E3 Ubiquitin Ligase TRIP12: Regulation, Structure, and Physiopathological Functions

Abstract
:1. Introduction
2. TRIP12 mRNA and Protein Expression
2.1. TRIP12 Gene Organization
2.2. TRIP12 mRNA Expression
2.3. TRIP12 Protein Expression and Cellular Localisation
3. TRIP12 Protein Structure and Domains
3.1. The HECT Domain
3.2. The WWE Domain
3.3. The Armadillo-Repeats Domain
3.4. The IDR Domain
3.5. Conservation of TRIP12 Protein Domains during Evolution
3.6. Post-Translational Modifications of TRIP12
4. Functions of TRIP12 in Ubiquitin-Mediated Proteolysis
4.1. Ubiquitination
4.2. Known TRIP12 Substrates
4.3. Role of TRIP12 in Ubiquitin Fusion Degradation and N-Degron Pathways
5. The Physio-Pathological Roles of TRIP12
5.1. TRIP12 Protein Interactors
5.2. Roles in Cell Cycle Progression
5.3. Roles in DNA Damage Response
5.3.1. Via the ARF/P53 Signaling Pathway
5.3.2. Via the Control of Histone Ubiquitination Spreading by RNF168
5.3.3. Via the Deubiquitinase USP7
5.4. The Roles of TRIP12 in Chromatin Remodeling
5.4.1. Via the Control of SWI/SNF Complex Integrity
5.4.2. Via the Maintenance of Silenced Genes by the Polycomb Complex
5.5. Roles in Cell Differentiation
6. TRIP12 in Pathologies
6.1. Alterations of TRIP12 Gene in Autism and Intellectual Disability
6.2. Alterations of TRIP12 Gene, mRNA and Protein Expression, and Function in Cancers
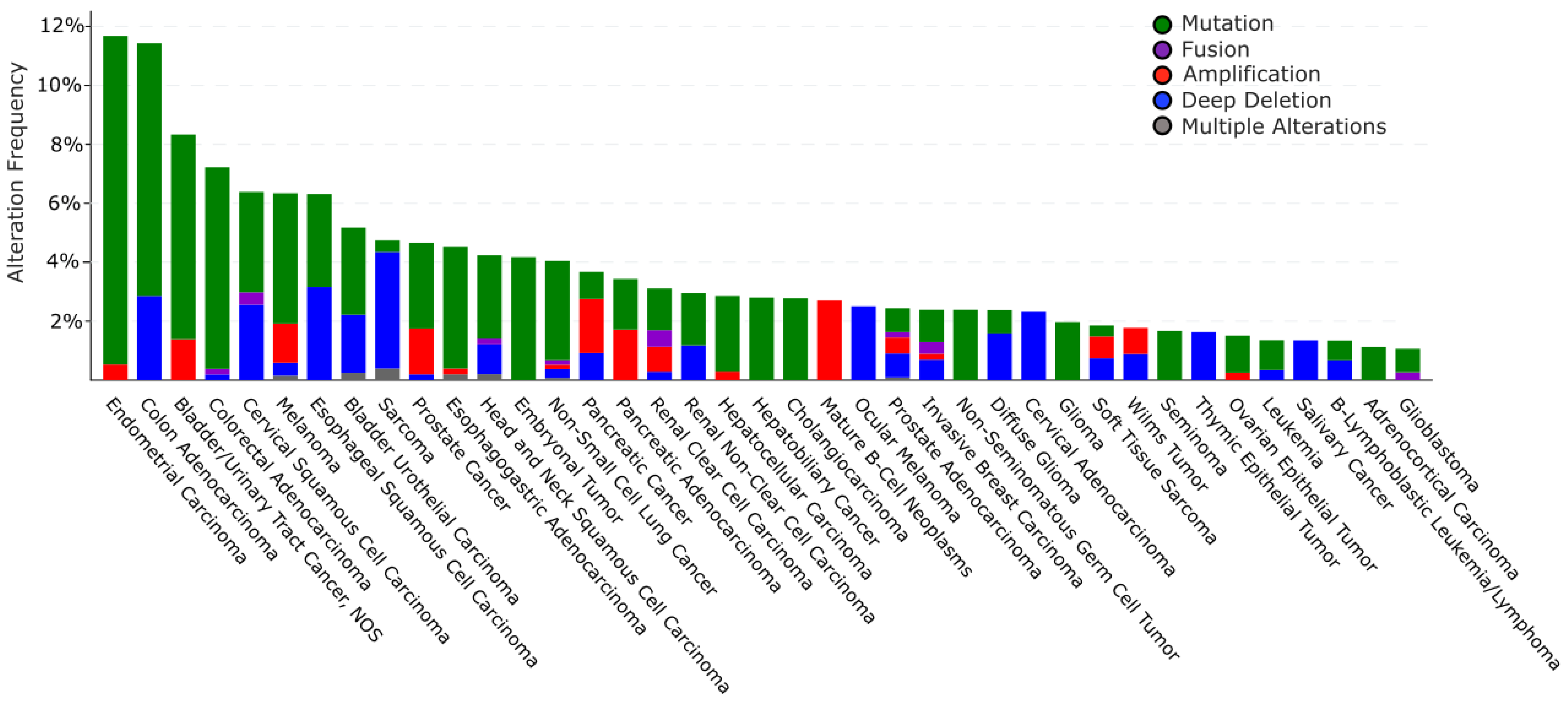
6.2.1. Alteration of TRIP12 Gene in Cancers
6.2.2. Alteration of TRIP12 mRNA Expression in Cancers
6.2.3. Alterations of TRIP12 Protein Expression in Cancers and Consequences
7. Conclusions and Perspectives
8. Software and Data Bases
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nomura, N.; Nagase, T.; Miyajima, N.; Sazuka, T.; Tanaka, A.; Sato, S.; Seki, N.; Kawarabayasi, Y.; Ishikawa, K.-I.; Tabata, S. Prediction of the Coding Sequences of Unidentified Human Genes. II. The Coding Sequences of 40 New Genes (KIAA0041-KIAA0080) Deduced by Analysis of cDNA Clones from Human Cell Line KG-1. DNA Res. 1994, 1, 223–229. [Google Scholar] [CrossRef]
- Huibregtse, J.M.; Scheffner, M.; Beaudenon, S.; Howley, P.M. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. USA 1995, 92, 2563–2567. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Choi, H.S.; Gyuris, J.; Brent, R.; Moore, D.D. Two classes of proteins dependent on either the presence or absence of thyroid hormone for interaction with the thyroid hormone receptor. Mol. Endocrinol. 1995, 9, 243–254. [Google Scholar]
- Park, Y.; Yoon, S.K.; Yoon, J.-B. TRIP12 functions as an E3 ubiquitin ligase of APP-BP1. Biochem. Biophys. Res. Commun. 2008, 374, 294–298. [Google Scholar] [CrossRef]
- Kajiro, M.; Tsuchiya, M.; Kawabe, Y.-I.; Furumai, R.; Iwasaki, N.; Hayashi, Y.; Katano, M.; Nakajima, Y.; Goto, N.; Watanabe, T.; et al. The E3 Ubiquitin Ligase Activity of Trip12 Is Essential for Mouse Embryogenesis. PLoS ONE 2011, 6, e25871. [Google Scholar] [CrossRef]
- Hanoun, N.; Fritsch, S.; Gayet, O.; Gigoux, V.; Cordelier, P.; Dusetti, N.; Torrisani, J.; Dufresne, M. The E3 Ubiquitin Ligase Thyroid Hormone Receptor-interacting Protein 12 Targets Pancreas Transcription Factor 1a for Proteasomal Degradation. J. Biol. Chem. 2014, 289, 35593–35604. [Google Scholar] [CrossRef] [Green Version]
- Larrieu, D.; Brunet, M.; Vargas, C.; Hanoun, N.; Ligat, L.; Dagnon, L.; Lulka, H.; Pommier, R.M.; Selves, J.; Jády, B.E.; et al. The E3 ubiquitin ligase TRIP12 participates in cell cycle progression and chromosome stability. Sci. Rep. 2020, 10, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinklein, N.D.; Aldred, S.F.; Hartman, S.J.; Schroeder, D.I.; Otillar, R.P.; Myers, R.M. An Abundance of Bidirectional Promoters in the Human Genome. Genome Res. 2003, 14, 62–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tekcham, D.S.; Chen, D.; Liu, Y.; Ling, T.; Zhang, Y.; Chen, H.; Wang, W.; Otkur, W.; Qi, H.; Xia, T.; et al. F-box proteins and cancer: An update from functional and regulatory mechanism to therapeutic clinical prospects. Theranostics 2020, 10, 4150–4167. [Google Scholar] [CrossRef] [PubMed]
- Bramswig, N.C.; Lüdecke, H.-J.; Pettersson, M.; Albrecht, B.; Bernier, R.A.; Cremer, K.; Eichler, E.E.; Falkenstein, D.; Gerdts, J.; Jansen, S.; et al. Identification of new TRIP12 variants and detailed clinical evaluation of individuals with non-syndromic intellectual disability with or without autism. Qual. Life Res. 2017, 136, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.-W.; Zhang, W.; Yeh, H.S.; De Jong, E.P.; Jun, S.; Kim, K.H.; Bae, S.S.; Beckman, K.; Hwang, T.H.; Kim, K.S.; et al. mRNA 3’-UTR shortening is a molecular signature of mTORC1 activation. Nat. Commun. 2015, 6, 7218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, C.-I.; Ganio, E.; Hagiwara, N. Trip12, a HECT domain E3 ubiquitin ligase, targets Sox6 for proteasomal degradation and affects fiber type-specific gene expression in muscle cells. Skelet. Muscle 2013, 3, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadkowski, T.; Jank, M.; Zwierzchowski, L.; Oprzadek, J.; Motyl, T. Comparison of skeletal muscle transcriptional profiles in dairy and beef breeds bulls. J. Appl. Genet. 2009, 50, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Gudjonsson, T.; Altmeyer, M.; Savic, V.; Toledo, L.; Dinant, C.; Grøfte, M.; Bartkova, J.; Poulsen, M.; Oka, Y.; Bekker-Jensen, S.; et al. TRIP12 and UBR5 Suppress Spreading of Chromatin Ubiquitylation at Damaged Chromosomes. Cell 2012, 150, 697–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Shan, J.; Zhu, W.-G.; Qin, J.; Gu, W. Transcription-independent ARF regulation in oncogenic stress-mediated p53 responses. Nat. Cell Biol. 2010, 464, 624–627. [Google Scholar] [CrossRef] [Green Version]
- Huber, A.H.; Nelson, W.J.; Weis, W.I. Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell 1997, 90, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Ries, L.K.; Liess, A.K.L.; Feiler, C.G.; Spratt, D.E.; Lowe, E.D.; Lorenz, S. Crystal structure of the catalytic C-lobe of the HECT-type ubiquitin ligase E6AP. Protein Sci. 2020, 29, 1550–1554. [Google Scholar] [CrossRef] [Green Version]
- Zweifel, M.E.; Leahy, D.J.; Barrick, D. Structure and Notch Receptor Binding of the Tandem WWE Domain of Deltex. Structure 2005, 13, 1599–1611. [Google Scholar] [CrossRef] [Green Version]
- Sluimer, J.; Distel, B. Regulating the human HECT E3 ligases. Cell. Mol. Life Sci. 2018, 75, 3121–3141. [Google Scholar] [CrossRef] [Green Version]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- Huang, L. Structure of an E6AP-UbcH7 Complex: Insights into Ubiquitination by the E2-E3 Enzyme Cascade. Science 1999, 286, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Wang, Y.; Argiles-Castillo, D.; Kane, E.I.; Zhou, A.; Spratt, D.E. HECT E3 ubiquitin ligases—Emerging insights into their biological roles and disease relevance. J. Cell Sci. 2020, 133, jcs228072. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L. The WWE domain: A common interaction module in protein ubiquitination and ADP ribosylation. Trends Biochem. Sci. 2001, 26, 273–275. [Google Scholar] [CrossRef]
- Wang, Z.; Michaud, G.A.; Cheng, Z.; Zhang, Y.; Hinds, T.R.; Fan, E.; Cong, F.; Xu, W. Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly(ADP-ribosyl)ation-dependent ubiquitination. Genes Dev. 2012, 26, 235–240. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.-Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2018, 47, D351–D360. [Google Scholar] [CrossRef] [Green Version]
- Gatti, M.; Imhof, R.; Huang, Q.; Baudis, M.; Altmeyer, M. The Ubiquitin Ligase TRIP12 Limits PARP1 Trapping and Constrains PARP Inhibitor Efficiency. Cell Rep. 2020, 32, 107985. [Google Scholar] [CrossRef]
- Tewari, R.; Bailes, E.; Bunting, K.A.; Coates, J.C. Armadillo-repeat protein functions: Questions for little creatures. Trends Cell Biol. 2010, 20, 470–481. [Google Scholar] [CrossRef]
- El Refy, A.; Perazza, D.; Zekraoui, L.; Valay, J.-G.; Bechtold, N.; Brown, S.; Hülskamp, M.; Herzog, M.; Bonneville, J.-M. The Arabidopsis KAKTUS gene encodes a HECT protein and controls the number of endoreduplication cycles. Mol. Genet. Genom. 2003, 270, 403–414. [Google Scholar] [CrossRef]
- Ju, D.; Wang, X.; Xu, H.; Xie, Y. The armadillo repeats of the Ufd4 ubiquitin ligase recognize ubiquitin-fusion proteins. FEBS Lett. 2006, 581, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Van Der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.W.; et al. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Toretsky, J.A.; Wright, P.E. Assemblages: Functional units formed by cellular phase separation. J. Cell Biol. 2014, 206, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Levy, Y. Intrinsically Disordered Regions as Affinity Tuners in Protein-DNA Interactions. Biophys. J. 2012, 102, 632a. [Google Scholar] [CrossRef] [Green Version]
- Watson, M.; Stott, K. Disordered domains in chromatin-binding proteins. Essays Biochem. 2019, 63, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Vuzman, D.; Levy, Y. DNA search efficiency is modulated by charge composition and distribution in the intrinsically disordered tail. Proc. Natl. Acad. Sci. USA 2010, 107, 21004–21009. [Google Scholar] [CrossRef] [Green Version]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef] [Green Version]
- Leach, B.I.; Kuntimaddi, A.; Schmidt, C.R.; Cierpicki, T.; Johnson, S.A.; Bushweller, J.H. Leukemia Fusion Target AF9 Is an Intrinsically Disordered Transcriptional Regulator that Recruits Multiple Partners via Coupled Folding and Binding. Structure 2013, 21, 176–183. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Cramer, P. Structure of the super-elongation complex subunit AFF4 C-terminal homology domain reveals requirements for AFF homo- and heterodimerization. J. Biol. Chem. 2019, 294, 10663–10673. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Yang, X.; Li, Y.; Zhao, S.; Li, C.; Ma, P.; Mao, B. Trip12 is an E3 ubiquitin ligase for USP7/HAUSP involved in the DNA damage response. FEBS Lett. 2016, 590, 4213–4222. [Google Scholar] [CrossRef] [Green Version]
- Banani, S.F.; Lee, H.O.; Hyman, H.O.L.A.A.; Rosen, S.F.B.M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Posey, A.E.; Holehouse, A.S.; Pappu, R.V. Phase Separation of Intrinsically Disordered Proteins. Methods Enzymol. 2018, 611, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Gallego, L.D.; Schneider, M.; Mittal, C.; Romanauska, A.; Carrillo, R.M.G.; Schubert, T.; Pugh, B.F.; Köhler, A. Phase separation directs ubiquitination of gene-body nucleosomes. Nat. Cell Biol. 2020, 579, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasius, M.; Forment, J.V.; Thakkar, N.; Wagner, S.A.; Choudhary, C.; Jackson, S.P. A phospho-proteomic screen identifies substrates of the checkpoint kinase Chk1. Genome Biol. 2011, 12, R78. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Petrone, A.; Adamo, M.E.; Cheng, C.; Kettenbach, A.N. Identification of Candidate Cyclin-dependent kinase 1 (Cdk1) Substrates in Mitosis by Quantitative Phosphoproteomics. Mol. Cell. Proteom. 2016, 15, 2448–2461. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.-B.; Shi, G.-M.; Dong, Z.-R.; Ke, A.-W.; Ma, H.-H.; Gao, Q.; Shen, Z.-Z.; Huang, X.-Y.; Chen, H.; Yu, D.-D.; et al. Ubiquitin-specific protease 7 accelerates p14 ARF degradation by deubiquitinating thyroid hormone receptor-interacting protein 12 and promotes hepatocellular carcinoma progression. Hepatology 2015, 61, 1603–1614. [Google Scholar] [CrossRef]
- Georges, A.; Marcon, E.; Greenblatt, J.; Frappier, L. Identification and Characterization of USP7 Targets in Cancer Cells. Sci. Rep. 2018, 8, 15833. [Google Scholar] [CrossRef] [Green Version]
- Hershko, A.; Ciechanover, A. The Ubiquitin System. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- McClellan, A.J.; Laugesen, S.H.; Ellgaard, L. Cellular functions and molecular mechanisms of non-lysine ubiquitination. Open Biol. 2019, 9, 190147. [Google Scholar] [CrossRef] [Green Version]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, K.D. Regulation of ubiquitin-dependent processes by deubiquitinating enzymes. FASEB J. 1997, 11, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Kweon, S.-M.; Chen, Y.; Moon, E.; Kvederaviciutė, K.; Klimasauskas, S.; Feldman, D.E. An Adversarial DNA N6-Methyladenine-Sensor Network Preserves Polycomb Silencing. Mol. Cell 2019, 74, 1138–1147.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keppler, B.R.; Archer, T.K. Ubiquitin-dependent and Ubiquitin-independent Control of Subunit Stoichiometry in the SWI/SNF Complex. J. Biol. Chem. 2010, 285, 35665–35674. [Google Scholar] [CrossRef] [Green Version]
- Hah, N.; Kolkman, A.; Ruhl, D.D.; Pijnappel, W.W.M.P.; Heck, A.J.R.; Timmers, H.T.M.; Kraus, W.L. A Role for BAF57 in Cell Cycle–Dependent Transcriptional Regulation by the SWI/SNF Chromatin Remodeling Complex. Cancer Res. 2010, 70, 4402–4411. [Google Scholar] [CrossRef] [Green Version]
- Kuo, M.-L.; Besten, W.D.; Sherr, C.J. N-Terminal Polyubiquitination of the ARF Tumor Suppressor, A Natural Lysine-Less Protein. Cell Cycle 2004, 3, 1367–1369. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Archer, T.K. Regulating SWI/SNF Subunit Levels via Protein-Protein Interactions and Proteasomal Degradation: BAF155 and BAF170 Limit Expression of BAF57. Mol. Cell. Biol. 2005, 25, 9016–9027. [Google Scholar] [CrossRef] [Green Version]
- Chio, I.I.C.; Sasaki, M.; Ghazarian, D.; Moreno, J.; Done, S.J.; Ueda, T.; Inoue, S.; Chang, Y.-L.; Chen, N.-J.; Mak, T.W. TRADD contributes to tumour suppression by regulating ULF-dependent p19Arf ubiquitylation. Nat. Cell Biol. 2012, 14, 625–633. [Google Scholar] [CrossRef]
- Johnson, E.S.; Ma, P.C.M.; Ota, I.M.; Varshavsky, A. A Proteolytic Pathway That Recognizes Ubiquitin as a Degradation Signal. J. Biol. Chem. 1995, 270, 17442–17456. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Yoon, S.K.; Yoon, J.-B. The HECT Domain of TRIP12 Ubiquitinates Substrates of the Ubiquitin Fusion Degradation Pathway. J. Biol. Chem. 2008, 284, 1540–1549. [Google Scholar] [CrossRef] [Green Version]
- Van Leeuwen, F.; De Kleijn, D.P.; Hurk, H.H.V.D.; Neubauer, A.; Sonnemans, M.A.; Sluijs, J.A.; Köycü, S.; Ramdjielal, R.D.; Salehi, A.; Martens, G.J.; et al. Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer’s and Down patients. Science 1998, 279, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, E.G.; Steinhauer, C.; Lees, M.; Lauridsen, A.-M.; Ellgaard, L.; Hartmann-Petersen, R. HUWE1 and TRIP12 Collaborate in Degradation of Ubiquitin-Fusion Proteins and Misframed Ubiquitin. PLoS ONE 2012, 7, e50548. [Google Scholar] [CrossRef] [PubMed]
- Setz, C.; Friedrich, M.; Hahn, S.; Dörrie, J.; Schaft, N.; Schuler, G.; Schubert, U. Just One Position-Independent Lysine Residue Can Direct MelanA into Proteasomal Degradation following N-Terminal Fusion of Ubiquitin. PLoS ONE 2013, 8, e55567. [Google Scholar] [CrossRef]
- Varshavsky, A. N-degron and C-degron pathways of protein degradation. Proc. Natl. Acad. Sci. USA 2019, 116, 358–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, C.-S.; Shemorry, A.; Auerbach, D.; Varshavsky, A. The N-end rule pathway is mediated by a complex of the RING-type Ubr1 and HECT-type Ufd4 ubiquitin ligases. Nat. Cell Biol. 2010, 12, 1177–1185. [Google Scholar] [CrossRef] [Green Version]
- Havugimana, P.C.; Hart, G.T.; Nepusz, T.; Yang, H.; Turinsky, A.L.; Li, Z.; Wang, P.I.; Boutz, D.R.; Fong, V.; Phanse, S.; et al. A Census of Human Soluble Protein Complexes. Cell 2012, 150, 1068–1081. [Google Scholar] [CrossRef] [Green Version]
- Arbogast, F.; Arnold, J.; Hammann, P.; Kuhn, L.; Chicher, J.; Murera, D.; Weishaar, J.; Muller, S.; Fauny, J.-D.; Gros, F. ATG5 is required for B cell polarization and presentation of particulate antigens. Autophagy 2018, 15, 280–294. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Ting, L.; Bruckner, R.J.; Gebreab, F.; Gygi, M.P.; Szpyt, J.; Tam, S.; Zarraga, G.; Colby, G.; Baltier, K.; et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 2015, 162, 425–440. [Google Scholar] [CrossRef] [Green Version]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nat. Cell Biol. 2017, 545, 505–509. [Google Scholar] [CrossRef]
- Nakayama, M.; Kikuno, R.; Ohara, O. Protein-Protein Interactions between Large Proteins: Two-Hybrid Screening Using a Functionally Classified Library Composed of Long cDNAs. Genome Res. 2002, 12, 1773–1784. [Google Scholar] [CrossRef] [Green Version]
- Hanson, D.; Stevens, A.; Murray, P.; Black, G.C.M.; Clayton, P.E. Identifying biological pathways that underlie primordial short stature using network analysis. J. Mol. Endocrinol. 2014, 52, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeronimo, C.; Forget, D.; Bouchard, A.; Li, Q.; Chua, G.; Poitras, C.; Thérien, C.; Bergeron, D.; Bourassa, S.; Greenblatt, J.; et al. Systematic Analysis of the Protein Interaction Network for the Human Transcription Machinery Reveals the Identity of the 7SK Capping Enzyme. Mol. Cell 2007, 27, 262–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Cho, Y.-E.; Kim, S.-H.; Kim, Y.-J.; Park, J.-H. GLTSCR2 promotes the nucleoplasmic translocation and subsequent degradation of nucleolar ARF. Oncotarget 2016, 8, 16293–16302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Hou, J.; Sun, L.; Wen, T.; Wang, L.; Zhao, X.; Xie, Q.; Zhang, S.Q. NMI mediates transcription-independent ARF regulation in response to cellular stresses. Mol. Biol. Cell 2012, 23, 4635–4646. [Google Scholar] [CrossRef] [PubMed]
- Lo, R.; Zhang, Y.; Dai, M.-S.; Sun, X.-X.; Zeng, S.X.; Lu, H. Nucleostemin stabilizes ARF by inhibiting the ubiquitin ligase ULF. Oncogene 2014, 34, 1688–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, G.D.; Coyaud, É.; Gonçalves, J.; Mojarad, B.A.; Liu, Y.; Wu, Q.; Gheiratmand, L.; Comartin, D.; Tkach, J.M.; Cheung, S.W.; et al. A Dynamic Protein Interaction Landscape of the Human Centrosome-Cilium Interface. Cell 2015, 163, 1484–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Venable, J.; Lapointe, P.; Hutt, D.M.; Koulov, A.V.; Coppinger, J.; Gurkan, C.; Kellner, W.; Matteson, J.; Plutner, H.; et al. Hsp90 Cochaperone Aha1 Downregulation Rescues Misfolding of CFTR in Cystic Fibrosis. Cell 2006, 127, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, T.; Masuda, T.; Canver, M.C.; Seiler, M.; Semba, Y.; Shboul, M.; Al-Raqad, M.; Maeda, M.; Schoonenberg, V.A.; Cole, M.A.; et al. Genome-wide CRISPR-Cas9 Screen Identifies Leukemia-Specific Dependence on a Pre-mRNA Metabolic Pathway Regulated by DCPS. Cancer Cell 2018, 33, 386–400.e5. [Google Scholar] [CrossRef] [Green Version]
- Guard, S.E.; Poss, Z.C.; Ebmeier, C.C.; Pagratis, M.; Simpson, H.; Taatjes, D.J.; Old, W.M. The nuclear interactome of DYRK1A reveals a functional role in DNA damage repair. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; Wang, X.; Zhao, M.; Yang, R.; Malik, R.; Qiao, Y.; Poliakov, A.; Yocum, A.K.; Li, Y.; Chen, W.; et al. The central role of EED in the orchestration of polycomb group complexes. Nat. Commun. 2014, 5, 1–13. [Google Scholar] [CrossRef]
- Wang, S.; Kollipara, R.K.; Humphries, C.G.; Ma, S.-H.; Hutchinson, R.; Li, R.; Siddiqui, J.; Tomlins, S.A.; Raj, G.V.; Kittler, R. The ubiquitin ligase TRIM25 targets ERG for degradation in prostate cancer. Oncotarget 2016, 7, 64921–64931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarallo, R.; Bamundo, A.; Nassa, G.; Nola, E.; Paris, O.; Ambrosino, C.; Facchiano, A.; Baumann, M.; Nyman, T.A.; Weisz, A. Identification of proteins associated with ligand-activated estrogen receptor α in human breast cancer cell nuclei by tandem affinity purification and nano LC-MS/MS. Proteom. Clin. Appl. 2011, 5, 470. [Google Scholar] [CrossRef]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A Human Interactome in Three Quantitative Dimensions Organized by Stoichiometries and Abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliviero, G.; Cagney, G.; Waston, A.; Streubel, G.; Jerman, E.; Andrews, D.; Doyle, B.; Munawar, N.; Wynne, K.; Crean, J.; et al. Dynamic Protein Interactions of the Polycomb Repressive Complex 2 during Differentiation of Pluripotent Cells. Mol. Cell. Proteom. 2016, 15, 3450–3460. [Google Scholar] [CrossRef] [Green Version]
- Gilmore, J.M.; Sardiu, M.E.; Groppe, B.D.; Thornton, J.L.; Liu, X.; Dayebgadoh, G.; Banks, C.A.; Slaughter, B.D.; Unruh, J.R.; Workman, J.L.; et al. WDR76 Co-Localizes with Heterochromatin Related Proteins and Rapidly Responds to DNA Damage. PLoS ONE 2016, 11, e0155492. [Google Scholar] [CrossRef]
- Lambert, J.-P.; Tucholska, M.; Go, C.; Knight, J.D.; Gingras, A.-C. Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J. Proteom. 2015, 118, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Saredi, G.; Becker, J.R.; Foster, B.M.; Nguyen, N.V.; Beyer, T.E.; Cesa, L.C.; Faull, P.A.; Lukauskas, S.; Frimurer, T.; et al. H4K20me0 recognition by BRCA1–BARD1 directs homologous recombination to sister chromatids. Nat. Cell Biol. 2019, 21, 311–318. [Google Scholar] [CrossRef]
- Diner, B.A.; Lum, K.K.; Javitt, A.; Cristea, I.M. Interactions of the Antiviral Factor Interferon Gamma-Inducible Protein 16 (IFI16) Mediate Immune Signaling and Herpes Simplex Virus-1 Immunosuppression. Mol. Cell. Proteom. 2015, 14, 2341–2356. [Google Scholar] [CrossRef] [Green Version]
- Yue, Y.; Liu, J.; Cui, X.; Cao, J.; Luo, G.; Zhang, Z.; Cheng, T.; Gao, M.; Shu, X.; Ma, H.; et al. VIRMA mediates preferential m6A mRNA methylation in 3′UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018, 4, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Hauri, S.; Comoglio, F.; Seimiya, M.; Gerstung, M.; Glatter, T.; Hansen, K.; Aebersold, R.; Paro, R.; Gstaiger, M.; Beisel, C. A High-Density Map for Navigating the Human Polycomb Complexome. Cell Rep. 2016, 17, 583–595. [Google Scholar] [CrossRef] [Green Version]
- Rosenbluh, J.; Mercer, J.; Shrestha, Y.; Oliver, R.; Tamayo, P.; Doench, J.G.; Tirosh, I.; Piccioni, F.; Hartenian, E.; Horn, H.; et al. Genetic and Proteomic Interrogation of Lower Confidence Candidate Genes Reveals Signaling Networks in β-Catenin-Active Cancers. Cell Syst. 2016, 3, 302–316.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.H.; Zhong, X.; McAlpine, W.; Liao, T.-C.; Zhang, D.-W.; Fang, B.; Russell, J.; Ludwig, S.; Nair-Gill, E.; Zhang, Z.; et al. LMBR1L regulates lymphopoiesis through Wnt/β-catenin signaling. Science 2019, 364, eaau0812. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Ma, S.; Shan, L.; Wang, Y.; Wang, Y.; Cao, C.; Liu, B.; Yang, C.; Wang, L.; Tian, S.; et al. Ubiquitin-specific protease 7 sustains DNA damage response and promotes cervical carcinogenesis. J. Clin. Investig. 2018, 128, 4280–4296. [Google Scholar] [CrossRef] [PubMed]
- Benleulmi-Chaachoua, A.; Chen, L.; Sokolina, K.; Wong, V.; Jurisica, I.; Emerit, M.B.; Darmon, M.; Espin, A.; Stagljar, I.; Tafelmeyer, P.; et al. Protein interactome mining defines melatonin MT1receptors as integral component of presynaptic protein complexes of neurons. J. Pineal Res. 2015, 60, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Heidelberger, J.B.; Voigt, A.; Borisova, M.E.; Petrosino, G.; Ruf, S.; Wagner, S.A.; Beli, P. Proteomic profiling of VCP substrates links VCP to K6-linked ubiquitylation and c-Myc function. EMBO Rep. 2018, 19, e44754. [Google Scholar] [CrossRef]
- Chen, D.; Yoon, J.-B.; Gu, W. Reactivating the ARF-p53 axis in AML cells by targeting ULF. Cell Cycle 2010, 9, 2946–2951. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Jearawiriyapaisarn, N.; Lee, M.P.; Hosoya, T.; Wu, Q.; Myers, G.; Lim, K.-C.; Kurita, R.; Nakamura, Y.; Vojtek, A.B.; et al. BAP1 regulation of the key adaptor protein NCoR1 is critical for γ-globin gene repression. Genes Dev. 2018, 32, 1537–1549. [Google Scholar] [CrossRef] [Green Version]
- Shami Shah, A.; Batrouni, A.G.; Kim, D.; Punyala, A.; Cao, W.; Han, C.; Goldberg, M.L.; Smolka, M.B.; Baskin, J.M. PLEKHA4/kramer Attenuates Dishevelled Ubiquitination to Modulate Wnt and Planar Cell Polarity Signaling. Cell Rep. 2019, 27, 2157–2170.e8. [Google Scholar] [CrossRef] [Green Version]
- Khanna, R.; Krishnamoorthy, V.; Parnaik, V.K. E3 ubiquitin ligase RNF 123 targets lamin B1 and lamin-binding proteins. FEBS J. 2018, 285, 2243–2262. [Google Scholar] [CrossRef] [Green Version]
- Maréchal, A.; Li, J.-M.; Ji, X.Y.; Wu, C.-S.; Yazinski, S.A.; Nguyen, H.D.; Liu, S.; Jiménez, A.E.; Jin, J.; Zou, L. PRP19 Transforms into a Sensor of RPA-ssDNA after DNA Damage and Drives ATR Activation via a Ubiquitin-Mediated Circuitry. Mol. Cell 2014, 53, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.-C.; Greco, T.M.; Boonmee, A.; Miteva, Y.; Cristea, I.M. Functional Proteomics Establishes the Interaction of SIRT7 with Chromatin Remodeling Complexes and Expands Its Role in Regulation of RNA Polymerase I Transcription. Mol. Cell. Proteom. 2012, 11, 60–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colland, F.; Jacq, X.; Trouplin, V.; Mougin, C.; Groizeleau, C.; Hamburger, A.; Meil, A.; Wojcik, J.; Legrain, P.; Gauthier, J.-M. Functional Proteomics Mapping of a Human Signaling Pathway. Genome Res. 2004, 14, 1324–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llères, D.; Denegri, M.; Biggiogera, M.; Ajuh, P.; Lamond, A.I. Direct interaction between hnRNP-M and CDC5L/PLRG1 proteins affects alternative splice site choice. EMBO Rep. 2010, 11, 445–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipp, J.J.; Marvin, M.C.; Shokat, K.M.; Guthrie, C. SR protein kinases promote splicing of nonconsensus introns. Nat. Struct. Mol. Biol. 2015, 22, 611–617. [Google Scholar] [CrossRef]
- Li, X.; Wang, W.; Wang, J.; Malovannaya, A.; Xi, Y.; Li, W.; Guerra, R.; Hawke, D.H.; Qin, J.; Chen, J. Proteomic analyses reveal distinct chromatin-associated and soluble transcription factor complexes. Mol. Syst. Biol. 2015, 11, 775. [Google Scholar] [CrossRef]
- Baranes-Bachar, K.; Levy-Barda, A.; Oehler, J.; Reid, D.A.; Soria-Bretones, I.; Voss, T.C.; Chung, D.; Park, Y.; Liu, C.; Yoon, J.B.; et al. The Ubiquitin E3/E4 Ligase UBE4A Adjusts Protein Ubiquitylation and Accumulation at Sites of DNA Damage, Facilitating Double-Strand Break Repair. Mol. Cell 2018, 69, 866–878.e7. [Google Scholar] [CrossRef] [Green Version]
- Mulder, M.P.; Witting, K.; Berlin, I.; Pruneda, J.N.; Wu, K.-P.; Chang, J.-G.; Merkx, R.; Bialas, J.; Groettrup, J.B.M.; Vertegaal, A.C.; et al. A cascading activity-based probe sequentially targets E1–E2–E3 ubiquitin enzymes. Nat. Chem. Biol. 2016, 12, 523–530. [Google Scholar] [CrossRef]
- Schwarz, S.E.; Rosa, J.L.; Scheffner, M. Characterization of Human hect Domain Family Members and Their Interaction with UbcH5 and UbcH7. J. Biol. Chem. 1998, 273, 12148–12154. [Google Scholar] [CrossRef] [Green Version]
- Pao, K.-C.; Wood, N.T.; Knebel, A.; Rafie, K.; Stanley, M.; Mabbitt, P.D.; Sundaramoorthy, R.; Hofmann, K.; Van Aalten, D.M.F.; Virdee, S. Activity-based E3 ligase profiling uncovers an E3 ligase with esterification activity. Nat. Cell Biol. 2018, 556, 381–385. [Google Scholar] [CrossRef] [Green Version]
- Sowa, M.E.; Bennett, E.J.; Gygi, S.P.; Harper, J.W. Defining the Human Deubiquitinating Enzyme Interaction Landscape. Cell 2009, 138, 389–403. [Google Scholar] [CrossRef] [Green Version]
- Georges, A.; Coyaud, E.; Marcon, E.; Greenblatt, J.; Raught, B.; Frappier, L. USP7 Regulates Cytokinesis through FBXO38 and KIF20B. Sci. Rep. 2019, 9, 2724. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-C.; Yang, J.-C.; Chang, Y.-C.; Chuang, J.-G.; Lin, C.-W.; Wu, M.-S.; Chow, L.-P. VCP Phosphorylation-Dependent Interaction Partners Prevent Apoptosis in Helicobacter pylori-Infected Gastric Epithelial Cells. PLoS ONE 2013, 8, e55724. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, S.; Schild-Poulter, C. Mapping the Ku Interactome Using Proximity-Dependent Biotin Identification in Human Cells. J. Proteome Res. 2018, 18, 1064–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junttila, M.R.; Evan, G.I. p53—A Jack of all trades but master of none. Nat. Rev. Cancer 2009, 9, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Kon, N.; Zhong, J.; Zhang, P.; Yu, L.; Gu, W. Differential Effects on ARF Stability by Normal versus Oncogenic Levels of c-Myc Expression. Mol. Cell 2013, 51, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Murphy, D.J.; Junttila, M.R.; Pouyet, L.; Karnezis, A.; Shchors, K.; Bui, D.A.; Brown-Swigart, L.; Johnson, L.; Evan, G.I. Distinct Thresholds Govern Myc’s Biological Output In Vivo. Cancer Cell 2008, 14, 447–457. [Google Scholar] [CrossRef] [Green Version]
- Ginno, P.A.; Burger, L.; Seebacher, J.; Iesmantavicius, V.; Schübeler, D. Cell cycle-resolved chromatin proteomics reveals the extent of mitotic preservation of the genomic regulatory landscape. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Velimezi, G.; Liontos, M.; Vougas, K.; Roumeliotis, T.; Bartkova, J.; Sideridou, M.; Dereli-Oz, A.; Kocylowski, M.; Pateras, I.S.; Evangelou, K.; et al. Functional interplay between the DNA-damage-response kinase ATM and ARF tumour suppressor protein in human cancer. Nat. Cell Biol. 2013, 15, 967–977. [Google Scholar] [CrossRef]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 Ubiquitylates Histones at DNA Double-Strand Breaks and Promotes Assembly of Repair Proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Brooks, C.L.; Kon, N.; Gu, W. A Dynamic Role of HAUSP in the p53-Mdm2 Pathway. Mol. Cell 2004, 13, 879–886. [Google Scholar] [CrossRef]
- Zhu, Q.; Sharma, N.; Qianzheng, Z.; Wani, G.; A Wani, A. USP7 deubiquitinase promotes ubiquitin-dependent DNA damage signaling by stabilizing RNF168. Cell Cycle 2015, 14, 1413–1425. [Google Scholar] [CrossRef] [Green Version]
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 Binds and Amplifies Ubiquitin Conjugates on Damaged Chromosomes to Allow Accumulation of Repair Proteins. Cell 2009, 136, 435–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenfeld, G.; Groudine, M. Controlling the double helix. Nat. Cell Biol. 2003, 421, 448–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, C.W.M.; Orkin, S.H. The SWI/SNF complex—Chromatin and cancer. Nat. Rev. Cancer 2004, 4, 133–142. [Google Scholar] [CrossRef]
- Vignali, M.; Hassan, A.H.; Neely, K.E.; Workman, J.L. ATP-Dependent Chromatin-Remodeling Complexes. Mol. Cell. Biol. 2000, 20, 1899–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Beaulaurier, J.; Deikus, G.; Wu, T.P.; Strahl, M.; Hao, Z.; Luo, G.; Gregory, J.A.; Chess, A.; He, C.; et al. Mapping and characterizing N6-methyladenine in eukaryotic genomes using single-molecule real-time sequencing. Genome Res. 2018, 28, 1067–1078. [Google Scholar] [CrossRef]
- Kirmizis, A. Silencing of human polycomb target genes is associated with methylation of histone H3 Lys 27. Genes Dev. 2004, 18, 1592–1605. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nat. Cell Biol. 2004, 431, 873–878. [Google Scholar] [CrossRef]
- Scheuermann, J.C.; Alonso, A.G.D.A.; Oktaba, K.; Ly-Hartig, N.; McGinty, R.K.; Fraterman, S.; Wilm, M.; Muir, T.W.; Müller, J. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nat. Cell Biol. 2010, 465, 243–247. [Google Scholar] [CrossRef] [Green Version]
- Kloet, S.L.; Makowski, M.M.; Baymaz, H.I.; Van Voorthuijsen, L.; Karemaker, I.D.; Santanach, A.; Jansen, P.W.; Di Croce, A.S.L.; Vermeulen, M. The dynamic interactome and genomic targets of Polycomb complexes during stem-cell differentiation. Nat. Struct. Mol. Biol. 2016, 23, 682–690. [Google Scholar] [CrossRef] [Green Version]
- Hoischen, A.; Van Bon, B.W.; Rodríguez-Santiago, B.; Gilissen, C.; Vissers, L.E.L.M.; De Vries, P.; Janssen, I.; Van Lier, B.; Hastings, R.; Smithson, S.F.; et al. De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat. Genet. 2011, 43, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.M.; Nisperos, S.V.; Weeks, J.; Ghulam, M.; Marín, I.; Zayas, R.M. Identification of HECT E3 ubiquitin ligase family genes involved in stem cell regulation and regeneration in planarians. Dev. Biol. 2015, 404, 21–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagiwara, N.; Yeh, M.; Liu, A. Sox6 is required for normal fiber type differentiation of fetal skeletal muscle in mice. Dev. Dyn. 2007, 236, 2062–2076. [Google Scholar] [CrossRef] [PubMed]
- Krah, N.M.; De La O, J.-P.; Swift, G.H.; Hoang, C.Q.; Willet, S.G.; Pan, F.C.; Cash, G.M.; Bronner, M.P.; Wright, C.V.; Macdonald, R.J.; et al. The acinar differentiation determinant PTF1A inhibits initiation of pancreatic ductal adenocarcinoma. eLife 2015, 4, e07125. [Google Scholar] [CrossRef]
- Borromeo, M.D.; Meredith, D.M.; Castro, D.S.; Chang, J.C.; Tung, K.-C.; Guillemot, F.; Johnson, J.E. A transcription factor network specifying inhibitory versus excitatory neurons in the dorsal spinal cord. Development 2014, 141, 2803–2812. [Google Scholar] [CrossRef] [Green Version]
- Lelièvre, E.; Lek, M.; Boije, H.; Houille-Vernes, L.; Brajeul, V.; Slembrouck, A.; Roger, J.; Sahel, J.-A.; Matter, J.-M.; Sennlaub, F.; et al. Ptf1a/Rbpj complex inhibits ganglion cell fate and drives the specification of all horizontal cell subtypes in the chick retina. Dev. Biol. 2011, 358, 296–308. [Google Scholar] [CrossRef]
- Millen, K.J.; Steshina, E.Y.; Iskusnykh, I.Y.; Chizhikov, V.V. Transformation of the cerebellum into more ventral brainstem fates causes cerebellar agenesis in the absence of Ptf1a function. Proc. Natl. Acad. Sci. USA 2014, 111, E1777–E1786. [Google Scholar] [CrossRef] [Green Version]
- Ilyas, M.; Mir, A.; Efthymiou, S.; Houlden, H. The genetics of intellectual disability: Advancing technology and gene editing. F1000Research 2020, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Mendicino, A.; Sabbadini, G.; Pergola, M.S. Clark—Baraitser syndrome: Report of a new case and review of the literature. Clin. Dysmorphol. 2005, 14, 133–135. [Google Scholar] [CrossRef]
- Louie, R.J.; Friez, M.J.; Skinner, C.; Baraitser, M.; Clark, R.D.; Schwartz, C.E.; Stevenson, R.E. Clark-Baraitser syndrome is associated with a nonsense alteration in the autosomal gene TRIP12. Am. J. Med Genet. Part A 2019, 182, 595–596. [Google Scholar] [CrossRef]
- Doco-Fenzy, M.; Landais, E.; Andrieux, J.; Schneider, A.; Delemer, B.; Sulmont, V.; Melin, J.-P.; Ploton, M.; Thevenard, J.; Monboisse, J.-C.; et al. Deletion 2q36.2q36.3 with multiple renal cysts and severe mental retardation. Eur. J. Med. Genet. 2008, 51, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Lelieveld, S.H.; Reijnders, M.R.F.; Pfundt, R.; Yntema, H.G.; Kamsteeg, E.-J.; De Vries, P.; De Vries, B.B.A.; Willemsen, M.H.; Kleefstra, T.; Löhner, K.; et al. Meta-analysis of 2104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 2016, 19, 1194–1196. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nat. Cell Biol. 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Roak, B.J.; Stessman, H.A.; Boyle, E.A.; Witherspoon, K.T.; Martin, B.; Lee, C.; Vives, L.; Baker, C.; Hiatt, J.B.; Nickerson, D.A.; et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat. Commun. 2014, 5, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Gambin, T.; Yuan, B.; Szafranski, P.; Rosenfeld, J.A.; Al Balwi, M.; Alswaid, A.; Al-Gazali, L.; Al Shamsi, A.M.; Komara, M.; et al. Haploinsufficiency of the E3 ubiquitin-protein ligase gene TRIP12 causes intellectual disability with or without autism spectrum disorders, speech delay, and dysmorphic features. Qual. Life Res. 2017, 136, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Takata, A.; Miyake, N.; Tsurusaki, Y.; Fukai, R.; Miyatake, S.; Koshimizu, E.; Kushima, I.; Okada, T.; Morikawa, M.; Uno, Y.; et al. Integrative Analyses of De Novo Mutations Provide Deeper Biological Insights into Autism Spectrum Disorder. Cell Rep. 2018, 22, 734–747. [Google Scholar] [CrossRef] [Green Version]
- Ruzzo, E.K.; Pérez-Cano, L.; Jung, J.Y.; Wang, L.K.; Kashef-Haghighi, D.; Hartl, C.; Singh, C.; Xu, J.; Hoekstra, J.N.; Leventhal, O.; et al. Inherited and De Novo Genetic Risk for Autism Impacts Shared Networks. Cell 2019, 178, 850–866.e26. [Google Scholar] [CrossRef] [Green Version]
- Stessman, H.A.F.; Xiong, B.; Coe, B.P.; Wang, T.; Hoekzema, K.; Fenckova, M.; Kvarnung, M.; Gerdts, J.; Trinh, S.; Cosemans, N.; et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 2017, 49, 515–526. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Yoo, N.J.; Park, S.W.; Lee, S.H. Frameshift mutations of ubiquitination-related genes HERC2, HERC3, TRIP12, UBE2Q1 and UBE4B in gastric and colorectal carcinomas with microsatellite instability. Pathology 2011, 43, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Hein, R.; Network, T.G.; Flesch-Janys, D.; Dahmen, N.; Beckmann, L.; Lindström, S.; Schoof, N.; Czene, K.; Mittelstraß, K.; Illig, T.; et al. A genome-wide association study to identify genetic susceptibility loci that modify ductal and lobular postmenopausal breast cancer risk associated with menopausal hormone therapy use: A two-stage design with replication. Breast Cancer Res. Treat. 2013, 138, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yi, S.; Yang, F.; Zhou, Y.; Ji, Q.; Cai, J.; Mei, Y. Identification of mutant genes with high-frequency, high-risk, and high-expression in lung adenocarcinoma. Thorac. Cancer 2014, 5, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Cacheux, W.; Dangles-Marie, V.; Rouleau, E.; Lazartigues, J.; Girard, E.; Briaux, A.; Mariani, P.; Richon, S.; Vacher, S.; Buecher, B.; et al. Exome sequencing reveals aberrant signalling pathways as hallmark of treatment-naive anal squamous cell carcinoma. Oncotarget 2017, 9, 464–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiba, N.; Yoshida, K.; Hara, Y.; Yamato, G.; Shiraishi, Y.; Matsuo, H.; Okuno, Y.; Chiba, K.; Tanaka, H.; Kaburagi, T.; et al. Transcriptome analysis offers a comprehensive illustration of the genetic background of pediatric acute myeloid leukemia. Blood Adv. 2019, 3, 3157–3169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, H.; Attiye, M.; Gerold, J.M.; Makohon-Moore, A.P.; Hayashi, A.; Hong, J.; Kappagantula, R.; Zhang, L.; Melchor, J.P.; Reiter, J.G.; et al. The Evolutionary Origins of Recurrent Pancreatic Cancer. Cancer Discov. 2020, 10, 792–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Jin, Z.; Cheng, Y.; Cao, X. RNA-Seq analysis identifies aberrant RNA splicing of TRIP12 in acute myeloid leukemia patients at remission. Tumor Biol. 2014, 35, 9585–9590. [Google Scholar] [CrossRef]
- Horvat, A.; Noto, J.M.; Ramatchandirin, B.; Zaika, E.; Palrasu, M.; Wei, J.; Schneider, B.G.; El-Rifai, W.; Peek, R.M.; Zaika, A.I. Helicobacter pylori pathogen regulates p14ARF tumor suppressor and autophagy in gastric epithelial cells. Oncogene 2018, 37, 5054–5065. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, P.-J.; Molkentine, D.P.; Chen, C.; Molkentine, J.; Piao, H.; Raju, U.; Zhang, J.; Valdecanas, D.R.; Tailor, R.C.; et al. TRIP12 as a mediator of human papillomavirus/p16-related radiation enhancement effects. Oncogene 2016, 36, 820–828. [Google Scholar] [CrossRef] [Green Version]
- Molkentine, J.M.; Molkentine, D.P.; Bridges, K.A.; Xie, T.; Yang, L.; Sheth, A.; Heffernan, T.P.; Clump, D.A.; Faust, A.Z.; Ferris, R.L.; et al. Targeting DNA damage response in head and neck cancers through abrogation of cell cycle checkpoints. Int. J. Radiat. Biol. 2020, 1–8. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
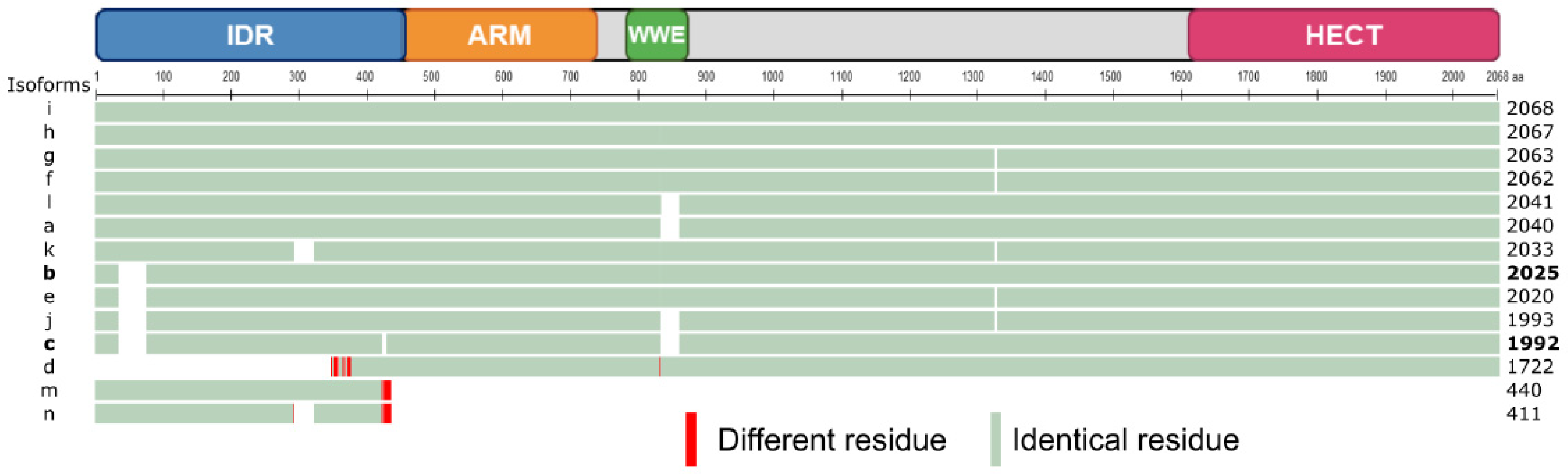
| mRNA Variant | Accession Number | mRNA Length | Exon Number | Protein Isoform | Protein ID | aa Number | Theoretical kDa |
|---|---|---|---|---|---|---|---|
| 1 | NM_001284214.2 | 10019 | 42 | a | NP_001271143.1 | 2040 | 224 |
| 2 | NM_001284215.2 | 9974 | 41 | b | NP_001271144.1 | 2025 | 223 |
| 3 | NM_004238.3 | 9875 | 41 | c | NP_004229.1 | 1992 | 219 |
| 4 | NM_001284216.2 | 9065 | 39 | d | NP_001271145.1 | 1722 | 189 |
| 5 | NM_001348315.2 | 10408 | 42 | a | NP_001335244.1 | 2040 | 224 |
| 6 | NM_001348316.2 | 10070 | 41 | b | NP_001335245.1 | 2025 | 223 |
| 7 | NM_001348317.1 | 9959 | 41 | e | NP_001335246.1 | 2020 | 222 |
| 8 | NM_001348318.2 | 10348 | 41 | e | NP_001335247.1 | 2020 | 222 |
| 9 | NM_001348319.1 | 10085 | 42 | f | NP_001335248.1 | 2062 | 227 |
| 10 | NM_001348320.2 | 10474 | 42 | f | NP_001335249.1 | 2062 | 227 |
| 11 | NM_001348321.1 | 10088 | 42 | g | NP_001335250.1 | 2063 | 227 |
| 12 | NM_001348322.1 | 10218 | 43 | h | NP_001335251.1 | 2067 | 227 |
| 13 | NM_001348323.3 | 10100 | 42 | h | NP_001335252.1 | 2067 | 227 |
| 14 | NM_001348324.2 | 10196 | 42 | h | NP_001335253.1 | 2067 | 227 |
| 15 | NM_001348325.2 | 10611 | 43 | h | NP_001335254.1 | 2067 | 227 |
| 16 | NM_001348326.2 | 10486 | 42 | h | NP_001335255.1 | 2067 | 227 |
| 17 | NM_001348327.2 | 10489 | 42 | h | NP_001335256.1 | 2067 | 227 |
| 18 | NM_001348328.1 | 10103 | 42 | i | NP_001335257.1 | 2068 | 227 |
| 19 | NM_001348329.2 | 10454 | 42 | i | NP_001335258.1 | 2068 | 227 |
| 20 | NM_001348330.2 | 10492 | 42 | i | NP_001335259.1 | 2068 | 227 |
| 21 | NM_001348331.1 | 9878 | 41 | j | NP_001335260.1 | 1993 | 219 |
| 22 | NM_001348332.1 | 9998 | 43 | k | NP_001335261.1 | 2033 | 224 |
| 23 | NM_001348333.1 | 10022 | 42 | l | NP_001335262.1 | 2041 | 225 |
| 24 | NM_001348335.1 | 5955 | 6 | m | NP_001335264.1 | 440 | 48 |
| 25 | NM_001348336.1 | 6052 | 6 | m | NP_001335265.1 | 440 | 48 |
| 26 | NM_001348334.1 | 5951 | 7 | n | NP_001335263.1 | 411 | 45 |
| # | Interactor Gene | Interactor Name | Bait and/or | Interaction Detection Method | Interaction Type | References |
|---|---|---|---|---|---|---|
| Hit | ||||||
| 1 | ALYREF | Aly/REF export factor | bait | aff tech | co-localization | [66] |
| 2 | ASXL1 | Additional Sex Combs-Like Protein 1 | bait | biochemical | ph ass | [53] |
| 3 | ATG16L1 | Autophagy-related protein 16 like1 | bait | aff tech | ph ass | [67] |
| 4 | BCAT1 | Branched-chain-amino-acid aminotransferase | bait | ph ass | ph ass | [68,69] |
| 5 | CAPN1 | Calpain-1 catalytic subunit | bait | aff tech | ph ass | [69] |
| 6 | CARNS1 | Carnosine synthase 1 | bait | two hybrid | dir int | [70] |
| 7 | CCDC8 | Coiled-coil domain-containing protein 8 | bait | aff tech | ph ass | [71] |
| 8 | CDK9 | Cyclin-dependent kinase 9 | bait, hit | aff tech | ph ass | [72] |
| 9 | CDKN2A | Cyclin-Dependent Kinase Inhibitor 2A/p14ARF | bait | aff tech, pd, ph ass | ph ass, dir int, ph int | [15,58,73,74,75] |
| 10 | CEP19 | Centrosomal protein of 19 kDa | bait | bioid | ph ass | [76] |
| 11 | CFTR | Cystic fibrosis transmembrane conductance regulator | bait | aff tech | ph ass | [77] |
| 12 | CLSTN1 | Calsyntenin-1 | bait | two hybrid | dir int | [70] |
| 13 | DCPS | Decapping scavenger enzyme | bait | aff tech | ph ass | [78] |
| 14 | DYRK1A | Dual specificity Tyrosine-phosphorylation-regulated kinase 1A | bait | aff tech | ph ass | [79] |
| 15 | EED | Embryonic ectoderm development protein | bait | aff tech | ph ass | [80] |
| 16 | EIF3I | Eukaryotic translation initiation factor 3 subunit I | bait | aff tech | ph ass | [68,69] |
| 17 | ERG | Transcriptional regulator ERG | bait | pd | dir int | [81] |
| 18 | ESR1 | Estrogen receptor alpha | bait | aff tech | ph ass | [82] |
| 19 | EXOSC9 | Exosome complex component RRP45 | bait | aff tech | ph ass | [83] |
| 20 | EZH2 | Histone-lysine N-methyltransferase | bait | aff tech | ph ass | [84] |
| 21 | FAM46A | HBV X-transactivated gene 11 protein) | bait | aff tech | ph ass | [83] |
| 22 | GNL3 | Guanine nucleotide-binding protein-like 3/Nucleostemin | hit | aff tech | ph ass, dir int, ph int | [75] |
| 23 | HIST1H2AB | Histone H2A | bait | aff tech | ph ass | [85] |
| 24 | HIST1H2BB | Histone H2B | bait | aff tech, bioid | ph ass | [85,86] |
| 25 | HIST1H4A | Histone H4 | bait | aff tech | ph ass | [85,87] |
| 26 | IFI16 | Gamma-interferon-inducible protein 16 | bait | aff tech | ph ass | [88] |
| 27 | KBTBD7 | Kelch repeat and BTB domain-containing protein 7 | bait | aff tech | ph ass | [69] |
| 28 | KIAA1429 | Protein virilizer homolog | bait | aff tech | ph ass | [89] |
| 29 | KPNA1 | Importin subunit alpha-5 (Karyopherin subunit alpha-1) | hit | aff tech | ph ass | [90] |
| 30 | KPNA5 | Importin subunit alpha-6 (Karyopherin subunit alpha-5) | hit | aff tech | ph ass | [90] |
| 31 | KPNA6 | Importin subunit alpha-7 (Karyopherin subunit alpha-6) | hit | aff tech | ph ass | [90] |
| 32 | KRAS | GTPase KRas | bait | aff tech | ph ass | [91] |
| 33 | LMBR1L | Limb development membrane protein 1-like | bait | aff tech | ph ass | [92] |
| 34 | MAP2K3 | Dual specificity mitogen-activated protein kinase kinase 3 | bait | aff tech | ph ass | [91] |
| 35 | MDC1 | Mediator of DNA damage checkpoint protein 1 | bait | aff tech | ph ass | [93] |
| 36 | MECP2 | Methyl-CpG-binding protein 2 | bait | aff tech | ph ass | [69] |
| 37 | MEPCE | 7SK snRNA methylphosphate capping enzyme | bait | aff tech | ph ass | [72] |
| 38 | MTNR1A | Melatonin receptor type 1A | bait | two hybrid | dir int | [94] |
| 39 | MTNR1B | Melatonin receptor type 1B | bait | two hybrid | dir int | [94] |
| 40 | MYC | Myc proto-oncogene protein | bait, hit | aff tech, pd | ph ass, dir int | [15,95] |
| 41 | NAE1 | NEDD8-activating enzyme E1 regulatory subunit/APP-BP1 | hit | aff tech, enz study | ph ass, dir int | [4] |
| 42 | NMI | N-myc-interactor | bait, hit | aff tech, pd | ph ass, dir int | [74] |
| 43 | NPM | Nucleophosmin | bait, hit | aff tech, pd | ph ass, dir int | [96] |
| 44 | NR2C2 | Nuclear receptor subfamily 2 group C member 2 | bait | aff tech | ph ass | [97] |
| 45 | OBSL1 | Obscurin-like protein 1 | bait | aff tech | ph ass | [71] |
| 46 | OPCML | Opioid-binding protein/cell adhesion molecule | bait | aff tech | ph ass | [69] |
| 47 | PARP1 | Poly-(ADP Ribose) polymerase 1 | bait, hit | aff tech, pd, enz study | ph ass, dir int | [27] |
| 48 | PHF8 | Histone lysine demethylase | bait | aff tech | ph ass | [83] |
| 49 | PLEKHA4 | Peckstrin homology domain containing, family member 4 | bait | aff tech | ph ass | [98] |
| 50 | PTF1A | Pancreas transcription factor 1 subunit alpha | bait, hit | aff tech, FRET, two hybrid | ph ass, dir int, dir int | [6] |
| 51 | RNF123 | E3 ring finger ubiquitin ligase 123 | bait | aff tech | ph ass | [99] |
| 52 | RNF168 | E3 ring finger ubiquitin ligase168 | bait | aff tech | ph ass | [14] |
| 53 | RP2 | Protein XRP2 | bait | aff tech | ph ass | [68,69] |
| 54 | RPA | Replication protein A | bait | aff tech | ph ass | [100] |
| 55 | RPL39 | 60S ribosomal protein L39 | hit | aff tech | ph ass | [90] |
| 56 | SIRT7 | Sirtuin 7 | bait | aff tech | ph ass | [101] |
| 57 | SMAD9 | SMAD family member 9 | bait | two hybrid | dir int | [102] |
| 58 | SMARCE1 | SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily E member 1/BAF57 | bait | aff tech | ph ass | [54] |
| 59 | SNW1 | SNW domain-containing protein 1 | bait | aff tech | ph ass | [103] |
| 60 | SOX6 | Transcription factor SOX-6 | bait, hit | enz study, aff tech, two hybrid | dir int, ph ass | [12] |
| 61 | SRPK2 | SRSF protein kinase 2 | bait | enz study | dir int | [104] |
| 62 | SUZ12 | Polycomb protein SUZ12 | bait | aff tech | ph ass | [84] |
| 63 | TEAD2 | Transcriptional enhancer factor TEF-4 | bait | aff tech | ph ass | [105] |
| 64 | TRADD | Tumor necrosis factor receptor type 1-associated DEATH domain | bait, hit | aff tech | ph ass | [58] |
| 65 | UBC | Ubiquitin-C | bait, hit | enz study, aff tech | dir int, ph ass | [60,106,107] |
| 66 | UBE2D1 | Ubiquitin-conjugating enzyme E2 D1 | hit | pd | dir int | [4,12,60,108] |
| 67 | UBE2D2 | Ubiquitin-conjugating enzyme E2 D2 | bait | pd | dir int | [109] |
| 68 | UBE2L3 | Ubiquitin-conjugating enzyme E2 L3 | bait, hit | pd | dir int | [108,109] |
| 69 | UBE2L6 | Ubiquitin-conjugating enzyme E2 L6 | bait | aff tech | ph ass | [91] |
| 70 | UBE4B | Ubiquitin conjugation factor E4 B | bait | pd | dir int | [4] |
| 71 | USP7 | Ubiquitin-specific-peptidase 7 | bait, hit | aff tech, ph ass | ph ass | [39,48,110,111] |
| 72 | USP11 | Ubiquitin-specific peptidase 11 | bait | aff tech | ph ass | [110] |
| 73 | VCP | Valosin-containing protein | bait | aff tech | ph ass | [112] |
| 74 | VHL | von Hippel-Lindau E3 ubiquitin ligase | bait | aff tech | ph ass | [83] |
| 75 | WDR76 | WD repeat-containing protein 76 | bait | aff tech | ph ass | [85] |
| 76 | XRCC6 | X-ray repair cross-complementing protein | bait | bioid | ph ass | [113] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brunet, M.; Vargas, C.; Larrieu, D.; Torrisani, J.; Dufresne, M. E3 Ubiquitin Ligase TRIP12: Regulation, Structure, and Physiopathological Functions. Int. J. Mol. Sci. 2020, 21, 8515. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228515
Brunet M, Vargas C, Larrieu D, Torrisani J, Dufresne M. E3 Ubiquitin Ligase TRIP12: Regulation, Structure, and Physiopathological Functions. International Journal of Molecular Sciences. 2020; 21(22):8515. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228515
Chicago/Turabian StyleBrunet, Manon, Claire Vargas, Dorian Larrieu, Jérôme Torrisani, and Marlène Dufresne. 2020. "E3 Ubiquitin Ligase TRIP12: Regulation, Structure, and Physiopathological Functions" International Journal of Molecular Sciences 21, no. 22: 8515. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228515