Platelet-Reactive Antibodies in Patients after Ischaemic Stroke—An Epiphenomenon or a Natural Protective Mechanism

 , , , and
, , , and 
Abstract
:1. Introduction
2. Results
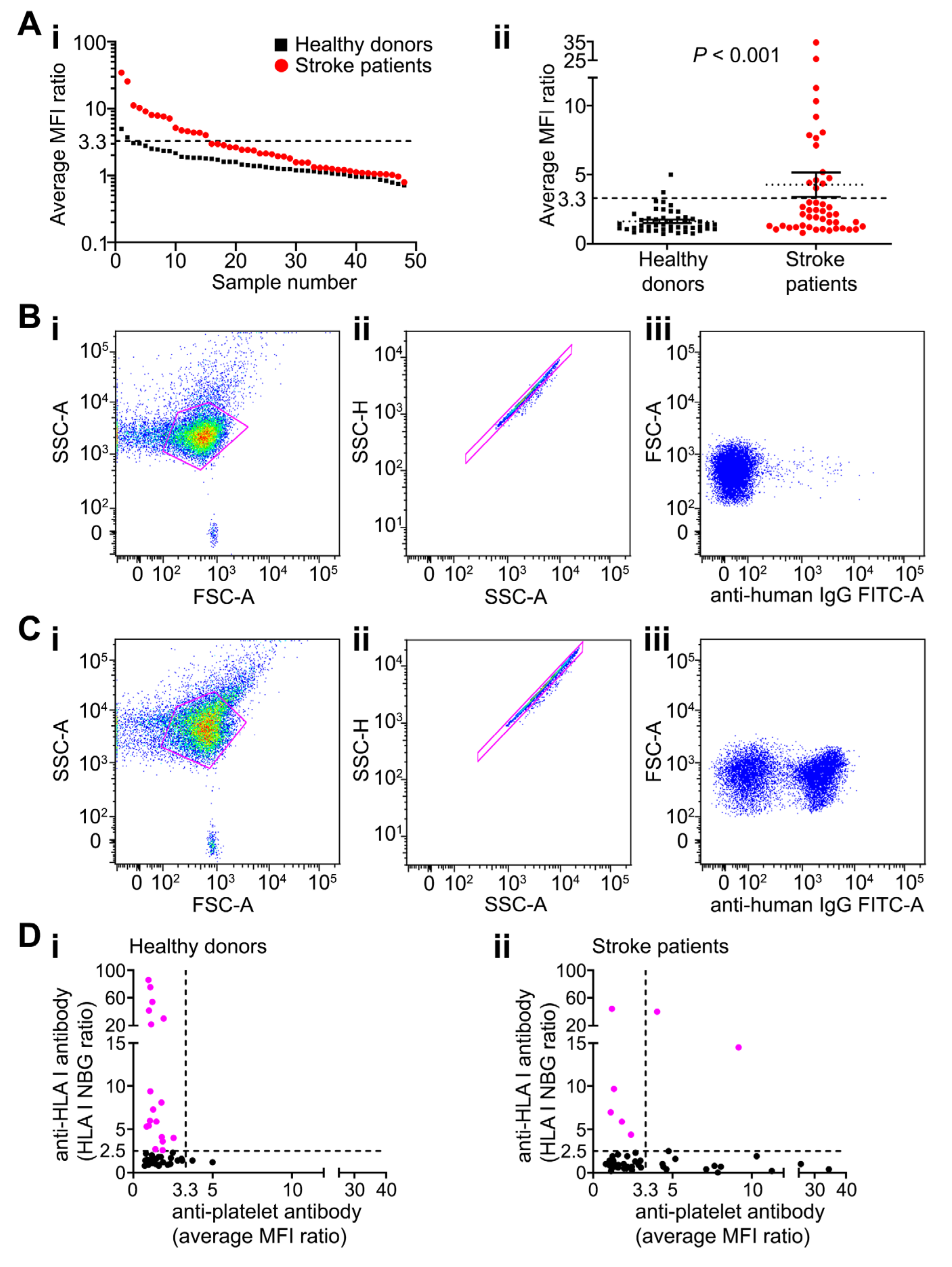
2.1. Platelet-Reactive Antibodies are Found in Peripheral Blood of Patients after Stroke
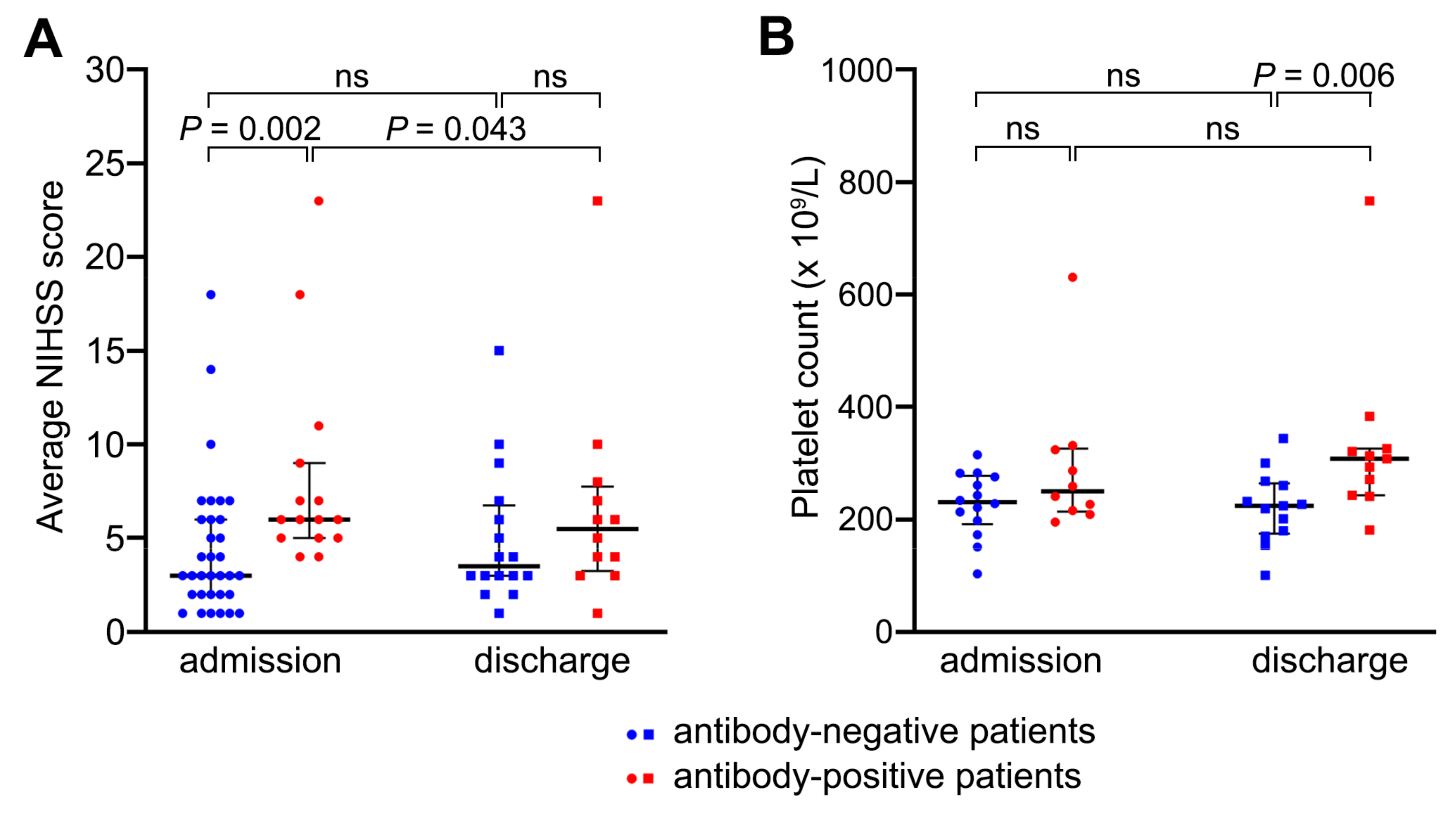
2.2. Patients with Anti-Platelet Antibodies Had Larger Strokes but Had Better Recovery from Neurological Dysfunction by Day 7 Compared with Antibody-Negative Patients
2.3. Anti-Platelet Antibodies Did Not Cause Platelet Consumption
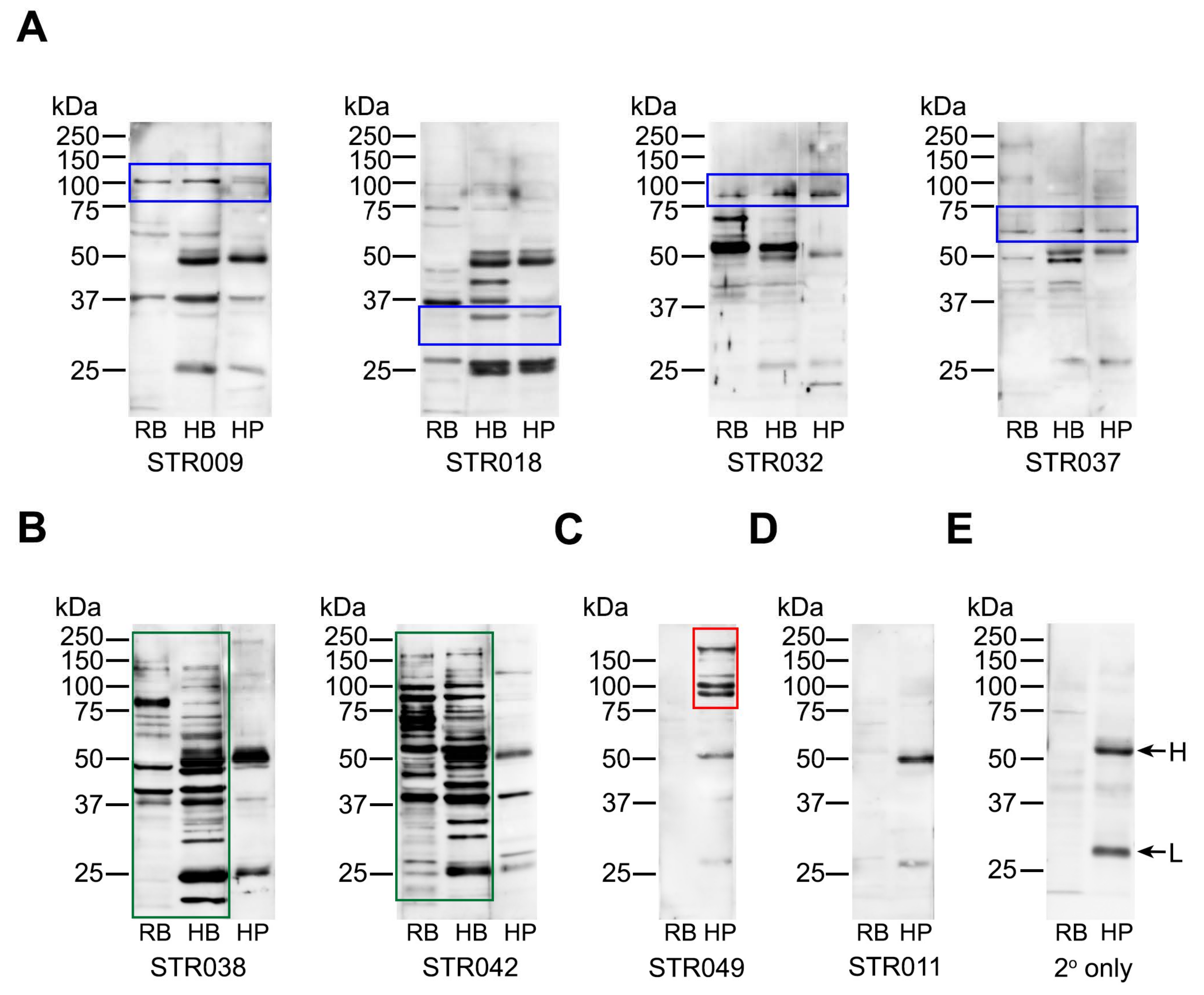
2.4. Anti-Platelet Antibody Targets are Heterogenous in Stroke Patients But Often Overlap with Brain Proteins
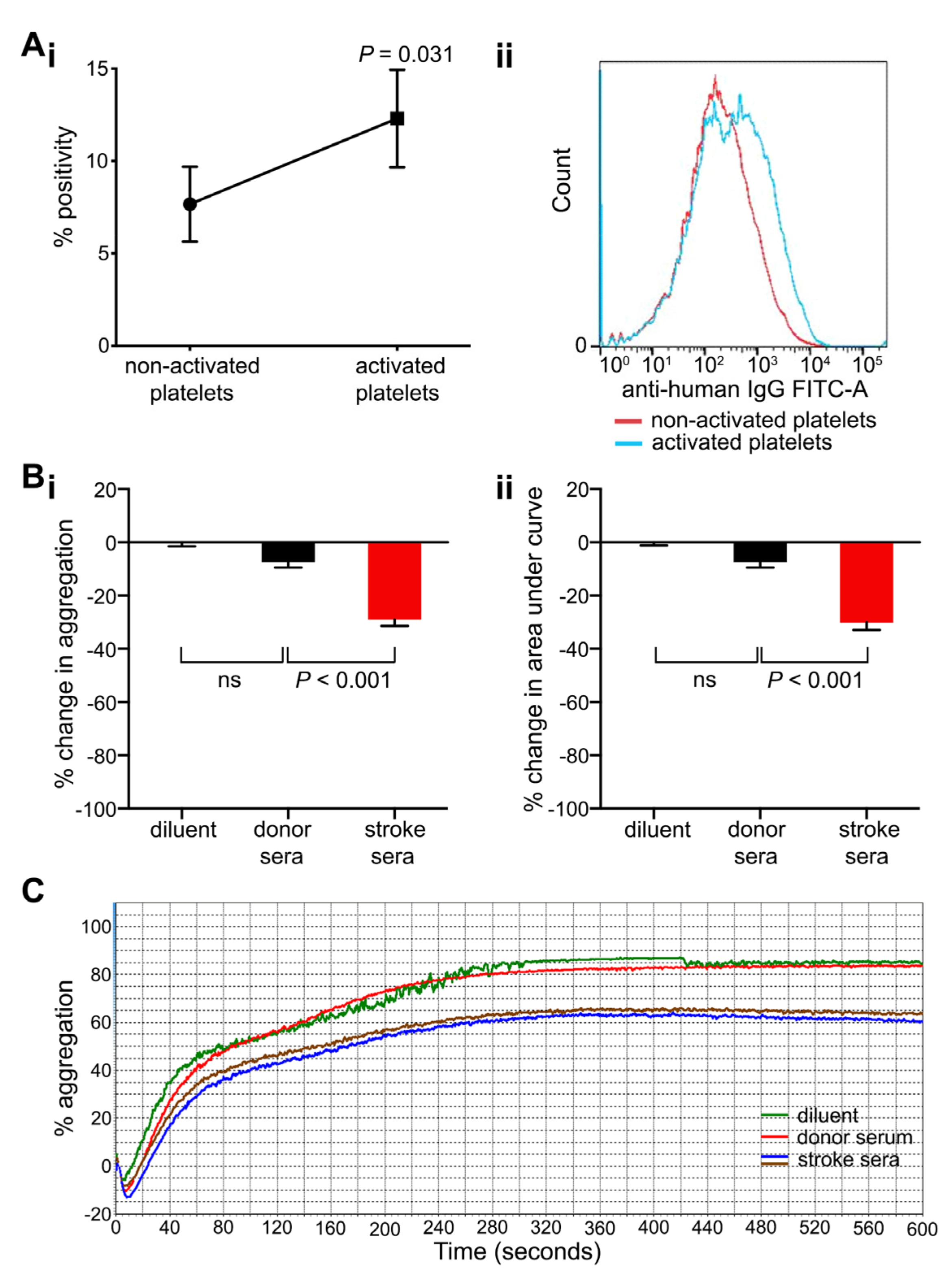
2.5. Anti-Platelet Antibodies Found in Stroke Patients Bind More Strongly to Activated Platelets and Inhibit Platelet Aggregation
3. Discussion
4. Materials and Methods
4.1. Patients and Controls
4.2. Testing for Anti-Platelet Antibodies
4.3. Platelet Activation
4.4. HLA Antibody Testing
4.5. Western Blotting
4.6. Light Transmission Aggregometry
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADP | adenosine diphosphate |
| ASPECT | Alberta Stroke Program Early Computed Tomography |
| BBB | blood–brain barrier |
| FITC | fluorescein isothiocyanate |
| FSC-A | forward scatter area |
| GP | glycoprotein |
| HLA | human leukocyte antigens |
| IgG | immunoglobulin G |
| ITP | immune thrombocytopenic purpura |
| LACI | lacunar infarct |
| MFI | mean fluorescence intensity |
| NBG | normalised background ratio |
| NIHSS | National Institutes of Health Stroke Scale |
| NMDA | N-methyl-D-aspartate |
| OCSP | Oxfordshire Community Stroke Project |
| PACI | partial anterior circulation infarct |
| POCI | posterior circulation infarct |
| PRP | platelet rich plasma |
| SD | standard deviation |
| SEM | standard error of the mean |
| TACI | total anterior circulation infarct |
| TIA | transient ischaemic attack |
| SSC-A | side scatter area |
| SSC-H | side scatter height |
| w/v | weight per volume |
References
- Javidi, E.; Magnus, T. Autoimmunity after Ischemic Stroke and Brain Injury. Front. Immunol. 2019, 10, 686. [Google Scholar] [CrossRef] [Green Version]
- Bornstein, N.; Aronovich, B.; Korczyn, A.; Shavit, S.; Michaelson, D.; Chapman, J. Antibodies to brain antigens following stroke. Neurology 2001, 56, 529–530. [Google Scholar] [CrossRef]
- Mecocci, P.; Parnetti, L.; Romano, G.; Scarelli, A.; Chionne, F.; Cecchetti, R.; Polidori, M.C.; Palumbo, B.; Cherubini, A.; Senin, U. Serum anti-GFAP and anti-S100 autoantibodies in brain aging, Alzheimer’s disease and vascular dementia. J. Neuroimmunol 1995, 57, 165–170. [Google Scholar] [CrossRef]
- Shibata, D.; Cain, K.; Tanzi, P.; Zierath, D.; Becker, K. Myelin basic protein autoantibodies, white matter disease and stroke outcome. J. Neuroimmunol. 2012, 252, 106–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dambinova, S.A.; Khounteev, G.A.; Skoromets, A.A. Multiple panel of biomarkers for TIA/stroke evaluation. Stroke 2002, 33, 1181–1182. [Google Scholar] [CrossRef] [PubMed]
- Kalev-Zylinska, M.L.; Symes, W.; Little, K.C.E.; Sun, P.; Wen, D.; Qiao, L.; Young, D.; During, M.J.; Barber, P.A. Stroke Patients Develop Antibodies That React With Components of N-Methyl-D-Aspartate Receptor Subunit 1 in Proportion to Lesion Size. Stroke 2013, 44, 2212–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dambinova, S.A.; Khounteev, G.A.; Izykenova, G.A.; Zavolokov, I.G.; Ilyukhina, A.Y.; Skoromets, A.A. Blood Test Detecting Autoantibodies to N-Methyl-d-aspartate Neuroreceptors for Evaluation of Patients with Transient Ischemic Attack and Stroke. Clin. Chem. 2003, 49, 1752–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, C.; Braunstein, Z.; Toomey, A.C.; Zhong, J.; Rao, X. S100 Proteins As an Important Regulator of Macrophage Inflammation. Front. Immunol. 2018, 8, 1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croce, K.; Gao, H.; Wang, Y.; Mooroka, T.; Sakuma, M.; Shi, C.; Sukhova, G.K.; Packard, R.R.S.; Hogg, N.; Libby, P.; et al. Myeloid-related protein-8/14 is critical for the biological response to vascular injury. Circulation 2009, 120, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Healy, A.M.; Pickard, M.D.; Pradhan, A.D.; Wang, Y.; Chen, Z.; Croce, K.; Sakuma, M.; Shi, C.; Zago, A.C.; Garasic, J.; et al. Platelet Expression Profiling and Clinical Validation of Myeloid-Related Protein-14 as a Novel Determinant of Cardiovascular Events. Circulation 2006, 113, 2278–2284. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Fang, C.; Gao, H.; Bilodeau, M.L.; Zhang, Z.; Croce, K.; Liu, S.; Morooka, T.; Sakuma, M.; Nakajima, K.; et al. Platelet-derived S100 family member myeloid-related protein-14 regulates thrombosis. J. Clin. Investig. 2014, 124, 2160–2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawano, T.; Shimamura, M.; Nakagami, H.; Iso, T.; Koriyama, H.; Takeda, S.; Baba, K.; Sasaki, T.; Sakaguchi, M.; Morishita, R.; et al. Therapeutic Vaccine Against S100A9 (S100 Calcium-Binding Protein A9) Inhibits Thrombosis Without Increasing the Risk of Bleeding in Ischemic Stroke in Mice. Hypertension 2018, 72, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Von Kummer, R.; Bourquain, H.; Bastianello, S.; Bozzao, L.; Manelfe, C.; Meier, D.; Hacke, W. Early Prediction of Irreversible Brain Damage after Ischemic Stroke at CT. Radiology 2001, 219, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Barber, P.A.; Demchuk, A.M.; Zhang, J.; Buchan, A.M. Validity and reliability of a quantitative computed tomography score in predicting outcome of hyperacute stroke before thrombolytic therapy. Lancet 2000, 355, 1670–1674. [Google Scholar] [CrossRef]
- Qin, X.; Akter, F.; Qin, L.; Cheng, J.; Guo, M.; Yao, S.; Jian, Z.; Liu, R.; Wu, S. Adaptive Immunity Regulation and Cerebral Ischemia. Front. Immunol. 2020, 11, 689. [Google Scholar] [CrossRef] [PubMed]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J. Neuroinflamm. 2019, 16, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Gill, D.; Veltkamp, R. Dynamics of T cell responses after stroke. Curr. Opin. Pharmacol. 2016, 26, 26–32. [Google Scholar] [CrossRef]
- Chen, Y.; Bodhankar, S.; Murphy, S.J.; Vandenbark, A.A.; Alkayed, N.J.; Offner, H. Intrastriatal B-cell administration limits infarct size after stroke in B-cell deficient mice. Metab. Brain Dis. 2012, 27, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Bodhankar, S.; Chen, Y.; Vandenbark, A.A.; Murphy, S.J.; Offner, H. IL-10-producing B-cells limit CNS inflammation and infarct volume in experimental stroke. Metab. Brain Dis. 2013, 28, 375–386. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Akiyoshi, K.; Dziennis, S.; Vandenbark, A.A.; Herson, P.S.; Hurn, P.D.; Offner, H. Regulatory B Cells Limit CNS Inflammation and Neurologic Deficits in Murine Experimental Stroke. J. Neurosci. 2011, 31, 8556–8563. [Google Scholar] [CrossRef]
- Ortega, S.B.; Torres, V.O.; Latchney, S.E.; Whoolery, C.W.; Noorbhai, I.Z.; Poinsatte, K.; Selvaraj, U.M.; Benson, M.A.; Meeuwissen, A.J.M.; Plautz, E.J.; et al. B cells migrate into remote brain areas and support neurogenesis and functional recovery after focal stroke in mice. Proc. Natl. Acad. Sci. USA 2020, 117, 4983–4993. [Google Scholar] [CrossRef] [Green Version]
- Woods, D.; Jiang, Q.; Chu, X.-P. Monoclonal antibody as an emerging therapy for acute ischemic stroke. Int. J. Physiol. Pathophysiol. Pharmacol. 2020, 12, 95–106. [Google Scholar] [PubMed]
- Kleinschnitz, C.; Schwab, N.; Kraft, P.; Hagedorn, I.; Dreykluft, A.; Schwarz, T.; Austinat, M.; Nieswandt, B.; Wiendl, H.; Stoll, G. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood 2010, 115, 3835–3842. [Google Scholar] [CrossRef]
- Schuhmann, M.K.; Langhauser, F.; Kraft, P.; Kleinschnitz, C. B cells do not have a major pathophysiologic role in acute ischemic stroke in mice. J. Neuroinflamm. 2017, 14, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Doyle, K.P.; Quach, L.N.; Solé, M.; Axtell, R.C.; Nguyen, T.-V.V.; Soler-Llavina, G.J.; Jurado, S.; Han, J.; Steinman, L.; Longo, F.M.; et al. B-Lymphocyte-Mediated Delayed Cognitive Impairment following Stroke. J. Neurosci. 2015, 35, 2133–2145. [Google Scholar] [CrossRef]
- Tomaiuolo, M.; Brass, L.F.; Stalker, T.J. Regulation of Platelet Activation and Coagulation and Its Role in Vascular Injury and Arterial Thrombosis. Interv. Cardiol. Clin. 2017, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Rose, S.R.; Petersen, N.J.; Gardner, T.J.; Hamill, R.J.; Trautner, B.W. Etiology of Thrombocytosis in a General Medicine Population: Analysis of 801 Cases With Emphasis on Infectious Causes. J. Clin. Med. Res. 2012, 4, 415–423. [Google Scholar] [CrossRef] [Green Version]
- Shim, R.; Wen, S.W.; Wanrooy, B.J.; Rank, M.; Thirugnanachandran, T.; Ho, L.; Sepehrizadeh, T.; de Veer, M.; Srikanth, V.K.; Ma, H.; et al. Stroke Severity, and Not Cerebral Infarct Location, Increases the Risk of Infection. Transl. Stroke Res. 2019, 11, 387–401. [Google Scholar] [CrossRef]
- Swinkels, M.; Rijkers, M.; Voorberg, J.; Vidarsson, G.; Leebeek, F.W.G.; Jansen, A.J.G. Emerging Concepts in Immune Thrombocytopenia. Front. Immunol. 2018, 9, 880. [Google Scholar] [CrossRef]
- Arnold, D.M.; Nazi, I.; Toltl, L.J.; Ross, C.; Ivetic, N.; Smith, J.W.; Liu, Y.; Kelton, J.G. Antibody binding to megakaryocytesin vivoin patients with immune thrombocytopenia. Eur. J. Haematol. 2015, 95, 532–537. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, S.; Nazy, I.; Smith, J.W.; Kelton, J.G.; Arnold, D.M. Platelet autoantibodies in the bone marrow of patients with immune thrombocytopenia. Blood Adv. 2020, 4, 2962–2966. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.-J.; Shan, N.-N. Megakaryocytic dysfunction in immune thrombocytopenia is linked to autophagy. Cancer Cell Int. 2019, 19, 59. [Google Scholar] [CrossRef] [PubMed]
- Grodzielski, M.; Goette, N.P.; Glembotsky, A.C.; Pietto, M.C.B.; Méndez-Huergo, S.P.; Pierdominici, M.S.; Montero, V.S.; Rabinovich, G.A.; Molinas, F.C.; Heller, P.G.; et al. Multiple concomitant mechanisms contribute to low platelet count in patients with immune thrombocytopenia. Sci. Rep. 2019, 9, 2208. [Google Scholar] [CrossRef]
- Greving, J.P.; Diener, H.-C.; Reitsma, J.B.; Bath, P.M.; Csiba, L.; Hacke, W.; Kappelle, L.J.; Koudstaal, P.J.; Leys, D.; Mas, J.-L.; et al. Antiplatelet Therapy After Noncardioembolic Stroke. Stroke 2019, 50, 1812–1818. [Google Scholar] [CrossRef]
- Kernan, W.N.; Ovbiagele, B.; Black, H.R.; Bravata, D.M.; Chimowitz, M.I.; Ezekowitz, M.D.; Fang, M.C.; Fisher, M.; Furie, K.; Heck, D.V.; et al. Guidelines for the Prevention of Stroke in Patients With Stroke and Transient Ischemic Attack: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 2014, 45, 2160–2236. [Google Scholar] [CrossRef]
- Wiśniewski, A.; Filipska, K.; Sikora, J.; Ślusarz, R.; Kozera, G. The Prognostic Value of High Platelet Reactivity in Ischemic Stroke Depends on the Etiology: A Pilot Study. J. Clin. Med. 2020, 9, 859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yip, H.-K.; Liou, C.-W.; Chang, H.-W.; Lan, M.-Y.; Liu, J.S.; Chen, M.-C. Link between Platelet Activity and Outcomes after an Ischemic Stroke. Cerebrovasc. Dis. 2005, 20, 120–128. [Google Scholar] [CrossRef]
- Voors-Pette, C.; Lebozec, K.; Dogterom, P.; Jullien, L.; Billiald, P.; Ferlan, P.; Renaud, L.; Favre-Bulle, O.; Avenard, G.; Machacek, M.; et al. Safety and Tolerability, Pharmacokinetics, and Pharmacodynamics of ACT017, an Antiplatelet GPVI (Glycoprotein VI) Fab. Arter. Thromb. Vasc. Biol. 2019, 39, 956–964. [Google Scholar] [CrossRef]
- Kleinschnitz, C.; Pozgajova, M.; Pham, M.; Bendszus, M.; Nieswandt, B.; Stoll, G. Targeting Platelets in Acute Experimental Stroke. Circulation 2007, 115, 2323–2330. [Google Scholar] [CrossRef] [Green Version]
- Serebruany, V.L.; Malinin, A.I.; Oshrine, B.R.; Sane, D.C.; Takserman, A.; Atar, D.; Hennekens, C.H. Lack of uniform platelet activation in patients after ischemic stroke and choice of antiplatelet therapy. Thromb. Res. 2004, 113, 197–204. [Google Scholar] [CrossRef]
- Jurk, K.; Jahn, U.-R.; Van Aken, H.; Schriek, C.; Droste, D.W.; Ritter, M.A.; Ringelstein, E.; Kehrel, B.E. Platelets in patients with acute ischemic stroke are exhausted and refractory to thrombin, due to cleavage of the seven-transmembrane thrombin receptor (PAR-1). Thromb. Haemost. 2004, 91, 334–344. [Google Scholar] [CrossRef]
- Weissman, J.D.; Khunteev, G.A.; Heath, R.; Dambinova, S.A. NR2 antibodies: Risk assessment of transient ischemic attack (TIA)/stroke in patients with history of isolated and multiple cerebrovascular events. J. Neurol. Sci. 2011, 300, 97–102. [Google Scholar] [CrossRef]
- Bokesch, P.M.; Izykenova, G.A.; Justice, J.B.; Easley, K.A.; Dambinova, S.A. NMDA Receptor Antibodies Predict Adverse Neurological Outcome After Cardiac Surgery in High-Risk Patients. Stroke 2006, 37, 1432–1436. [Google Scholar] [CrossRef] [Green Version]
- Vermeer, S.E.; Longstreth, W.T.; Koudstaal, P.J. Silent brain infarcts: A systematic review. Lancet Neurol. 2007, 6, 611–619. [Google Scholar] [CrossRef]
- Gupta, A.; Giambrone, A.E.; Gialdini, G.; Finn, C.; Delgado, D.; Gutierrez, J.; Wright, C.; Beiser, A.S.; Seshadri, S.; Pandya, A.; et al. Silent Brain Infarction and Risk of Future Stroke. Stroke 2016, 47, 719–725. [Google Scholar] [CrossRef] [Green Version]
- Samol, A.; Hahne, K.; Mönnig, G. Atrial fibrillation and silent stroke: Links, risks, and challenges. Vasc. Heal. Risk Manag. 2016, 12, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Fanning, J.P.; Wong, A.A.; Fraser, J.F. The epidemiology of silent brain infarction: A systematic review of population-based cohorts. BMC Med. 2014, 12, 119. [Google Scholar] [CrossRef]
- Mead, G.E.; Lewis, S.C.; Wardlaw, J.M.; Dennis, M.S.; Warlow, C.P. How well does the Oxfordshire Community Stroke Project classification predict the site and size of the infarct on brain imaging? J. Neurol. Neurosurg. Psychiatry 2000, 68, 558–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, M.-D.; de Veld, J.C.; van der Holt, B.; Veer, M.B.V. Screening for alloantibodies in the serum of patients receiving platelet transfusions: A comparison of the ELISA, lymphocytotoxicity, and the indirect immunofluorescence method. Transfusion 2003, 43, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Kalev-Zylinska, M.L.; Green, T.N.; Morel-Kopp, M.-C.; Sun, P.P.; Park, Y.-E.; Lasham, A.; During, M.J.; Ward, C.M. N-methyl-d-aspartate receptors amplify activation and aggregation of human platelets. Thromb. Res. 2014, 133, 837–847. [Google Scholar] [CrossRef]
- Green, T.N.; Hamilton, J.R.; Morel-Kopp, M.-C.; Zheng, Z.; Chen, T.-Y.T.; Hearn, J.I.; Sun, P.P.; Flanagan, J.U.; Young, D.; Barber, P.A.; et al. Inhibition of NMDA receptor function with an anti-GluN1-S2 antibody impairs human platelet function and thrombosis. Platelets 2017, 28, 799–811. [Google Scholar] [CrossRef] [Green Version]
- Colombo, M.B.; Haworth, S.E.; Poli, F.; Nocco, A.; Puglisi, G.; Innocente, A.; Serafini, M.; Messa, P.; Scalamogna, M. Luminex technology for anti-HLA antibody screening: Evaluation of performance and of impact on laboratory routine. Cytom. Part B Clin. Cytom. 2007, 72, 465–471. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | All Stroke Patients (n = 48) | Anti-Platelet Antibodies | p-Value | |
|---|---|---|---|---|
| Present (n = 15) | Absent (n = 33) | |||
| Age, mean ± SD in years | 70 ± 17 | 74 ± 12 | 67 ± 18 | 0.193 ‡ |
| Females, n (%) | 25 (52) | 9 (60) | 16 (48) | 0.459 * |
| Previous stroke, n (%) | 14 (29) | 6 (40) | 8 (24) | 0.266 * |
| Previous TIA, n (%) | 6 (13) | 2 (13) | 4 (12) | 1.000 † |
| Risk factors, n (%) | ||||
| Hypertension | 33 (69) | 11 (73) | 22 (67) | 0.644 * |
| Atrial fibrillation | 14 (29) | 6 (40) | 8 (24) | 0.266 * |
| Ischaemic heart disease | 12 (25) | 3 (20) | 9 (27) | 0.728 † |
| Hyperlipidaemia | 11 (23) | 4 (27) | 7 (21) | 0.720 † |
| Current smoker | 11 (23) | 1 (7) | 10 (30) | 0.136 † |
| Peripheral vascular disease | 6 (13) | 3 (20) | 3 (9) | 0.360 † |
| NIHSS score, median (range) | ||||
| On admission (day 0) | 5 (1–23) | 6 (4–23) | 3 (1–18) | 0.002 § |
| At discharge (day 7 ± 2) | 4 (1–23) | 6 (1–23) | 4 (1–15) | 0.291 § |
| OCSP, n (%) | 0.022 * | |||
| TACI | 5 (10) | 4 (27) | 1 (3) | 0.028 † |
| PACI | 21 (44) | 8 (53) | 13 (39) | 0.367 * |
| LACI | 15 (31) | 2 (13) | 13 (9) | 0.098 † |
| POCI | 7 (15) | 1 (7) | 6 (18) | 0.409 † |
| ASPECT score, median (range) | 9 (3–10) | 9 (5–10) | 10 (3–10) | 0.074 § |
| Anti-Platelet Antibody Levels | General Linear Model | ||
|---|---|---|---|
| β-Coefficient Estimate | 95% Confidence Interval | p-Value | |
| NIHSS score on admission (day 0) | −1.280 | −2.480−(−0.080) | 0.040 |
| NIHSS score at discharge (day 7 ± 2) | 1.185 | 0.003–2.367 | 0.050 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, Y.E.; Penumarthy, R.; Sun, P.P.; Kang, C.Y.; Morel-Kopp, M.-C.; Downing, J.; Green, T.N.; Immanuel, T.; Ward, C.M.; Young, D.; et al. Platelet-Reactive Antibodies in Patients after Ischaemic Stroke—An Epiphenomenon or a Natural Protective Mechanism. Int. J. Mol. Sci. 2020, 21, 8398. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218398
Park YE, Penumarthy R, Sun PP, Kang CY, Morel-Kopp M-C, Downing J, Green TN, Immanuel T, Ward CM, Young D, et al. Platelet-Reactive Antibodies in Patients after Ischaemic Stroke—An Epiphenomenon or a Natural Protective Mechanism. International Journal of Molecular Sciences. 2020; 21(21):8398. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218398
Chicago/Turabian StylePark, Young Eun, Rushi Penumarthy, Paul P. Sun, Caroline Y. Kang, Marie-Christine Morel-Kopp, Jonathan Downing, Taryn N. Green, Tracey Immanuel, Christopher M. Ward, Deborah Young, and et al. 2020. "Platelet-Reactive Antibodies in Patients after Ischaemic Stroke—An Epiphenomenon or a Natural Protective Mechanism" International Journal of Molecular Sciences 21, no. 21: 8398. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218398