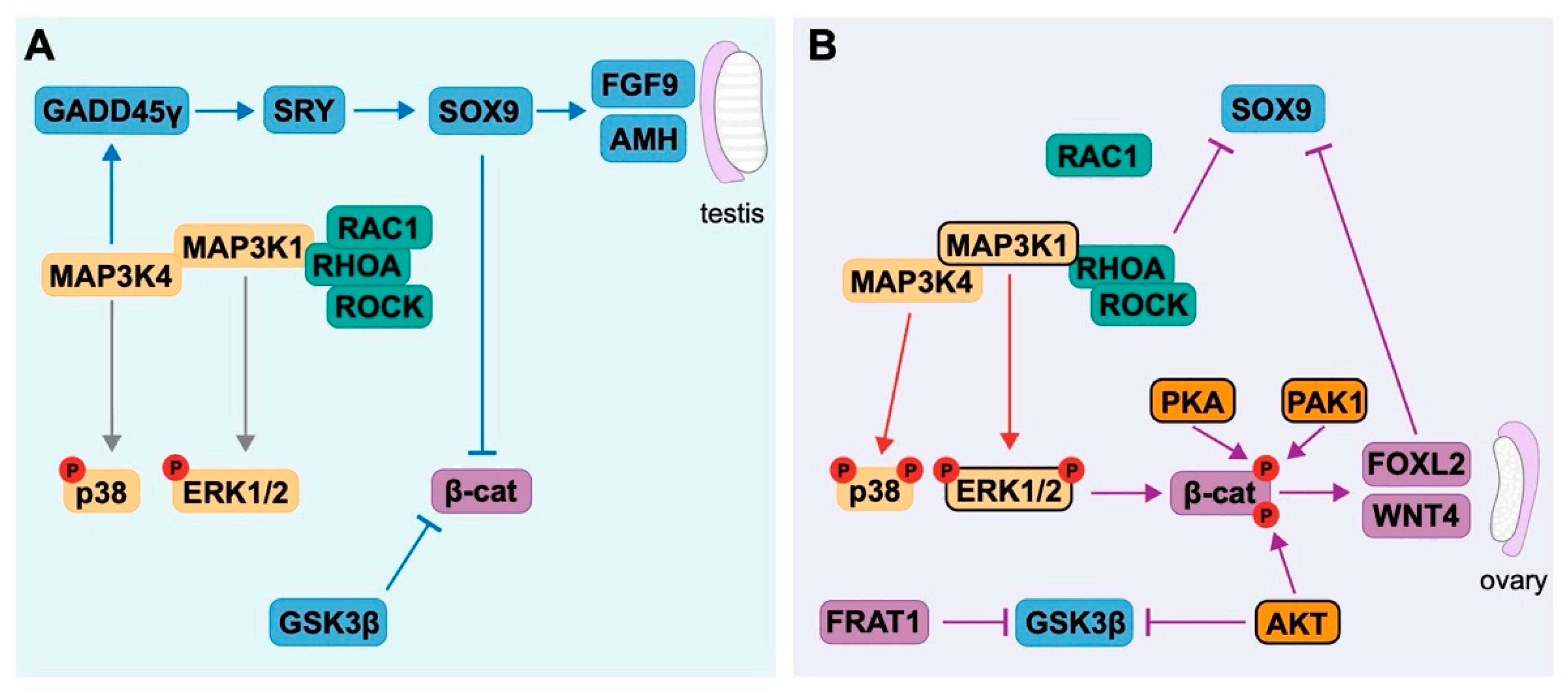
4.3. Antagonism between Pro-Testis and Pro-Ovarian Factors Drives Sex Determination
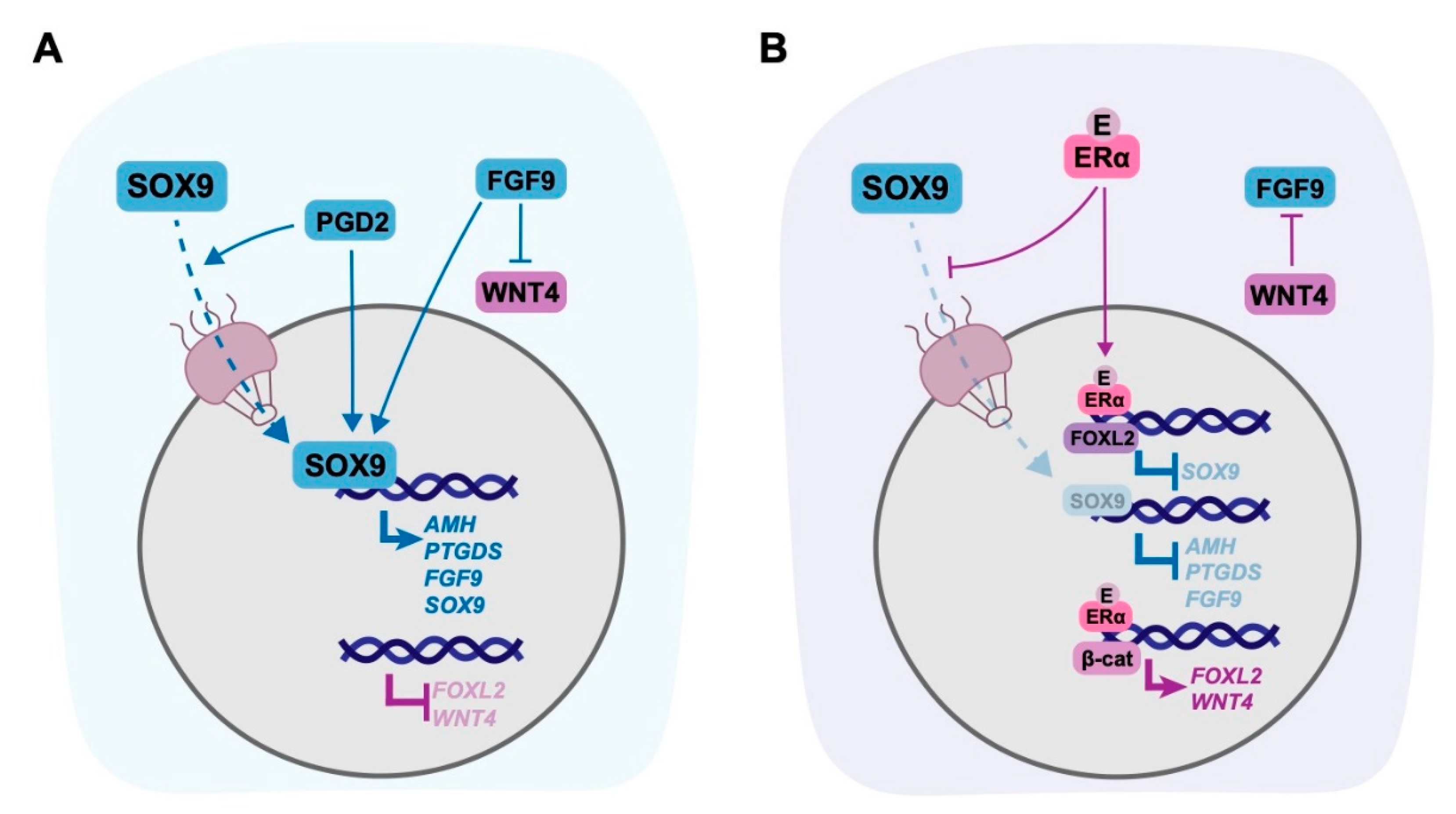
5. Targets of Oestrogen in the Gonad
5.1. Non-Genomic Targets of Oestrogen in the Gonad
5.2. Genomic Targets of Oestrogen in the Gonad
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| DSDs | Differences of sexual development |
| EDCs | Endocrine disrupting chemicals |
| TDS | Testicular dysgenesis syndrome |
| POI | Premature ovarian insufficiency |
| PCOS | Polycystic ovary syndrome |
| ER | Oestrogen receptor |
| NT2/D1 | NTERA-2 clone D1 |
| SOX9 | Sex-determining region Y box transcription factor 9 |
| SRY | Sex-determining region Y |
| AMH | Anti-Mullerian hormone |
| FGF9 | Fibroblast growth factor |
| PTGDS | Prostaglandin D synthase |
| SF1 | Steroidogenic factor 1 |
| FOXL2 | Forkhead box L2 |
| RSPO1 | R-spondin 1 |
| WNT4 | Wnt family member 4 |
| FST | Follistatin |
| BMP2 | Bone morphogenetic protein 2 |
| ERK1/2 | Extracellular regulated kinases 1/2 |
| MAP3K | Mitogen-activated protein kinase kinase kinase |
| GATA4 | GATA binding protein 4 |
| RHOA | Ras homolog family member A |
| ROCK | Rho-associated coiled coil containing protein kinase |
| RAC1 | Rac family small GTPase 1 |
| GSK3β | Glycogen synthase kinase 3β |
| FRAT1 | FRAT regulator of Wnt signalling pathway 1 |
| GADD45γ | Growth arrest and DNA damage-inducible protein γ |
| PKA | Protein kinase A |
| AKT | AKT serine/threonine kinase |
| PAK1 | p21 (RAC1) activated kinase 1 |
References
- Brennan, J.; Capel, B. One tissue, two fates: Molecular genetic events that underlie testis versus ovary development. Nat. Rev. Genet. 2004, 5, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Stévant, I.; Nef, S. Genetic control of gonadal sex determination and development. Trends Genet. 2019, 35, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Britt, K.L.; Drummond, A.E.; Dyson, M.; Wreford, N.G.; Jones, M.E.E.; Simpson, E.R.; Findlay, J.K. The ovarian phenotype of the aromatase knockout (ArKO) mouse. J. Steroid Biochem. Mol. Biol. 2001, 79, 181–185. [Google Scholar] [CrossRef]
- Maatouk, D.M.; DiNapoli, L.; Alvers, A.; Parker, K.L.; Taketo, M.M.; Capel, B. Stabilization of β-catenin in XY gonads causes male-to-female sex-reversal. Hum. Mol. Gen. 2008, 17, 2949–2955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, C.E.; Whitworth, D.J.; Qin, Y.J.; Agoulnik, A.I.; Agoulnik, I.U.; Harrison, W.R.; Behringer, R.R.; Overbeek, P.A. A transgenic insertion upstream of Sox9 is associated with dominant XX sex reversal in the mouse. Nat. Genet. 2000, 26, 490–494. [Google Scholar] [CrossRef]
- Vidal, V.P.; Chaboissier, M.C.; de Rooij, D.G.; Schedl, A. Sox9 induces testis development in XX transgenic mice. Nat. Genet. 2001, 28, 216–217. [Google Scholar] [CrossRef]
- Chaboissier, M.-C.; Kobayashi, A.; Vidal, V.I.P.; Lützkendorf, S.; van de Kant, H.J.G.; Wegner, M.; de Rooij, D.G.; Behringer, R.R.; Schedl, A. Functional analysis of Sox8 and Sox9 during sex determination in the mouse. Development 2004, 131, 1891–1901. [Google Scholar] [CrossRef] [Green Version]
- Barrionuevo, F.; Bagheri-Fam, S.; Klattig, J.; Kist, R.; Taketo, M.M.; Englert, C.; Scherer, G. Homozygous inactivation of Sox9 causes complete XY sex reversal in mice. Biol. Reprod. 2006, 74, 195–201. [Google Scholar] [CrossRef]
- Uhlenhaut, N.H.; Jakob, S.; Anlag, K.; Eisenberger, T.; Sekido, R.; Kress, J.; Treier, A.-C.; Klugmann, C.; Klasen, C.; Holter, N.I.; et al. Somatic sex reprogramming of adult ovaries to testes by FOXL2 ablation. Cell 2009, 139, 1130–1142. [Google Scholar] [CrossRef] [Green Version]
- Chassot, A.-A.; Ranc, F.; Gregoire, E.P.; Roepers-Gajadien, H.L.; Taketo, M.M.; Camerino, G.; de Rooij, D.G.; Schedl, A.; Chaboissier, M.-C. Activation of beta-catenin signaling by Rspo1 controls differentiation of the mammalian ovary. Hum. Mol. Gen. 2008, 17, 1264–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, M.K.; Mattiske, D.M.; Pask, A.J. Estrogen suppresses SOX9 and activates markers of female development in a human testis-derived cell line. BMC Mol. Cell Biol. 2020, 21, 158. [Google Scholar] [CrossRef] [PubMed]
- Pask, A.J.; Calatayud, N.E.; Shaw, G.; Wood, W.M.; Renfree, M.B. Oestrogen blocks the nuclear entry of SOX9 in the developing gonad of a marsupial mammal. BMC Biol. 2010, 8, 113–123. [Google Scholar] [CrossRef] [Green Version]
- Skakkebaek, N.E.; Jørgensen, N.; Andersson, A.-M.; Juul, A.; Main, K.M.; Jensen, T.K.; Toppari, J. Populations, decreasing fertility, and reproductive health. Lancet 2019, 393, 1500–1501. [Google Scholar] [CrossRef] [Green Version]
- Levine, H.; Jørgensen, N.; Martino-Andrade, A.; Mendiola, J.; Weksler-Derri, D.; Mindlis, I.; Pinotti, R.; Swan, S.H. Temporal trends in sperm count: A systematic review and meta-regression analysis. Hum. Reprod. Update 2017, 23, 646–659. [Google Scholar] [CrossRef] [PubMed]
- Richiardi, L.; Bellocco, R.; Adami, H.-O.; Torrång, A.; Barlow, L.; Hakulinen, T.; Rahu, M.; Stengrevics, A.; Storm, H.; Tretli, S.; et al. Testicular cancer incidence in eight northern European countries: Secular and recent trends. Cancer Epidemiol. Biomarkers Prev. 2004, 13, 2157–2166. [Google Scholar]
- Paulozzi, L.J. International trends in rates of hypospadias and cryptorchidism. Environ. Health Perspect. 1999, 107, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Nassar, N.; Bower, C.; Barker, A. Increasing prevalence of hypospadias in Western Australia, 1980–2000. Arch. Dis. Child. 2007, 92, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.A.; Nordenström, A.; Houk, C.P.; Ahmed, S.F.; Auchus, R.; Baratz, A.; Baratz Dalke, K.; Liao, L.-M.; Lin-Su, K.; Looijenga, L.H.J.; et al. Global DSD update consortium global disorders of sex development update since 2006: Perceptions, approach and care. Horm. Res. Paediatr. 2016, 85, 158–180. [Google Scholar] [CrossRef]
- Skakkebaek, N.E.; Rajpert-De Meyts, E.; Main, K.M. Testicular dysgenesis syndrome: An increasingly common developmental disorder with environmental aspects. Hum. Reprod. 2001, 16, 972–978. [Google Scholar] [CrossRef]
- Veje, C.W.; Main, K.M.; Skakkebæk, N.E. Testicular dysgenesis syndrome: Foetal origin of adult reproductive problems. Clin. Endocrinol. 2009, 71, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Schneuer, F.J.; Holland, A.J.A.; Pereira, G.; Bower, C.; Nassar, N. Prevalence, repairs and complications of hypospadias: An Australian population-based study. Arch. Dis. Child. 2015, 100, 1038–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagodi, L.; Kiss, A.; Kiss-Toth, E.; Barkai, L. Prevalence and possible causes of hypospadias. Orv. Hetil. 2014, 155, 978–985. [Google Scholar] [CrossRef]
- Fisher, J.S. Environmental anti-androgens and male reproductive health: Focus on phthalates and testicular dysgenesis syndrome. Reproduction 2004, 127, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Giordano, F.; Abballe, A.; De Felip, E.; di Domenico, A.; Ferro, F.; Grammatico, P.; Ingelido, A.M.; Marra, V.; Marrocco, G.; Vallasciani, S.; et al. Maternal exposures to endocrine disrupting chemicals and hypospadias in offspring. Birth Defects Res. Part A Clin. Mol. Teratol. 2010, 88, 241–250. [Google Scholar] [CrossRef]
- Morales-Suárez-Varela, M.M.; Toft, G.V.; Jensen, M.S.; Ramlau-Hansen, C.; Kaerlev, L.; Thulstrup, A.-M.; Llopis-González, A.; Olsen, J.; Bonde, J.P. Parental occupational exposure to endocrine disrupting chemicals and male genital malformations: A study in the Danish National Birth Cohort study. Environ. Health 2011, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Kalfa, N.; Paris, F.; Philibert, P.; Orsini, M.; Broussous, S.; Fauconnet-Servant, N.; Audran, F.; Gaspari, L.; Lehors, H.; Haddad, M.; et al. Is hypospadias associated with prenatal exposure to endocrine disruptors? A French collaborative controlled study of a cohort of 300 consecutive children without genetic defect. Eur. Urol. 2015, 68, 1023–1030. [Google Scholar] [CrossRef]
- Skakkebaek, N.E.; Rajpert-De Meyts, E.; Buck Louis, G.M.; Toppari, J.; Andersson, A.M.; Eisenberg, M.L.; Jensen, T.K.; Jorgensen, N.; Swan, S.H.; Sapra, K.J.; et al. Male reproductive disorders and fertility trends: Influences of environment and genetic susceptibility. Physiol. Rev. 2015, 96, 55–97. [Google Scholar] [CrossRef] [Green Version]
- Winston, J.J.; Emch, M.; Meyer, R.E.; Langlois, P.; Weyer, P.; Mosley, B.; Olshan, A.F.; Band, L.E.; Luben, T.J. National birth defects prevention study hypospadias and maternal exposure to atrazine via drinking water in the national birth defects prevention study. Environ. Health 2016, 15, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damstra, T.; Barlow, S.; Bergman, A.; Kavlock, R.; Van Der Kraak, G. Global Assessment of the State-of-the-Science of Endocrine Disruptors; World Health Organisation: Geneva, Switzerland, 2002. [Google Scholar]
- Diamanti-Kandarakis, E.; Bourguignon, J.P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-disrupting chemicals: An endocrine society scientific statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef]
- Josh, M.K.S.; Pradeep, S.; Adarsh, V.K.; Amma, K.S.V.; Devi, R.S.; Balachandran, S.; Sreejith, M.N.; Jaleel, U.C.A.; Benjamin, S. In silico evidences for the binding of phthalates onto human estrogen receptor α, β subtypes and human estrogen-related receptor γ. Mol. Simul. 2014, 40, 408–417. [Google Scholar] [CrossRef]
- Takeuchi, S.; Iida, M.; Kobayashi, S.; Jin, K.; Matsuda, T.; Kojima, H. Differential effects of phthalate esters on transcriptional activities via human estrogen receptors α and β, and androgen receptor. Toxicology 2005, 210, 223–233. [Google Scholar] [CrossRef]
- Luo, Q.; Liu, Z.-H.; Yin, H.; Dang, Z.; Wu, P.-X.; Zhu, N.-W.; Lin, Z.; Liu, Y. Global review of phthalates in edible oil: An emerging and nonnegligible exposure source to human. Sci. Total Environ. 2020, 704, 135369. [Google Scholar] [CrossRef]
- Zambrano, E.; Guzmán, C.; Rodríguez-González, G.L.; Durand-Carbajal, M.; Nathanielsz, P.W. Fetal programming of sexual development and reproductive function. Mol. Cell Endocrinol. 2014, 382, 538–549. [Google Scholar] [CrossRef]
- Cripps, S.M.; Mattiske, D.M.; Black, J.R.; Risbridger, G.P.; Govers, L.C.; Phillips, T.R.; Pask, A.J. A loss of estrogen signaling in the aromatase deficient mouse penis results in mild hypospadias. Differentiation 2019, 109, 42–52. [Google Scholar] [CrossRef]
- Govers, L.C.; Phillips, T.R.; Mattiske, D.M.; Rashoo, N.; Black, J.R.; Sinclair, A.; Baskin, L.S.; Risbridger, G.P.; Pask, A.J. A critical role for estrogen signaling in penis development. FASEB J. 2019, 33, 10383–10392. [Google Scholar] [CrossRef] [Green Version]
- Lemmen, J.G.; Broekhof, J.L.; Kuiper, G.G.; Gustafsson, J.A.; van der Saag, P.T.; van der Burg, B. Expression of estrogen receptor alpha and beta during mouse embryogenesis. Mech. Dev. 1999, 81, 163–167. [Google Scholar] [CrossRef]
- Fowler, P.A.; Anderson, R.A.; Saunders, P.T.; Kinnell, H.; Mason, J.I.; Evans, D.B.; Bhattacharya, S.; Flannigan, S.; Franks, S.; Monteiro, A.; et al. Development of steroid signaling pathways during primordial follicle formation in the human fetal ovary. J. Clin. Endocrinol. Metab. 2011, 96, 1754–1762. [Google Scholar] [CrossRef] [Green Version]
- Fietz, D.; Ratzenböck, C.; Hartmann, K.; Raabe, O.; Kliesch, S.; Weidner, W.; Klug, J.; Bergmann, M. Expression pattern of estrogen receptors α and β and G-protein-coupled estrogen receptor 1 in the human testis. Histochem. Cell Biol. 2014, 142, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.T.; Sharpe, R.M.; Anderson, R.A.; McKinnell, C.; Macpherson, S.; Smith, L.B.; Wallace, W.H.B.; Kelnar, C.J.H.; van den Driesche, S. Diethylstilboestrol exposure does not reduce testosterone production in human fetal testis xenografts. PLoS ONE 2013, 8, e61726–e61727. [Google Scholar] [CrossRef] [Green Version]
- Stewart, M.K.; Mattiske, D.M.; Pask, A.J. In utero exposure to both high- and low-dose diethylstilbestrol disrupts mouse genital tubercle development. Biol. Reprod. 2018, 99, 1184–1193. [Google Scholar] [CrossRef]
- Mahawong, P.; Sinclair, A.; Li, Y.; Schlomer, B.; Rodriguez, E.; Ferretti, M.M.; Liu, B.; Baskin, L.S.; Cunha, G.R. Prenatal diethylstilbestrol induces malformation of the external genitalia of male and female mice and persistent second-generation developmental abnormalities of the external genitalia in two mouse strains. Differentiation 2014, 88, 51–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, C.L.; Christiansen, S.; Vinggaard, A.M.; Axelstad, M.; Hass, U.; Svingen, T. Anogenital distance as a toxicological or clinical marker for fetal androgen action and risk for reproductive disorders. Arch Toxicol. 2019, 93, 253–272. [Google Scholar] [CrossRef] [Green Version]
- Thankamony, A.; Pasterski, V.; Ong, K.K.; Acerini, C.L.; Hughes, I.A. Anogenital distance as a marker of androgen exposure in humans. Andrology 2016, 4, 616–625. [Google Scholar] [CrossRef] [Green Version]
- N’Tumba-Byn, T.; Moison, D.; Lacroix, M.; Lecureuil, C.; Lesage, L.; Prud’homme, S.M.; Pozzi-Gaudin, S.; Frydman, R.; Benachi, A.; Livera, G.; et al. Differential effects of bisphenol a and diethylstilbestrol on human, rat and mouse fetal Leydig cell function. PLoS ONE 2012, 7, e51579. [Google Scholar] [CrossRef] [Green Version]
- Desdoits-Lethimonier, C.; Lesné, L.; Gaudriault, P.; Zalko, D.; Antignac, J.P.; Deceuninck, Y.; Platel, C.; Dejucq-Rainsford, N.; Mazaud-Guittot, S.; Jégou, B. Parallel assessment of the effects of bisphenol A and several of its analogs on the adult human testis. Hum. Reprod. 2017, 32, 1465–1473. [Google Scholar] [CrossRef]
- Lymperi, S.; Giwercman, A. Endocrine disruptors and testicular function. Metabolism 2018, 86, 79–90. [Google Scholar] [CrossRef]
- Carmichael, S.L.; Cogswell, M.E.; Ma, C.; Gonzalez-Feliciano, A.; Olney, R.S.; Correa, A.; Shaw, G.M. The national birth defects prevention study hypospadias and maternal intake of phytoestrogens. Am. J. Epidemiol. 2013, 178, 434–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalfa, N.; Philibert, P.; Baskin, L.S.; Sultan, C. Hypospadias: Interactions between environment and genetics. Mol. Cell Endocrinol. 2011, 335, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Lassen, T.H.; Frederiksen, H.; Jensen, T.K.; Petersen, J.H.; Joensen, U.N.; Main, K.M.; Skakkebaek, N.E.; Juul, A.; Jørgensen, N.; Andersson, A.-M. Urinary bisphenol A levels in young men: Association with reproductive hormones and semen quality. Environ. Health Perspect. 2014, 122, 478–484. [Google Scholar] [CrossRef] [Green Version]
- Li, D.-K.; Zhou, Z.; Miao, M.; He, Y.; Wang, J.; Ferber, J.; Herrinton, L.J.; Gao, E.; Yuan, W. Urine bisphenol-A (BPA) level in relation to semen quality. Fertil. Steril. 2011, 95, 625.e304–630.e304. [Google Scholar] [CrossRef]
- Leavy, M.; Trottmann, M.; Liedl, B.; Reese, S.; Stief, C.; Freitag, B.; Baugh, J.; Spagnoli, G.; Kölle, S. Effects of elevated β-estradiol levels on the functional morphology of the testis—New insights. Sci. Rep. 2017, 7, 39931. [Google Scholar] [CrossRef]
- Jiang, D.D.; Swenson, E.; Mason, M.; Turner, K.R.; Dugi, D.D.; Hedges, J.C.; Hecht, S.L. Effects of estrogen on spermatogenesis in transgender women. Urology 2019, 132, 117–122. [Google Scholar] [CrossRef]
- Cunha, G.R.; Robboy, S.J.; Kurita, T.; Isaacson, D.; Shen, J.; Cao, M.; Baskin, L.S. Development of the human female reproductive tract. Differentiation 2018, 103, 46–65. [Google Scholar] [CrossRef] [Green Version]
- Johansson, H.K.L.; Svingen, T.; Fowler, P.A.; Vinggaard, A.M.; Boberg, J. Environmental influences on ovarian dysgenesis—developmental windows sensitive to chemical exposures. Nat. Rev. Endocrinol. 2017, 13, 400–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klip, H.; Verloop, J.; van Gool, J.D.; Koster, M.E.; Burger, C.W.; van Leeuwen, F.E. Hypospadias in sons of women exposed to diethylstilbestrol in utero: A cohort study. Lancet 2002, 359, 1102–1107. [Google Scholar] [CrossRef]
- Brouwers, M.M.; Feitz, W.F.; Roelofs, L.A.; Kiemeney, L.A.; de Gier, R.P.; Roeleveld, N. Hypospadias: A transgenerational effect of diethylstilbestrol? Hum. Reprod. 2006, 21, 666–669. [Google Scholar] [CrossRef] [Green Version]
- IARC Pharmaceuticals. A Review of Human Carcinogens; IARC Monographs on the Evaluation of Carcinogenic Risks to Humans/World Health Organization/International Agency for Research on Cancer: Lyon, France, 2012; Volume 100, pp. 1–401. [Google Scholar]
- Jishi, A.T.; Sergi, C. Current perspective of diethylstilbestrol (DES) exposure in mothers and offspring. Reprod. Toxicol. 2017, 71, 71–77. [Google Scholar] [CrossRef]
- Giusti, R.M.; Iwamoto, K.; Hatch, E.E. Diethylstilbestrol revisited: A review of the long-term health effects. Ann. Intern. Med. 1995, 122, 778–788. [Google Scholar] [CrossRef]
- Biro, F.M.; Pajak, A.; Wolff, M.S.; Pinney, S.M.; Windham, G.C.; Galvez, M.P.; Greenspan, L.C.; Kushi, L.H.; Teitelbaum, S.L. Age of menarche in a longitudinal US cohort. J. Pediatr. Adolesc. Gynecol. 2018, 31, 339–345. [Google Scholar] [CrossRef]
- Meng, X.; Li, S.; Duan, W.; Sun, Y.; Jia, C. Secular trend of age at menarche in chinese adolescents born from 1973 to 2004. Pediatrics 2017, 140, e20170085. [Google Scholar] [CrossRef] [Green Version]
- Pathak, P.K.; Tripathi, N.; Subramanian, S.V. Secular trends in menarcheal age in India-evidence from the Indian human development survey. PLoS ONE 2014, 9, e111027. [Google Scholar] [CrossRef] [Green Version]
- Aksglaede, L.; Sørensen, K.; Petersen, J.H.; Skakkebaek, N.E.; Juul, A. Recent decline in age at breast development: The Copenhagen puberty study. Pediatrics 2009, 123, e932–e939. [Google Scholar] [CrossRef] [Green Version]
- Fudvoye, J.; Lopez-Rodriguez, D.; Franssen, D.; Parent, A.-S. Endocrine disrupters and possible contribution to pubertal changes. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101300. [Google Scholar] [CrossRef] [PubMed]
- Palioura, E.; Diamanti-Kandarakis, E. Polycystic ovary syndrome (PCOS) and endocrine disrupting chemicals (EDCs). Rev. Endocr. Metab. Disord. 2016, 16, 365–371. [Google Scholar] [CrossRef]
- Patel, S.; Zhou, C.; Rattan, S.; Flaws, J.A. Effects of endocrine-disrupting chemicals on the ovary. Biol. Reprod. 2015, 93, 20. [Google Scholar] [CrossRef]
- Ge, W.; Li, L.; Dyce, P.W.; De Felici, M.; Shen, W. Establishment and depletion of the ovarian reserve: Physiology and impact of environmental chemicals. Cell. Mol. Life Sci. 2019, 76, 1729–1746. [Google Scholar] [CrossRef]
- Sirmans, S.M.; Pate, K.A. Epidemiology, diagnosis, and management of polycystic ovary syndrome. Clin. Epidemiol. 2013, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Golezar, S.; Tehrani, F.R.; Khazaei, S.; Ebadi, A.; Keshavarz, Z. The global prevalence of primary ovarian insufficiency and early menopause: A meta-analysis. Climacteric 2019, 22, 403–411. [Google Scholar] [CrossRef]
- Sowers, M.R.; La Pietra, M.T. Menopause: Its epidemiology and potential association with chronic diseases. Epidemiol. Rev. 1995, 17, 287–302. [Google Scholar] [CrossRef]
- Bagur, A.C.; Mautalen, C.A. Risk for developing osteoporosis in untreated premature menopause. Calcif. Tissue Int. 1992, 51, 4–7. [Google Scholar] [CrossRef]
- Britt, K.L.; Kerr, J.; O’Donnell, L.; Jones, M.E.E.; Drummond, A.E.; Davis, S.R.; Simpson, E.R.; Findlay, J.K. Estrogen regulates development of the somatic cell phenotype in the eutherian ovary. FASEB J. 2002, 16, 1389–1397. [Google Scholar] [CrossRef] [Green Version]
- Mathew, H.; Mahalingaiah, S. Do prenatal exposures pose a real threat to ovarian function? Bisphenol A as a case study. Reproduction 2019, 157, R143–R157. [Google Scholar] [CrossRef] [Green Version]
- Kandaraki, E.; Chatzigeorgiou, A.; Livadas, S.; Palioura, E.; Economou, F.; Koutsilieris, M.; Palimeri, S.; Panidis, D.; Diamanti-Kandarakis, E. Endocrine disruptors and polycystic ovary syndrome (PCOS): Elevated serum levels of bisphenol A in women with PCOS. J. Clin. Endocrinol. Metab. 2011, 96, E480–E484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, M.; Bourguignon, N.; Lux-Lantos, V.; Libertun, C. Neonatal exposure to bisphenol a and reproductive and endocrine alterations resembling the polycystic ovarian syndrome in adult rats. Environ. Health Perspect. 2010, 118, 1217–1222. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Hafner, K.S.; Flaws, J.A. In utero bisphenol A exposure disrupts germ cell nest breakdown and reduces fertility with age in the mouse. Toxicol. Appl. Pharmacol. 2014, 276, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Johansson, H.K.L.; Jacobsen, P.R.; Hass, U.; Svingen, T.; Vinggaard, A.M.; Isling, L.K.; Axelstad, M.; Christiansen, S.; Boberg, J. Perinatal exposure to mixtures of endocrine disrupting chemicals reduces female rat follicle reserves and accelerates reproductive aging. Reprod. Toxicol. 2016, 61, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Medigović, I.; Ristić, N.; Trifunović, S.; Manojlović-Stojanoski, M.; Milošević, V.; Zikić, D.; Nestorović, N. Genistein affects ovarian folliculogenesis: A stereological study. Microsc. Res. Tech. 2012, 75, 1691–1699. [Google Scholar] [CrossRef]
- Peretz, J.; Flaws, J.A. Bisphenol A down-regulates rate-limiting Cyp11a1 to acutely inhibit steroidogenesis in cultured mouse antral follicles. Toxicol. Appl. Pharmacol. 2013, 271, 249–256. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Ohno, S.; Nakajin, S. Effects of bisphenol A on the expression of cytochrome P450 aromatase (CYP19) in human fetal osteoblastic and granulosa cell-like cell lines. Toxicol. Lett. 2012, 210, 95–99. [Google Scholar] [CrossRef]
- Rice, S.; Mason, H.D.; Whitehead, S.A. Phytoestrogens and their low dose combinations inhibit mRNA expression and activity of aromatase in human granulosa-luteal cells. J. Steroid Biochem. Mol. Biol. 2006, 101, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Holleley, C.E.; Sarre, S.D.; O’Meally, D.; Georges, A. Sex reversal in reptiles: Reproductive oddity or powerful driver of evolutionary change? Sex Dev. 2016, 10, 279–287. [Google Scholar] [CrossRef]
- Flament, S. Sex reversal in amphibians. Sex Dev. 2016, 10, 267–278. [Google Scholar] [CrossRef]
- Major, A.T.; Smith, C.A. Sex reversal in birds. Sex Dev. 2016, 10, 288–300. [Google Scholar] [CrossRef]
- Kobayashi, H.; Iwamatsu, T. Sex reversal in the medaka Oryzias latipes by brief exposure of early embryos to estradiol-17β. Zool. Sci. 2005, 22, 1163–1167. [Google Scholar] [CrossRef]
- Burns, R.K. Experimental reversal of sex in the gonads of the opossum didelphis virginiana. Proc. Natl. Acad. Sci. USA 1955, 41, 669–676. [Google Scholar] [CrossRef] [Green Version]
- Coveney, D.; Shaw, G.; Renfree, M.B. Estrogen-induced gonadal sex reversal in the tammar wallaby. Biol. Reprod 2001, 65, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Pannetier, M.; Mandon-Pepin, B.; Copelli, S.; Fellous, M. Molecular aspects of female and male gonadal development in mammals. Pediatr. Endocrinol. Rev. 2004, 1, 274–287. [Google Scholar]
- Payen, E.; Pailhoux, E.; Abou Merhi, R.; Gianquinto, L.; Kirszenbaum, M.; Locatelli, A.; Cotinot, C. Characterization of ovine SRY transcript and developmental expression of genes involved in sexual differentiation. Int. J. Dev. Biol. 1996, 40, 567–575. [Google Scholar]
- Juengel, J.L.; Heath, D.A.; Quirke, L.D.; McNatty, K.P. Oestrogen receptor alpha and beta, androgen receptor and progesterone receptor mRNA and protein localisation within the developing ovary and in small growing follicles of sheep. Reproduction 2006, 131, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Garverick, H.A.; Juengel, J.L.; Smith, P.; Heath, D.A.; Burkhart, M.N.; Perry, G.A.; Smith, M.F.; McNatty, K.P. Development of the ovary and ontongeny of mRNA and protein for P450 aromatase (arom) and estrogen receptors (ER) α and β during early fetal life in cattle. Anim. Reprod. Sci. 2010, 117, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Schomberg, D.W.; Couse, J.F.; Mukherjee, A.; Lubahn, D.B.; Sar, M.; Mayo, K.E.; Korach, K.S. Targeted disruption of the estrogen receptor-alpha gene in female mice: Characterization of ovarian responses and phenotype in the adult. Endocrinology 1999, 140, 2733–2744. [Google Scholar] [CrossRef]
- Capel, B. Vertebrate sex determination: Evolutionary plasticity of a fundamental switch. Nat. Rev. Genet. 2017, 18, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Song, G.; Lim, W. A mechanism for the effect of endocrine disrupting chemicals on placentation. Chemosphere 2019, 231, 326–336. [Google Scholar] [CrossRef]
- Nef, S.; Stévant, I.; Greenfield, A. Characterizing the bipotential mammalian gonad. Curr. Top. Dev. Biol. 2019, 134, 167–194. [Google Scholar]
- Eggers, S.; Ohnesorg, T.; Sinclair, A. Genetic regulation of mammalian gonad development. Nat. Rev. Endocrinol. 2014, 10, 673–683. [Google Scholar] [CrossRef]
- Svingen, T.; Koopman, P. Building the mammalian testis: Origins, differentiation, and assembly of the component cell populations. Genes Dev. 2013, 27, 2409–2426. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.B.; O’Shaughnessy, P.J.; Rebourcet, D. Cell-specific ablation in the testis: What have we learned? Andrology 2015, 3, 1035–1049. [Google Scholar] [CrossRef] [Green Version]
- Rebourcet, D.; Darbey, A.; Monteiro, A.; Soffientini, U.; Tsai, Y.T.; Handel, I.; Pitetti, J.-L.; Nef, S.; Smith, L.B.; O’Shaughnessy, P.J. Sertoli cell number defines and predicts germ and Leydig cell population sizes in the adult mouse testis. Endocrinology 2017, 158, 2955–2969. [Google Scholar] [CrossRef] [Green Version]
- Hacker, A.; Capel, B.; Goodfellow, P.; Lovell-Badge, R. Expression of Sry, the mouse sex determining gene. Development 1995, 121, 1603–1614. [Google Scholar]
- Sinclair, A.H.; Berta, P.; Palmer, M.S.; Hawkins, J.R.; Griffiths, B.L.; Smith, M.J.; Foster, J.W.; Frischauf, A.-M.; Lovell-Badge, R.; Goodfellow, P.N. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 1990, 346, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Hiramatsu, R.; Matoba, S.; Kanai-Azuma, M.; Tsunekawa, N.; Katoh-Fukui, Y.; Kurohmaru, M.; Morohashi, K.I.; Wilhelm, D.; Koopman, P.; Kanai, Y. A critical time window of Sry action in gonadal sex determination in mice. Development 2008, 136, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Nagamine, C.M.; Morohashi, K.; Carlisle, C.; Chang, D.K. Sex reversal caused by Mus musculus domesticus Y chromosomes linked to variant expression of the testis-determining gene Sry. Dev. Biol. 1999, 216, 182–194. [Google Scholar] [CrossRef]
- Morais da Silva, S.; Hacker, A.; Harley, V.; Goodfellow, P.; Swain, A.; Lovell-Badge, R. Sox9 expression during gonadal development implies a conserved role for the gene in testis differentiation in mammals and birds. Nat. Genet. 1996, 14, 62–68. [Google Scholar] [CrossRef]
- de Santa Barbara, P.; Moniot, B.; Poulat, F.; Berta, P. Expression and subcellular localization of SF-1, SOX9, WT1, and AMH proteins during early human testicular development. Dev. Dyn. 2000, 217, 293–298. [Google Scholar] [CrossRef]
- Qin, Y.; Bishop, C.E. Sox9 is sufficient for functional testis development producing fertile male mice in the absence of Sry. Hum. Mol. Gen. 2005, 14, 1221–1229. [Google Scholar] [CrossRef] [Green Version]
- Wagner, T.; Wirth, J.; Meyer, J.; Zabel, B.; Held, M.; Zimmer, J.; Pasantes, J.; Bricarelli, F.D.; Keutel, J.; Hustert, E.; et al. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell 1994, 79, 1111–1120. [Google Scholar] [CrossRef]
- Foster, J.W.; Dominguez-Steglich, M.A.; Guioli, S.; Kwok, C.; Weller, P.A.; Stevanovic, M.; Weissenbach, J.; Mansour, S.; Young, I.D.; Goodfellow, P.N. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature 1994, 372, 525–530. [Google Scholar] [CrossRef]
- Colvin, J.S.; Feldman, B.; Nadeau, J.H.; Goldfarb, M.; Ornitz, D.M. Genomic organization and embryonic expression of the mouse fibroblast growth factor 9 gene. Dev. Dyn. 1999, 216, 72–88. [Google Scholar] [CrossRef]
- Schmahl, J.; Kim, Y.; Colvin, J.S.; Ornitz, D.M.; Capel, B. Fgf9 induces proliferation and nuclear localization of FGFR2 in Sertoli precursors during male sex determination. Development 2004, 131, 3627–3636. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Kobayashi, A.; Sekido, R.; DiNapoli, L.; Brennan, J.; Chaboissier, M.-C.; Poulat, F.; Behringer, R.R.; Lovell-Badge, R.; Capel, B. Fgf9 and Wnt4 act as antagonistic signals to regulate mammalian sex determination. PLoS Biol. 2006, 4, e187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colvin, J.S.; Green, R.P.; Schmahl, J.; Capel, B.; Ornitz, D.M. Male-to-female sex reversal in mice lacking fibroblast growth factor 9. Cell 2001, 104, 875–889. [Google Scholar] [CrossRef]
- Bagheri-Fam, S.; Ono, M.; Li, L.; Zhao, L.; Ryan, J.; Lai, R.; Katsura, Y.; Rossello, F.J.; Koopman, P.; Scherer, G.; et al. FGFR2 mutation in 46,XY sex reversal with craniosynostosis. Hum. Mol. Gen. 2015, 24, 6699–6710. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, D.; Martinson, F.; Bradford, S.; Wilson, M.J.; Combes, A.N.; Beverdam, A.; Bowles, J.; Mizusaki, H.; Koopman, P. Sertoli cell differentiation is induced both cell-autonomously and through prostaglandin signaling during mammalian sex determination. Dev. Biol. 2005, 287, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, D.; Hiramatsu, R.; Mizusaki, H.; Widjaja, L.; Combes, A.N.; Kanai, Y.; Koopman, P. SOX9 regulates prostaglandin D synthase gene transcription in vivo to ensure testis development. J. Biol. Chem. 2007, 282, 10553–10560. [Google Scholar] [CrossRef] [Green Version]
- Adams, I.R.; McLaren, A. Sexually dimorphic development of mouse primordial germ cells: Switching from oogenesis to spermatogenesis. Development 2002, 129, 1155–1164. [Google Scholar]
- Moniot, B.; Declosmenil, F.; Barrionuevo, F.; Scherer, G.; Aritake, K.; MALKI, S.; Marzi, L.; Cohen-Solal, A.; Georg, I.; Klattig, J.; et al. The PGD2 pathway, independently of FGF9, amplifies SOX9 activity in Sertoli cells during male sexual differentiation. Development 2009, 136, 1813–1821. [Google Scholar] [CrossRef] [Green Version]
- de Santa Barbara, P.; Bonneaud, N.; Boizet, B.; Desclozeaux, M.; Moniot, B.; Sudbeck, P.; Scherer, G.; Poulat, F.; Berta, P. Direct interaction of SRY-related protein SOX9 and steroidogenic factor 1 regulates transcription of the human anti-Müllerian hormone gene. Mol. Cell. Biol. 1998, 18, 6653–6665. [Google Scholar] [CrossRef] [Green Version]
- Arango, N.A.; Lovell-Badge, R.; Behringer, R.R. Targeted mutagenesis of the endogenous mouse Mis gene promoter: In vivo definition of genetic pathways of vertebrate sexual development. Cell 1999, 99, 409–419. [Google Scholar] [CrossRef] [Green Version]
- Josso, N.; Cate, R.L.; Picard, J.Y.; Vigier, B.; di Clemente, N.; Wilson, C.; Imbeaud, S.; Pepinsky, R.B.; Guerrier, D.; Boussin, L. Anti-müllerian hormone: The Jost factor. Recent Prog. Horm. Res. 1993, 48, 1–59. [Google Scholar]
- Behringer, R.R.; Cate, R.L.; Froelick, G.J.; Palmiter, R.D.; Brinster, R.L. Abnormal sexual development in transgenic mice chronically expressing Müllerian inhibiting substance. Nature 1990, 345, 167–170. [Google Scholar] [CrossRef]
- Vainio, S.; Heikkilä, M.; Kispert, A.; Chin, N.; McMahon, A.P. Female development in mammals is regulated by Wnt-4 signalling. Nature 1999, 397, 405–409. [Google Scholar] [CrossRef]
- Parma, P.; Radi, O.; Vidal, V.; Chaboissier, M.-C.; Dellambra, E.; Valentini, S.; Guerra, L.; Schedl, A.; Camerino, G. R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat. Genet 2006, 38, 1304–1309. [Google Scholar] [CrossRef]
- Kim, K.-A.; Zhao, J.; Andarmani, S.; Kakitani, M.; Oshima, T.; Binnerts, M.E.; Abo, A.; Tomizuka, K.; Funk, W.D. R-Spondin proteins: A novel link to beta-catenin activation. Cell Cycle 2006, 5, 23–26. [Google Scholar] [CrossRef]
- Manuylov, N.L.; Smagulova, F.O.; Leach, L.; Tevosian, S.G. Ovarian development in mice requires the GATA4-FOG2 transcription complex. Development 2008, 135, 3731–3743. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, D.; Ovitt, C.E.; Anlag, K.; Fehsenfeld, S.; Gredsted, L.; Treier, A.-C.; Treier, M. The murine winged-helix transcription factor Foxl2 is required for granulosa cell differentiation and ovary maintenance. Development 2004, 131, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, L.; Hu, Y.; Chen, M.; Han, F.; Qin, Y.; Chen, M.; Cui, X.; Duo, S.; Tang, F.; et al. β-Catenin directs the transformation of testis Sertoli cells to ovarian granulosa-like cells by inducing Foxl2 expression. J. Biol. Chem. 2017, 292, 17577–17586. [Google Scholar] [CrossRef] [Green Version]
- Nicol, B.; Grimm, S.A.; Gruzdev, A.; Scott, G.J.; Ray, M.K.; Yao, H.H.C. Genome-wide identification of FOXL2 binding and characterization of FOXL2 feminizing action in the fetal gonads. Hum. Mol. Gen. 2018, 27, 4273–4287. [Google Scholar] [CrossRef]
- Crisponi, L.; Deiana, M.; Loi, A.; Chiappe, F.; Uda, M.; Amati, P.; Bisceglia, L.; Zelante, L.; Nagaraja, R.; Porcu, S.; et al. The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat. Genet. 2001, 27, 159–166. [Google Scholar] [CrossRef]
- Pailhoux, E.; Vigier, B.; Chaffaux, S.; Servel, N.; Taourit, S.; Furet, J.P.; Fellous, M.; Grosclaude, F.; Cribiu, E.P.; Cotinot, C.; et al. A 11.7-kb deletion triggers intersexuality and polledness in goats. Nat. Genet. 2001, 29, 453–458. [Google Scholar] [CrossRef]
- Georges, A.; L’Hôte, D.; Todeschini, A.L.; Auguste, A.; Legois, B.; Zider, A.; Veitia, R.A. The transcription factor FOXL2 mobilizes estrogen signaling to maintain the identity of ovarian granulosa cells. eLife 2014, 3, e85545. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Sonenshein, G.E. Forkhead box transcription factor FOXO3a regulates estrogen receptor alpha expression and is repressed by the Her-2/neu/phosphatidylinositol 3-kinase/Akt signaling pathway. Mol. Cell. Biol. 2004, 24, 8681–8690. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, K.; Horie-Inoue, K.; Inoue, S. Identification of estrogen-responsive genes based on the DNA binding properties of estrogen receptors using high-throughput sequencing technology. Acta Pharmacol. Sin. 2015, 36, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Hurtado, A.; Holmes, K.A.; Ross-Innes, C.S.; Schmidt, D.; Carroll, J.S. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat. Genet. 2011, 43, 27–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, B.K.; Shen, J.H.-C.; Olaso, R.; Ingraham, H.A.; Vilain, E. Wnt4 overexpression disrupts normal testicular vasculature and inhibits testosterone synthesis by repressing steroidogenic factor 1/beta-catenin synergy. Proc. Natl. Acad. Sci. USA 2003, 100, 10866–10871. [Google Scholar] [CrossRef] [Green Version]
- Ottolenghi, C.; Pelosi, E.; Tran, J.; Colombino, M.; Douglass, E.; Nedorezov, T.; Cao, A.; Forabosco, A.; Schlessinger, D. Loss of Wnt4 and Foxl2 leads to female-to-male sex reversal extending to germ cells. Hum. Mol. Gen. 2007, 16, 2795–2804. [Google Scholar] [CrossRef] [Green Version]
- Bernard, P.; Sim, H.; Knower, K.; Vilain, E.; Harley, V. Human SRY inhibits β-catenin-mediated transcription. Int. J. Biochem. Cell Biol. 2008, 40, 2889–2900. [Google Scholar] [CrossRef] [Green Version]
- Topol, L.; Chen, W.; Song, H.; Day, T.F.; Yang, Y. Sox9 inhibits Wnt signaling by promoting beta-catenin phosphorylation in the nucleus. J. Biol. Chem. 2009, 284, 3323–3333. [Google Scholar] [CrossRef] [Green Version]
- Bernard, P.; Ryan, J.; Sim, H.; Czech, D.P.; Sinclair, A.H.; Koopman, P.; Harley, V.R. Wnt signaling in ovarian development inhibits Sf1 activation of Sox9 via the Tesco enhancer. Endocrinol 2012, 153, 901–912. [Google Scholar] [CrossRef] [Green Version]
- Ostrer, H. Disorders of sex development (DSDs): An update. J. Clin. Endocrinol. Metab. 2014, 99, 1503–1509. [Google Scholar] [CrossRef] [Green Version]
- Bogani, D.; Siggers, P.; Brixey, R.; Warr, N.; Beddow, S.; Edwards, J.; Williams, D.; Wilhelm, D.; Koopman, P.; Flavell, R.A.; et al. Loss of mitogen-activated protein kinase kinase kinase 4 (MAP3K4) reveals a requirement for mapk signalling in mouse sex determination. PLoS Biol. 2009, 7, e1000196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, Z.; Takekawa, M.; Ge, Q.; Saito, H. Activation of MTK1/MEKK4 by GADD45 through induced N-C dissociation and dimerization-mediated trans autophosphorylation of the MTK1 kinase domain. Mol. Cell Biol. 2007, 27, 2765–2776. [Google Scholar] [CrossRef] [Green Version]
- Gierl, M.S.; Gruhn, W.H.; von Seggern, A.; Maltry, N.; Niehrs, C. GADD45G functions in male sex determination by promoting p38 signaling and sry expression. Dev. Cell 2012, 23, 1032–1042. [Google Scholar] [CrossRef] [Green Version]
- Warr, N.; Carre, G.-A.; Siggers, P.; Faleato, J.V.; Brixey, R.; Pope, M.; Bogani, D.; Childers, M.; Wells, S.; Scudamore, C.L.; et al. Gadd45γ and Map3k4 interactions regulate mouse testis determination via p38 MAPK-mediated control of Sry expression. Dev. Cell 2012, 23, 1020–1031. [Google Scholar] [CrossRef] [Green Version]
- Warr, N.; Bogani, D.; Siggers, P.; Brixey, R.; Tateossian, H.; Dopplapudi, A.; Wells, S.; Cheeseman, M.; Xia, Y.; Ostrer, H.; et al. Minor abnormalities of testis development in mice lacking the gene encoding the MAPK signalling component, MAP3K1. PLoS ONE 2011, 6, e19572. [Google Scholar] [CrossRef] [Green Version]
- Loke, J.; Pearlman, A.; Radi, O.; Zuffardi, O.; Giussani, U.; Pallotta, R.; Camerino, G.; Ostrer, H. Mutations in MAP3K1 tilt the balance from SOX9/FGF9 to WNT/beta-catenin signaling. Hum. Mol. Gen. 2014, 23, 1073–1083. [Google Scholar] [CrossRef] [Green Version]
- Pearlman, A.; Loke, J.; Le Caignec, C.; White, S.; Chin, L.; Friedman, A.; Warr, N.; Willan, J.; Brauer, D.; Farmer, C.; et al. Mutations in MAP3K1 cause 46,XY disorders of sex development and implicate a common signal transduction pathway in human testis determination. Am. J. Hum. Genet. 2010, 87, 898–904. [Google Scholar] [CrossRef]
- Chamberlin, A.; Huether, R.; Machado, A.Z.; Groden, M.; Liu, H.-M.; Upadhyay, K.; Vivian, O.; Gomes, N.L.; Lerario, A.M.; Nishi, M.Y.; et al. Mutations in MAP3K1 that cause 46,XY disorders of sex development disrupt distinct structural domains in the protein. Hum. Mol. Gen. 2019, 28, 1620–1628. [Google Scholar] [CrossRef]
- Trukhina, A.V.; Lukina, N.A.; Wackerow-Kouzova, N.D.; Smirnov, A.F. The variety of vertebrate mechanisms of sex determination. Biomed. Res. Int. 2013, 2013, 587460. [Google Scholar] [CrossRef] [PubMed]
- Pask, A.J. A role for estrogen in somatic cell fate of the mammalian gonad. Chromosome Res. 2012, 20, 239–245. [Google Scholar] [CrossRef] [Green Version]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29, 2905–2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björnström, L.; Sjöberg, M. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, S.; Mäkelä, S.; Treuter, E.; Tujague, M.; Thomsen, J.; Andersson, G.; Enmark, E.; Pettersson, K.; Warner, M.; Gustafsson, J.A. Mechanisms of estrogen action. Physiol. Rev. 2001, 81, 1535–1565. [Google Scholar] [CrossRef]
- Stefkovich, M.L.; Arao, Y.; Hamilton, K.J.; Korach, K.S. Experimental models for evaluating non-genomic estrogen signaling. Steroids 2018, 133, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Meroni, S.B.; Galardo, M.N.; Rindone, G.; Gorga, A.; Riera, M.F.; Cigorraga, S.B. Molecular mechanisms and signaling pathways involved in Sertoli cell proliferation. Front. Endocrinol. 2019, 10, 241. [Google Scholar] [CrossRef]
- Jessop, H.L.; Sjöberg, M.; Cheng, M.Z.; Zaman, G.; Wheeler-Jones, C.P.; Lanyon, L.E. Mechanical strain and estrogen activate estrogen receptor alpha in bone cells. J. Bone Miner. Res. 2001, 16, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Endoh, H.; Sasaki, H.; Maruyama, K.; Takeyama, K.; Waga, I.; Shimizu, T.; Kato, S.; Kawashima, H. Rapid activation of MAP kinase by estrogen in the bone cell line. Biochem. Biophys. Res. 1997, 235, 99–102. [Google Scholar] [CrossRef]
- Chen, Z.; Yuhanna, I.S.; Galcheva-Gargova, Z.; Karas, R.H.; Mendelsohn, M.E.; Shaul, P.W. Estrogen receptor α mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J. Clin. Invest. 1999, 103, 401–406. [Google Scholar] [CrossRef] [Green Version]
- Watters, J.J.; Campbell, J.S.; Cunningham, M.J.; Krebs, E.G.; Dorsa, D.M. Rapid membrane effects of steroids in neuroblastoma cells: Effects of estrogen on mitogen activated protein kinase signalling cascade and c-fos immediate early gene transcription. Endocrinology 1997, 138, 4030–4033. [Google Scholar] [CrossRef]
- Stewart, M.K.; Mattiske, D.M.; Pask, A.J. Oestrogen regulates SOX9 bioavailability by rapidly activating ERK1/2 and stabilising microtubules in a human testis-derived cell line. Exp. Cell Res. 2020. (under review). [Google Scholar]
- Eblen, S.T. Extracellular-regulated kinases: Signaling from RAS to ERK substrates to control biological outcomes. Adv. Cancer Res. 2018, 138, 99–142. [Google Scholar]
- Paranjpe, M.; Yu, H.; Frankenberg, S.; Pask, A.J.; Shaw, G.; Renfree, M.B. Transcriptomic analysis of MAP3K1 and MAP3K4 in the developing marsupial gonad. Sex Dev. 2019, 13, 195–204. [Google Scholar] [CrossRef]
- Nanjappa, M.K.; Medrano, T.I.; Mesa, A.M.; Ortega, M.T.; Caldo, P.D.; Mao, J.; Kinkade, J.A.; Levin, E.R.; Rosenfeld, C.S.; Cooke, P.S. Mice lacking membrane estrogen receptor 1 are protected from reproductive pathologies resulting from developmental estrogen exposure. Biol. Reprod. 2019, 101, 392–404. [Google Scholar] [CrossRef]
- Varea, O.; Garrido, J.J.; Dopazo, A.; Mendez, P.; Garcia-Segura, L.M.; Wandosell, F. Estradiol activates beta-catenin dependent transcription in neurons. PLoS ONE 2009, 4, e5153. [Google Scholar] [CrossRef] [Green Version]
- Kouzmenko, A.P.; Takeyama, K.-I.; Ito, S.; Furutani, T.; Sawatsubashi, S.; Maki, A.; Suzuki, E.; Kawasaki, Y.; Akiyama, T.; Tabata, T.; et al. Wnt/beta-catenin and estrogen signaling converge in vivo. J. Biol. Chem. 2004, 279, 40255–40258. [Google Scholar] [CrossRef] [Green Version]
- Cardona-Gomez, P.; Pérez, M.; Avila, J.; Garcia-Segura, L.M.; Wandosell, F. Estradiol inhibits GSK3 and regulates interaction of estrogen receptors, GSK3, and beta-catenin in the hippocampus. Mol. Cell. Neurosci. 2004, 25, 363–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, D.; Hawke, D.; Zheng, Y.; Xia, Y.; Meisenhelder, J.; Nika, H.; Mills, G.B.; Kobayashi, R.; Hunter, T.; Lu, Z. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J. Biol. Chem. 2007, 282, 11221–11229. [Google Scholar] [CrossRef] [Green Version]
- Kawagoe, J.; Ohmichi, M.; Takahashi, T.; Ohshima, C.; Mabuchi, S.; Takahashi, K.; Igarashi, H.; Mori-Abe, A.; Saitoh, M.; Du, B.; et al. Raloxifene inhibits estrogen-induced up-regulation of telomerase activity in a human breast cancer cell line. J. Biol. Chem. 2003, 278, 43363–43372. [Google Scholar] [CrossRef] [Green Version]
- Prossnitz, E.R. GPER modulators: Opportunity Nox on the heels of a class Akt. J. Steroid Biochem. Mol. Biol. 2018, 176, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [Green Version]
- Taurin, S.; Sandbo, N.; Qin, Y.; Browning, D.; Dulin, N.O. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase. J. Biol. Chem. 2006, 281, 9971–9976. [Google Scholar] [CrossRef] [Green Version]
- Sassone-Corsi, P. The cyclic AMP pathway. Cold. Spring Harb. Perspect Biol. 2012, 4, a011148. [Google Scholar] [CrossRef] [PubMed]
- Aronica, S.M.; Kraus, W.L.; Katzenellenbogen, B.S. Estrogen action via the cAMP signaling pathway: Stimulation of adenylate cyclase and cAMP-regulated gene transcription. Proc. Natl. Acad. Sci. USA 1994, 91, 8517–8521. [Google Scholar] [CrossRef] [Green Version]
- Bennesch, M.A.; Segala, G.; Wider, D.; Picard, D. LSD1 engages a corepressor complex for the activation of the estrogen receptor α by estrogen and cAMP. Nucleic Acids Res. 2016, 44, 8655–8670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carascossa, S.; Dudek, P.; Cenni, B.; Briand, P.-A.; Picard, D. CARM1 mediates the ligand-independent and tamoxifen-resistant activation of the estrogen receptor alpha by cAMP. Genes Dev. 2010, 24, 708–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.; Wang, Y.; Huang, B.; Liang, J.; Ding, Y.; Xu, A.; Wu, W. A Rac1/PAK1 cascade controls β-catenin activation in colon cancer cells. Oncogene 2012, 31, 1001–1012. [Google Scholar] [CrossRef] [Green Version]
- Mazumdar, A.; Kumar, R. Estrogen regulation of Pak1 and FKHR pathways in breast cancer cells. FEBS Lett. 2003, 535, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Raghavan, S.; Venkatraman, G.; Rayala, S.K. Cloning and functional characterization of human Pak1 promoter by steroid hormones. Gene 2018, 646, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Ni, F.-D.; Hao, S.-L.; Yang, W.-X. Molecular insights into hormone regulation via signaling pathways in Sertoli cells: With discussion on infertility and testicular tumor. Gene 2020, 753, 144812. [Google Scholar] [CrossRef]
- Barske, L.A.; Capel, B. Estrogen represses SOX9 during sex determination in the red-eared slider turtle Trachemys scripta. Dev. Biol. 2010, 341, 305–314. [Google Scholar] [CrossRef]
- Lambeth, L.S.; Cummins, D.M.; Doran, T.J.; Sinclair, A.H.; Smith, C.A. Overexpression of aromatase alone is sufficient for ovarian development in genetically male chicken embryos. PLoS ONE 2013, 8, e68362. [Google Scholar] [CrossRef] [Green Version]
- Durando, M.; Cocito, L.; Rodriguez, H.A.; Varayoud, J.; Ramos, J.G.; Luque, E.H.; Muñoz-de-Toro, M. Neonatal expression of amh, sox9 and sf-1 mRNA in Caiman latirostris and effects of in ovo exposure to endocrine disrupting chemicals. Gen. Comp. Endocrinol. 2013, 191, 31–38. [Google Scholar] [CrossRef]
- Sim, H.; Argentaro, A.; Harley, V.R. Boys, girls and shuttling of SRY and SOX9. Trends Endocrinol. Metab. 2008, 19, 213–222. [Google Scholar] [CrossRef]
- Piasecka, D.; Braun, M.; Kitowska, K.; Mieczkowski, K.; Kordek, R.; Sadej, R.; Romanska, H. FGFs/FGFRs-dependent signalling in regulation of steroid hormone receptors—implications for therapy of luminal breast cancer. J. Exp. Clin. Cancer Res. 2019, 38, 1171. [Google Scholar] [CrossRef] [Green Version]
- Bourdeau, V.; Deschênes, J.; Métivier, R.; Nagai, Y.; Nguyen, D.; Bretschneider, N.; Gannon, F.; White, J.H.; Mader, S. Genome-wide identification of high-affinity estrogen response elements in human and mouse. Mol. Endocrinol. 2004, 18, 1411–1427. [Google Scholar] [CrossRef] [Green Version]
- Grynberg, M.; Pierre, A.; Rey, R.; Leclerc, A.; Arouche, N.; Hesters, L.; Catteau-Jonard, S.; Frydman, R.; Picard, J.-Y.; Fanchin, R.; et al. Differential regulation of ovarian anti-müllerian hormone (AMH) by estradiol through α- and β-estrogen receptors. J. Clin. Endocrinol. Metab. 2012, 97, E1649–E1657. [Google Scholar] [CrossRef] [Green Version]
- Weenen, C.; Laven, J.S.E.; Von Bergh, A.R.M.; Cranfield, M.; Groome, N.P.; Visser, J.A.; Kramer, P.; Fauser, B.C.J.M.; Themmen, A.P.N. Anti-Müllerian hormone expression pattern in the human ovary: Potential implications for initial and cyclic follicle recruitment. Mol. Hum. Reprod. 2004, 10, 77–83. [Google Scholar] [CrossRef]
- Durlinger, A.L.L.; Gruijters, M.J.G.; Kramer, P.; Karels, B.; Ingraham, H.A.; Nachtigal, M.W.; Uilenbroek, J.T.J.; Grootegoed, J.A.; Themmen, A.P.N. Anti-Müllerian hormone inhibits initiation of primordial follicle growth in the mouse ovary. Endocrinology 2002, 143, 1076–1084. [Google Scholar] [CrossRef]
- Lv, Y.; Li, L.; Fang, Y.; Chen, P.; Wu, S.; Chen, X.; Ni, C.; Zhu, Q.; Huang, T.; Lian, Q.; et al. In utero exposure to bisphenol A disrupts fetal testis development in rats. Environ. Pollut. 2019, 246, 217–224. [Google Scholar] [CrossRef]
- Jones, S.; Boisvert, A.; Duong, T.B.; Francois, S.; Thrane, P.; Culty, M. Disruption of rat testis development following combined in utero exposure to the phytoestrogen genistein and antiandrogenic plasticizer di-(2-ethylhexyl) phthalate. Biol. Reprod. 2014, 91, 64. [Google Scholar] [CrossRef] [PubMed]
- Strauss, L.; Kallio, J.; Desai, N.; Pakarinen, P.; Miettinen, T.; Gylling, H.; Albrecht, M.; Mäkelä, S.; Mayerhofer, A.; Poutanen, M. Increased exposure to estrogens disturbs maturation, steroidogenesis, and cholesterol homeostasis via estrogen receptor alpha in adult mouse Leydig cells. Endocrinology 2009, 150, 2865–2872. [Google Scholar] [CrossRef] [Green Version]
- Sakakibara, M.; Uenoyama, Y.; Minabe, S.; Watanabe, Y.; Deura, C.; Nakamura, S.; Suzuki, G.; Maeda, K.-I.; Tsukamura, H. Microarray analysis of perinatal-estrogen-induced changes in gene expression related to brain sexual differentiation in mice. PLoS ONE 2013, 8, e79437. [Google Scholar] [CrossRef] [Green Version]
- Nicol, B.; Grimm, S.A.; Chalmel, F.; Lecluze, E.; Pannetier, M.; Pailhoux, E.; Dupin-De-Beyssat, E.; Guiguen, Y.; Capel, B.; Yao, H.H.C. RUNX1 maintains the identity of the fetal ovary through an interplay with FOXL2. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Mulligan, W.A.; Wegner, K.A.; Keil, K.P.; Mehta, V.; Taketo, M.M.; Vezina, C.M. Beta-catenin and estrogen signaling collaborate to drive cyclin D1 expression in developing mouse prostate. Differentiation 2017, 93, 66–71. [Google Scholar] [CrossRef] [Green Version]
- Ray, S.; Xu, F.; Wang, H.; Das, S.K. Cooperative control via lymphoid enhancer factor 1/T cell factor 3 and estrogen receptor-α for uterine gene regulation by estrogen. Mol. Endocrinol. 2008, 22, 1125–1140. [Google Scholar] [CrossRef] [Green Version]
- Chimge, N.-O.; Little, G.H.; Baniwal, S.K.; Adisetiyo, H.; Xie, Y.; Zhang, T.; O’Laughlin, A.; Liu, Z.Y.; Ulrich, P.; Martin, A.; et al. RUNX1 prevents oestrogen-mediated AXIN1 suppression and β-catenin activation in ER-positive breast cancer. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Miyakoshi, T.; Kajiya, H.; Miyajima, K.; Takei, M.; Tobita, M.; Takekoshi, S.; Osamura, R.Y. The expression of Wnt4 is regulated by estrogen via an estrogen receptor alpha-dependent pathway in rat pituitary growth hormone-producing cells. Acta Histochem. Cytochem. 2009, 42, 205–213. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stewart, M.K.; Mattiske, D.M.; Pask, A.J. Exogenous Oestrogen Impacts Cell Fate Decision in the Developing Gonads: A Potential Cause of Declining Human Reproductive Health. Int. J. Mol. Sci. 2020, 21, 8377. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218377
Stewart MK, Mattiske DM, Pask AJ. Exogenous Oestrogen Impacts Cell Fate Decision in the Developing Gonads: A Potential Cause of Declining Human Reproductive Health. International Journal of Molecular Sciences. 2020; 21(21):8377. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218377
Chicago/Turabian StyleStewart, Melanie K., Deidre M. Mattiske, and Andrew J. Pask. 2020. "Exogenous Oestrogen Impacts Cell Fate Decision in the Developing Gonads: A Potential Cause of Declining Human Reproductive Health" International Journal of Molecular Sciences 21, no. 21: 8377. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218377