Platelet-Activating Factor-Receptor Signaling Mediates Targeted Therapies-Induced Microvesicle Particles Release in Lung Cancer Cells

 and
and 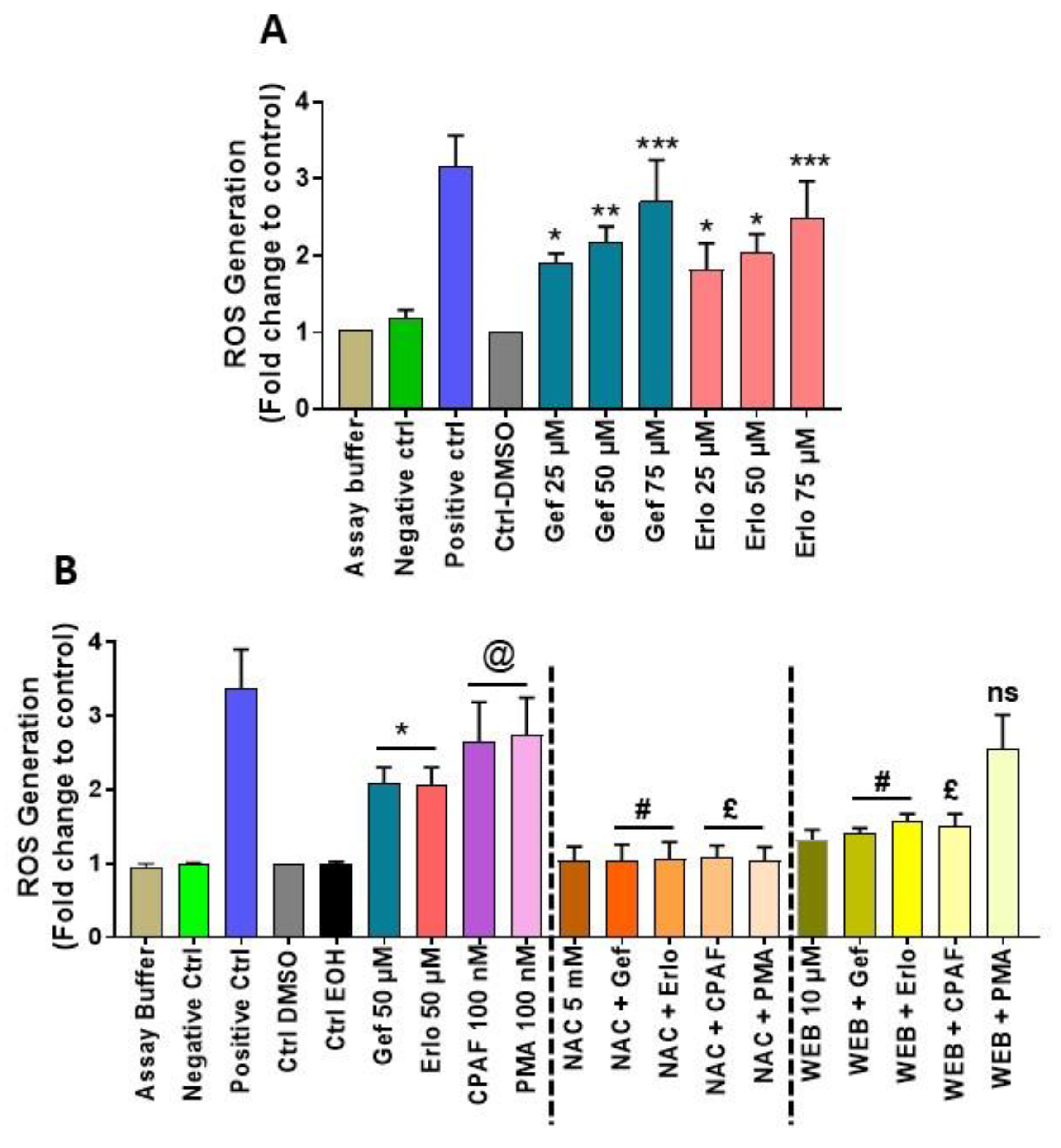{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Exposure to Gefitinib and Erlotinib Generates ROS
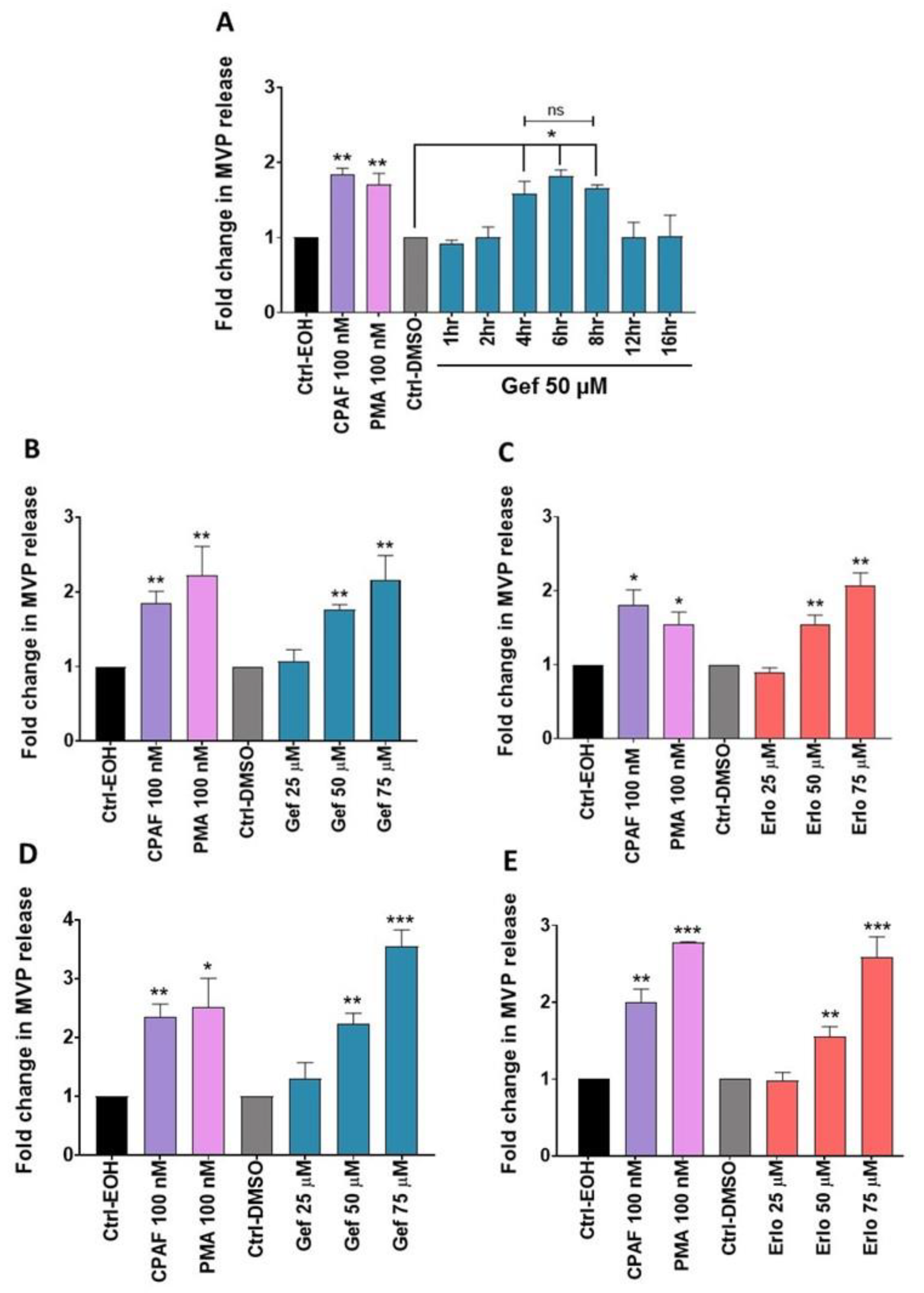
2.2. Gefitinib and Erlotinib Treatments Induce MVP Release from NSCLC Cell Lines in a Time- and Dose-Dependent Manner
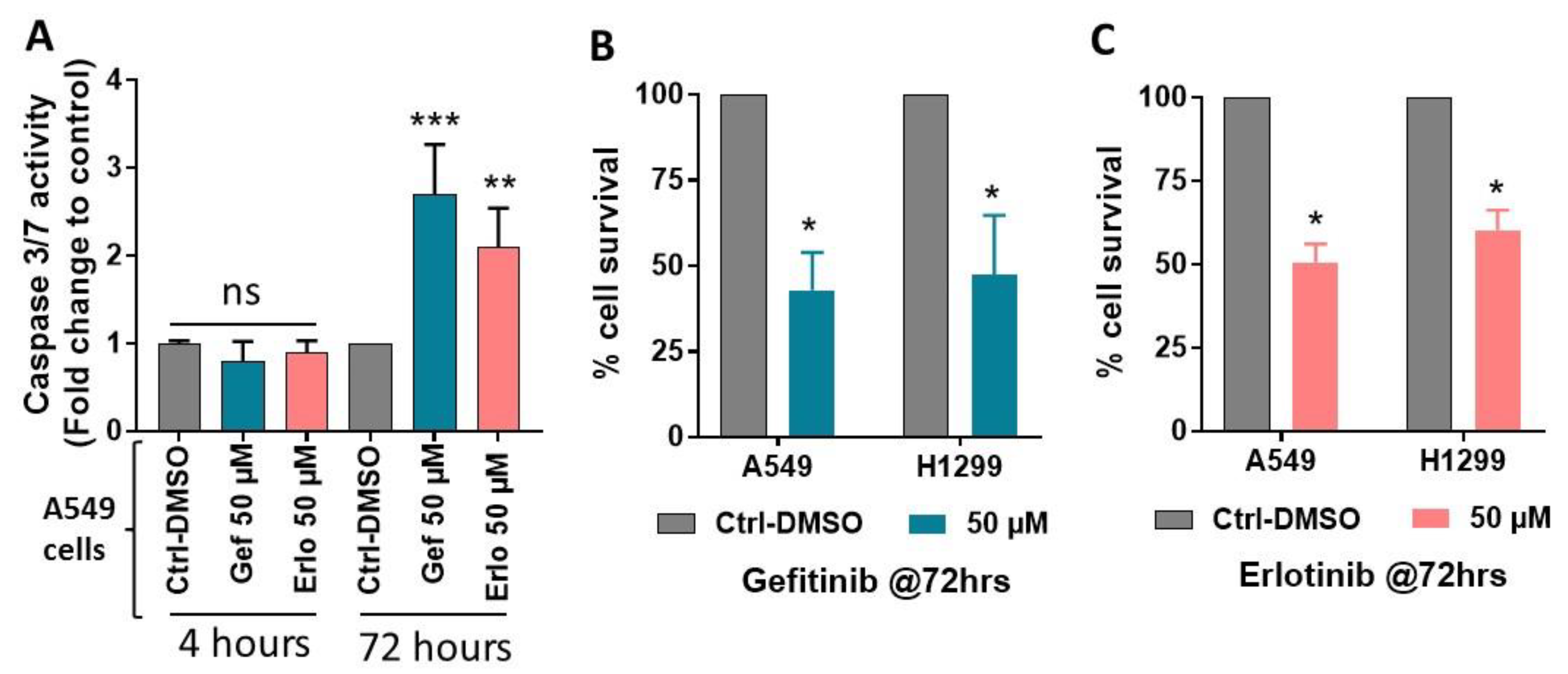
2.3. Effect of Gefitinib and Erlotinib Treatments on Apoptosis Induction
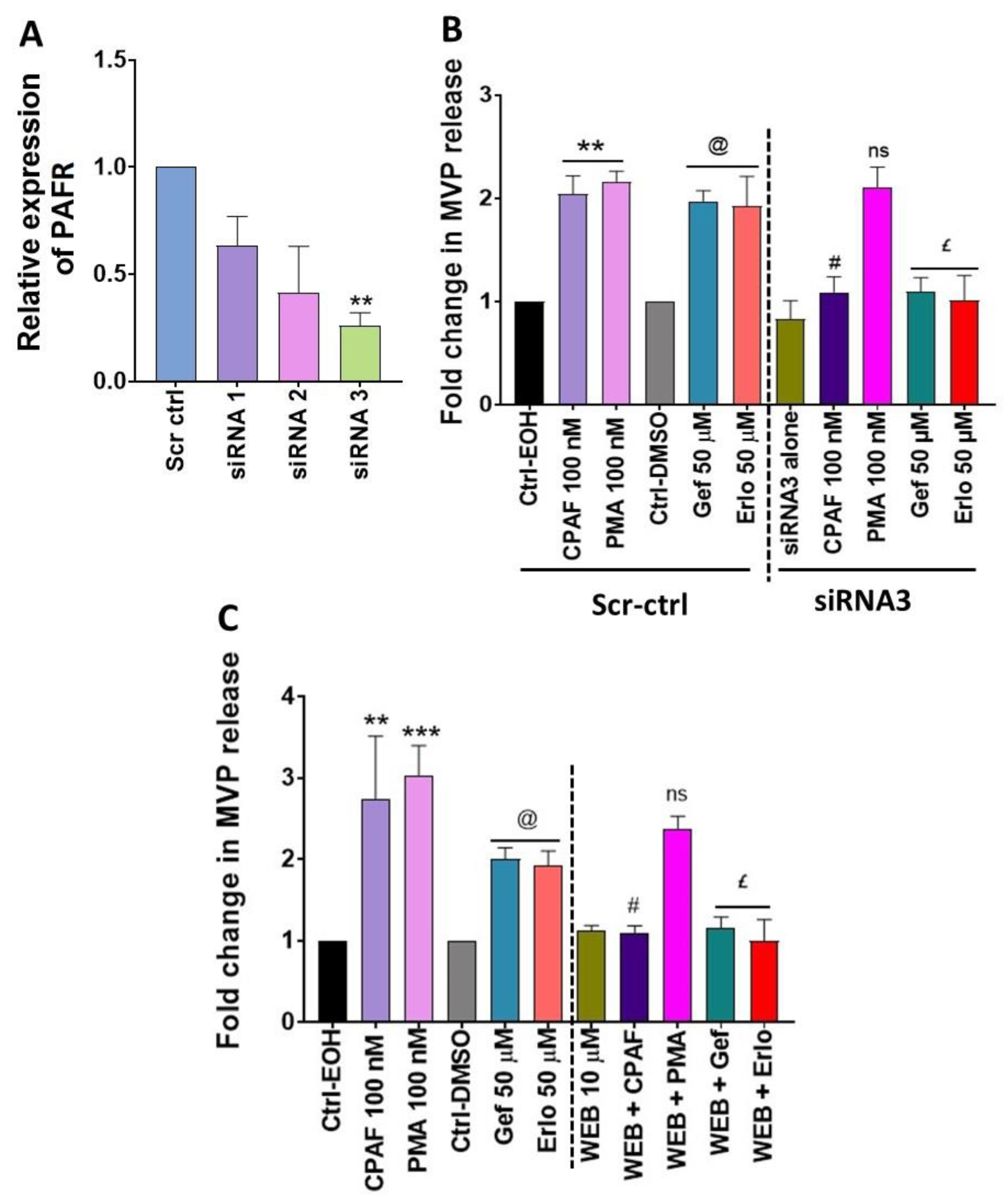
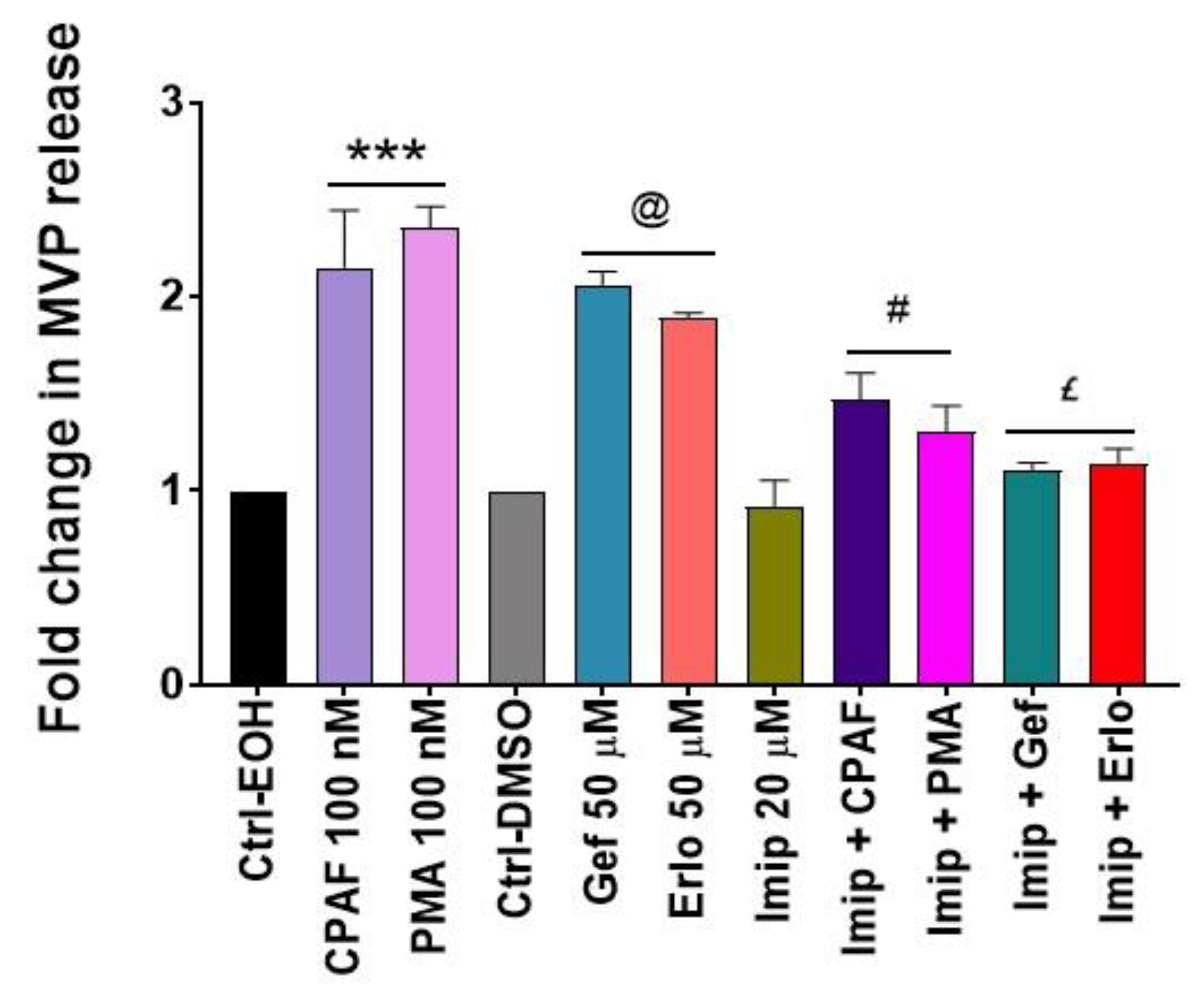
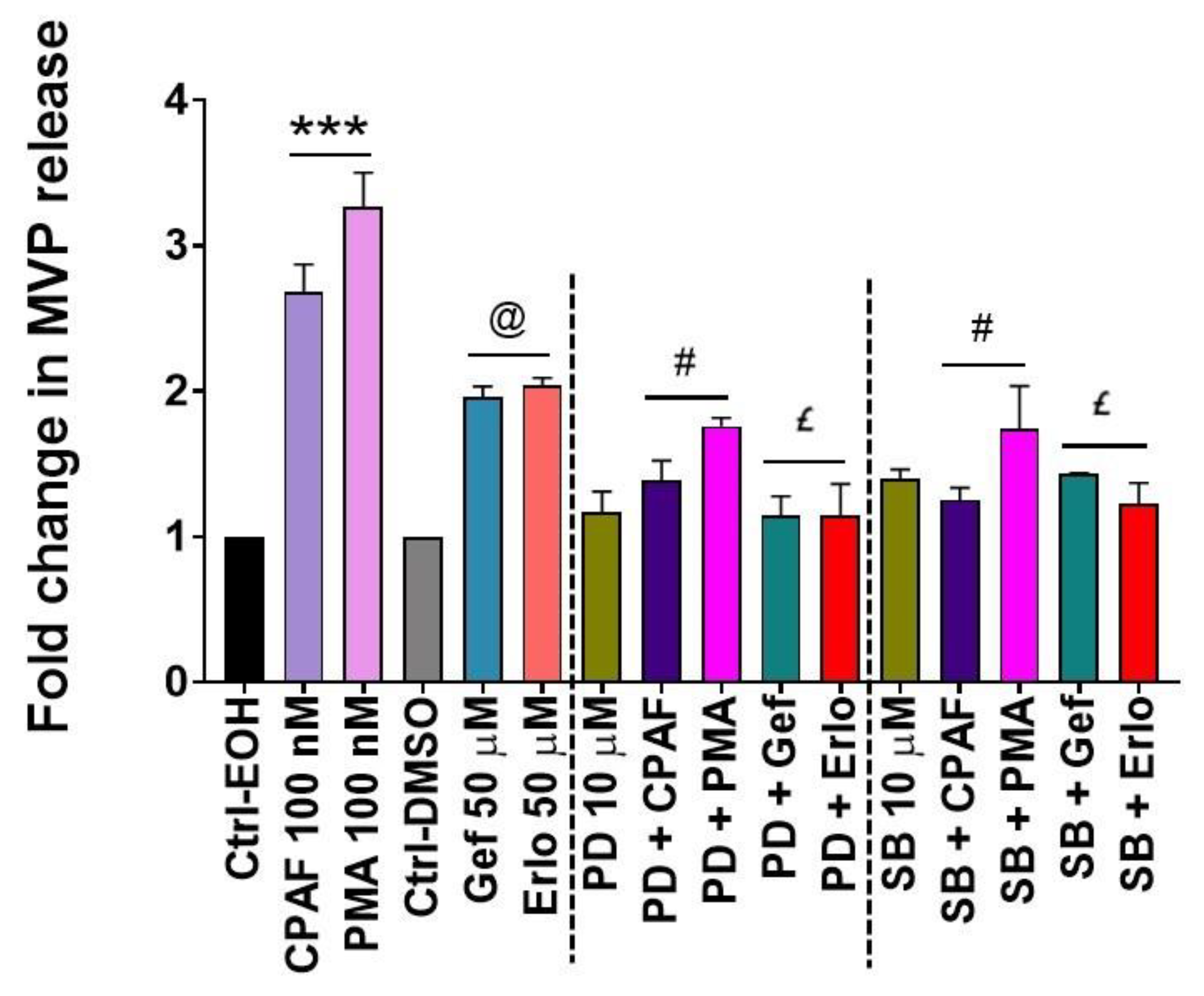
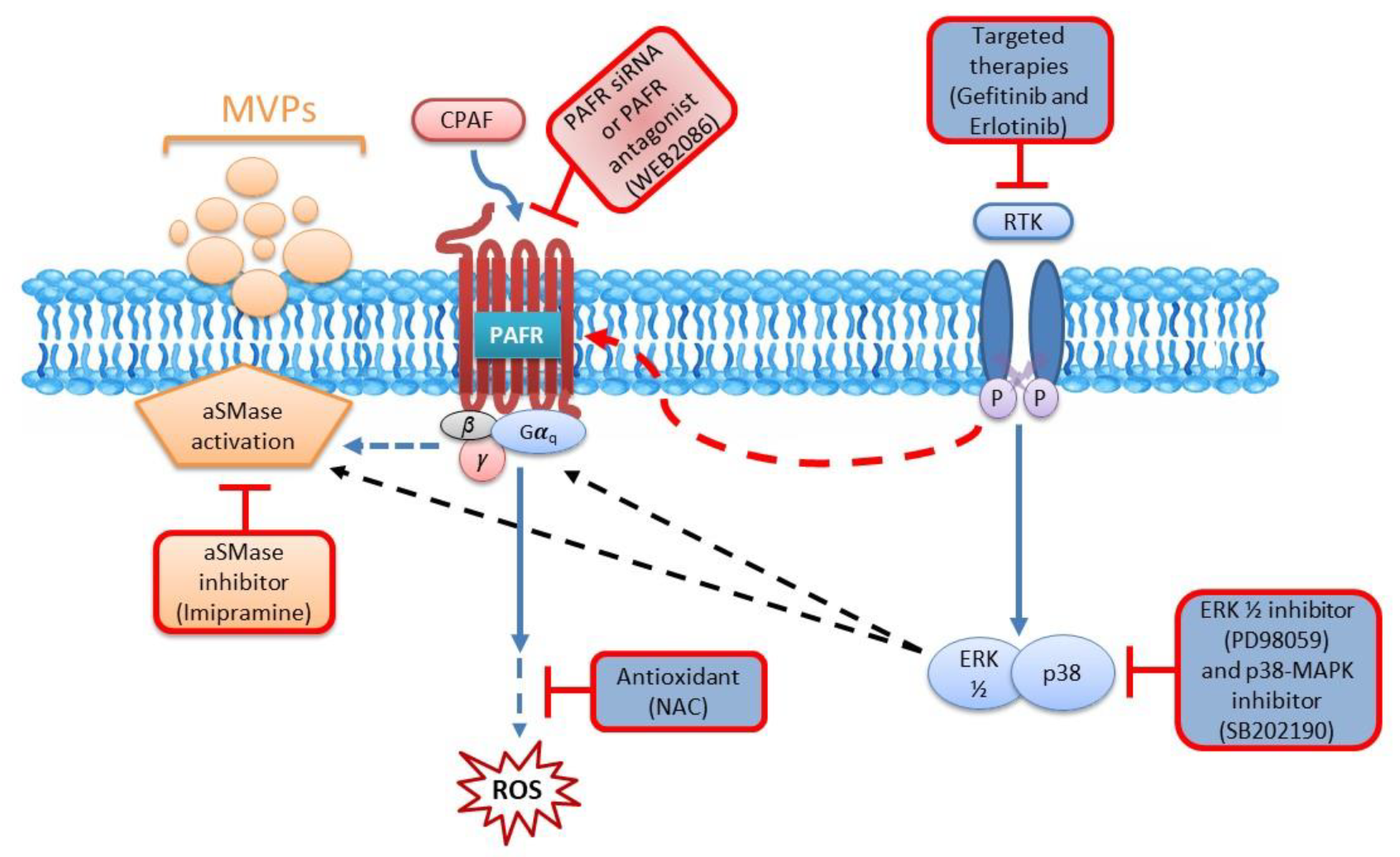
2.4. Blockade of the PAFR Attenuates Erlotinib and Gefitinib-Induced MVP Release
2.5. Inhibition of aSMase Blocks MVP Release
2.6. MAPK Pathway Mediates Erlotinib and Gefitinib-Mediated MVP Release
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Measurement of ROS Generation
4.4. Cell Survival Assay
4.5. Apoptosis Assay
4.6. siRNA Transfection, RNA Extraction and Quantitative Real-Time PCR (qRT-PCR) Analysis
4.7. Microvesicle Particles (MVP) Extraction and Analysis
4.8. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PAF | Platelet-activating factor |
| PAFR | Platelet-activating factor-receptor |
| Ox-GPCs | Oxidized glycerophosphocholines |
| MVP | Microvesicle particles |
| ROS | Reactive oxygen species |
| MAPK | Mitogen-activated protein kinase |
| STAT3 | Signal transduction and activator of transcription |
| NSCLC | Non-small cell lung cancer |
| SCLC | Small-cell lung cancer |
| aSMase | Acid sphingomyelinase |
| Imip | Imipramine |
| PMA | Phorbol myristate acetate |
| CPAF | Carbamoyl-PAF |
| COX-2 | Cyclooxygenase type 2 |
| PAF-AH | PAF-acetyl hydrolase |
| Tregs | Regulatory T cells |
| EGFR | Epidermal growth factor receptor |
| Gef | Gefitinib |
| Erlo | Erlotinib |
| ERK PLC PI3K | Extracellular signal-regulated kinase Phospholipase C Phosphoinositide 3-kinase |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- McIntyre, A.; Ganti, A.K. Lung cancer-A global perspective. J. Surg. Oncol. 2017, 115, 550–554. [Google Scholar] [CrossRef]
- Mao, Y.; Yang, D.; He, J.; Krasna, M.J. Epidemiology of Lung Cancer. Surg. Oncol. Clin. N. Am. 2016, 25, 439–445. [Google Scholar] [CrossRef]
- Soza-Ried, C.; Bustamante, E.; Caglevic, C.; Rolfo, C.; Sirera, R.; Marsiglia, H. Oncogenic role of arsenic exposure in lung cancer: A forgotten risk factor. Crit. Rev. Oncol. Hematol. 2019, 139, 128–133. [Google Scholar] [CrossRef]
- Malhotra, J.; Malvezzi, M.; Negri, E.; La Vecchia, C.; Boffetta, P. Risk factors for lung cancer worldwide. Eur. Respir. J. 2016, 48, 889–902. [Google Scholar] [CrossRef] [Green Version]
- Bastian, L.A.; Gray, K.E.; DeRycke, E.; Mirza, S.; Gierisch, J.M.; Haskell, S.G.; Magruder, K.M.; Wakelee, H.A.; Wang, A.; Ho, G.Y.; et al. Differences in Active and Passive Smoking Exposures and Lung Cancer Incidence Between Veterans and Non-Veterans in the Women’s Health Initiative. Gerontologist 2016, 56 (Suppl. 1), S102–S111. [Google Scholar] [CrossRef] [Green Version]
- Corrales, L.; Rosell, R.; Cardona, A.F.; Martín, C.; Zatarain-Barrón, Z.L.; Arrieta, O. Lung cancer in never smokers: The role of different risk factors other than tobacco smoking. Crit. Rev. Oncol. Hematol. 2020, 148, 102895. [Google Scholar] [CrossRef]
- Akhtar, N.; Bansal, J.G. Risk factors of Lung Cancer in nonsmoker. Curr. Probl. Cancer 2017, 41, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Rivera, G.A.; Wakelee, H. Lung Cancer in Never Smokers. Adv. Exp. Med. Biol. 2016, 893, 43–57. [Google Scholar] [CrossRef]
- Nasim, F.; Sabath, B.F.; Eapen, G.A. Lung cancer. Med. Clin. N. Am. 2019, 103, 463–473. [Google Scholar] [CrossRef]
- Latimer, K.M.; Mott, T.F. Lung cancer: Diagnosis, treatment principles, and screening. Am. Fam. Physician 2015, 91, 250–256. [Google Scholar] [PubMed]
- Rodriguez-Canales, J.; Parra-Cuentas, E.; Wistuba, I.I. Diagnosis and Molecular Classification of Lung Cancer. Cancer Treat. Res. 2016, 170, 25–46. [Google Scholar] [CrossRef] [PubMed]
- Varella-Garcia, M. Chromosomal and genomic changes in lung cancer. Cell Adh. Migr. 2010, 4, 100–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current Challenges in Cancer Treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemjabbar-Alaoui, H.; Hassan, O.U.; Yang, Y.W.; Buchanan, P. Lung cancer: Biology and treatment options. Biochim. Biophys. Acta 2015, 1856, 189–210. [Google Scholar] [CrossRef] [Green Version]
- Pallis, A.G.; Gridelli, C.; Wedding, U.; Faivre-Finn, C.; Veronesi, G.; Jaklitsch, M.; Luciani, A.; O’Brien, M. Management of elderly patients with NSCLC; updated expert’s opinion paper: EORTC Elderly Task Force, Lung Cancer Group and International Society for Geriatric Oncology. Ann. Oncol. 2014, 25, 1270–1283. [Google Scholar] [CrossRef]
- Nagasaka, M.; Gadgeel, S.M. Role of chemotherapy and targeted therapy in early-stage non-small cell lung cancer. Expert Rev. Anticancer Ther. 2018, 18, 63–70. [Google Scholar] [CrossRef]
- Shroff, G.S.; de Groot, P.M.; Papadimitrakopoulou, V.A.; Truong, M.T.; Carter, B.W. Targeted Therapy and Immunotherapy in the Treatment of Non-Small Cell Lung Cancer. Radiol. Clin. N. Am. 2018, 56, 485–495. [Google Scholar] [CrossRef]
- Reck, M.; Heigener, D.; Reinmuth, N. Immunotherapy for small-cell lung cancer: Emerging evidence. Future Oncol. 2016, 12, 931–943. [Google Scholar] [CrossRef]
- Wang, S.; Song, Y.; Liu, D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017, 385, 51–54. [Google Scholar] [CrossRef]
- Rosell, R.; Dafni, U.; Felip, E.; Curioni-Fontecedro, A.; Gautschi, O.; Peters, S.; Massutí, B.; Palmero, R.; Aix, S.P.; Carcereny, E.; et al. Erlotinib and bevacizumab in patients with advanced non-small-cell lung cancer and activating EGFR mutations (BELIEF): An international, multicentre, single-arm, phase 2 trial. Lancet Respir. Med. 2017, 5, 435–444. [Google Scholar] [CrossRef]
- Chung, C. Tyrosine kinase inhibitors for epidermal growth factor receptor gene mutation-positive non-small cell lung cancers: An update for recent advances in therapeutics. J. Oncol. Pharm. Pract. 2016, 22, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Teppo, H.R.; Soini, Y.; Karihtala, P. Reactive Oxygen Species-Mediated Mechanisms of Action of Targeted Cancer Therapy. Oxid. Med. Cell Longev. 2017, 2017, 1485283. [Google Scholar] [CrossRef] [PubMed]
- Okon, I.S.; Coughlan, K.A.; Zhang, M.; Wang, Q.; Zou, M.-H. Gefitinib-mediated reactive oxygen specie (ROS) instigates mitochondrial dysfunction and drug resistance in lung cancer cells. J. Biol. Chem. 2015, 290, 9101–9110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, F.; Shao, Z.; Jiang, S.; Cheng, Z. Erlotinib induces the human non–small-cell lung cancer cells apoptosis via activating ROS-dependent JNK pathways. Cancer Med. 2016, 5, 3166–3175. [Google Scholar] [CrossRef]
- Marcar, L.; Bardhan, K.; Gheorghiu, L.; Dinkelborg, P.; Pfäffle, H.; Liu, Q.; Wang, M.; Piotrowska, Z.; Sequist, L.V.; Borgmann, K.; et al. Acquired Resistance of EGFR-Mutated Lung Cancer to Tyrosine Kinase Inhibitor Treatment Promotes PARP Inhibitor Sensitivity. Cell Rep. 2019, 27, 3422–3432. [Google Scholar] [CrossRef] [Green Version]
- Krall, E.B.; Wang, B.; Munoz, D.M.; Ilic, N.; Raghavan, S.; Niederst, M.J.; Yu, K.; Ruddy, D.A.; Aguirre, A.J.; Kim, J.W.; et al. KEAP1 loss modulates sensitivity to kinase targeted therapy in lung cancer. eLife 2017, 6, e18970. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Schmid-Bindert, G.; Wang, D.; Zhao, Y.; Yang, X.; Su, B.; Zhou, C. Blocking the PI3K/AKT and MEK/ERK signaling pathways can overcome gefitinib-resistance in non-small cell lung cancer cell lines. Adv. Med. Sci. 2011, 56, 275–284. [Google Scholar] [CrossRef]
- da Silva, A., Jr.; Chammas, R.; Lepique, A.P.; Jancar, S. Platelet-activating factor (PAF) receptor as a promising target for cancer cell repopulation after radiotherapy. Oncogenesis 2017, 6, e296. [Google Scholar] [CrossRef] [Green Version]
- Sahu, R.P.; Ocana, J.A.; Harrison, K.A.; Ferracini, M.; Touloukian, C.E.; Al-Hassani, M.; Sun, L.; Loesch, M.; Murphy, R.C.; Althouse, S.K.; et al. Chemotherapeutic agents subvert tumor immunity by generating agonists of platelet-activating factor. Cancer Res. 2014, 74, 7069–7078. [Google Scholar] [CrossRef] [Green Version]
- Sahu, R.P.; Harrison, K.A.; Weyerbacher, J.; Murphy, R.C.; Konger, R.L.; Garrett, J.E.; Chin-Sinex, H.J.; Johnston, M.E., 2nd; Dynlacht, J.R.; Mendonca, M.; et al. Radiation therapy generates platelet-activating factor agonists. Oncotarget 2016, 7, 20788–20800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, R.P.; Petrache, I.; Van Demark, M.J.; Rashid, B.M.; Ocana, J.A.; Tang, Y.; Yi, Q.; Turner, M.J.; Konger, R.L.; Travers, J.B. Cigarette smoke exposure inhibits contact hypersensitivity via the generation of platelet-activating factor agonists. J. Immunol. 2013, 190, 2447–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackler, P.C.; Reuss, S.; Konger, R.L.; Travers, J.B.; Sahu, R.P. Systemic Platelet-activating Factor Receptor Activation Augments Experimental Lung Tumor Growth and Metastasis. Cancer Growth Metastasis 2014, 7, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thyagarajan, A.; Kadam, S.M.; Liu, L.; Kelly, L.E.; Rapp, C.M.; Chen, Y.; Sahu, R.P. Gemcitabine Induces Microvesicle Particle Release in a Platelet-Activating Factor-Receptor-Dependent Manner via Modulation of the MAPK Pathway in Pancreatic Cancer Cells. Int. J. Mol. Sci. 2018, 20, 32. [Google Scholar] [CrossRef] [Green Version]
- Fahy, K.; Liu, L.; Rapp, C.M.; Borchers, C.; Bihl, J.C.; Chen, Y.; Simman, R.; Travers, J.B. UVB-generated Microvesicle Particles: A Novel Pathway by Which a Skin-specific Stimulus Could Exert Systemic Effects. Photochem. Photobiol. 2017, 93, 937–942. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Fahy, K.E.; Awoyemi, A.A.; Thapa, P.; Kelly, L.E.; Chen, J.; Bihl, J.C.; Cool, D.R.; Chen, Y.; Rapp, C.M.; et al. Thermal Burn Injury Generates Bioactive Microvesicles: Evidence for a Novel Transport Mechanism for the Lipid Mediator Platelet-Activating Factor (PAF) That Involves Subcellular Particles and the PAF Receptor. J. Immunol. 2020, 205, 193–201. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, M.; Zhang, X.; Cai, Q.; Zhu, Z.; Jiang, W.; Xu, C. Transactivation of epidermal growth factor receptor through platelet-activating factor/receptor in ovarian cancer cells. J. Exp. Clin. Cancer Res. 2014, 33, 85. [Google Scholar] [CrossRef]
- Chen, J.; Lan, T.; Zhang, W.; Dong, L.; Kang, N.; Zhang, S.; Fu, M.; Liu, B.; Liu, K.; Zhan, Q. Feed-Forward Reciprocal Activation of PAFR and STAT3 Regulates Epithelial-Mesenchymal Transition in Non-Small Cell Lung Cancer. Cancer Res. 2015, 75, 4198–4210. [Google Scholar] [CrossRef] [Green Version]
- Marques, S.A.; Dy, L.C.; Southall, M.D.; Yi, Q.; Smietana, E.; Kapur, R.; Marques, M.; Travers, J.B.; Spandau, D.F. The Platelet-Activating Factor Receptor Activates the Extracellular Signal-Regulated Kinase Mitogen-Activated Protein Kinase and Induces Proliferation of Epidermal Cells through an Epidermal Growth Factor-Receptor-Dependent Pathway. J. Pharmacol. Exp. Ther. 2002, 300, 1026–1035. [Google Scholar] [CrossRef] [Green Version]
- Steen, N.V.D.; Potze, L.; Giovannetti, E.; Cavazzoni, A.; Ruijtenbeek, R.; Rolfo, C.; Pauwels, P.; Peters, G.J. Molecular mechanism underlying the pharmacological interactions of the protein kinase C-β inhibitor enzastaurin and erlotinib in non-small cell lung cancer cells. Am. J. Cancer Res. 2017, 7, 816–830. [Google Scholar]
- Howe, G.A.; Xiao, B.; Zhao, H.; Al-Zahrani, K.N.; Hasin, M.S.; Villeneuve, J.; Sekhon, H.S.; Goss, G.D.; Sabourin, L.A.; Dimitroulakos, J.; et al. Focal Adhesion Kinase Inhibitors in Combination with Erlotinib Demonstrate Enhanced Anti-Tumor Activity in Non-Small Cell Lung Cancer. PLoS ONE 2016, 11, e0150567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Qin, Y.; Zhang, H.; Gao, M.Y.; Wang, Y.F. EGF upregulates RFPL3 and hTERT via the MEK signaling pathway in non-small cell lung cancer cells. Oncol. Rep. 2018, 40, 29–38. [Google Scholar] [CrossRef]
- Choi, J.; Kang, M.; Nam, S.H.; Lee, G.H.; Kim, H.J.; Ryu, J.; Cheong, J.G.; Jung, J.W.; Kim, T.Y.; Lee, H.Y.; et al. Bidirectional signaling between TM4SF5 and IGF1R promotes resistance to EGFR kinase inhibitors. Lung Cancer 2015, 90, 22–31. [Google Scholar] [CrossRef]
- Neri, T.; Pergoli, L.; Petrini, S.; Gravendonk, L.; Balia, C.; Scalise, V.; Amoruso, A.; Pedrinelli, R.; Paggiaro, P.; Bollati, V.; et al. Particulate matter induces prothrombotic microparticle shedding by human mononuclear and endothelial cells. Toxicol. In Vitro 2016, 32, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Tricarico, C.; Clancy, J.; D’Souza-Schorey, C. Biology and biogenesis of shed microvesicles. Small GTPases 2017, 8, 220–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albouz, S.; Hauw, J.J.; Berwald-Netter, Y.; Boutry, J.M.; Bourdon, R.; Baumann, N. Tricyclic antidepressants induce sphingomyelinase deficiency in fibroblast and neuroblastoma cell cultures. Biomedicine 1981, 35, 218–220. [Google Scholar]
- Catalano, M.; O’Driscoll, L. Inhibiting extracellular vesicles formation and release: A review of EV inhibitors. J. Extracell. Vesicles 2019, 9, 1703244. [Google Scholar] [CrossRef] [Green Version]
- Landis, M.; Yi, Q.; Hyatt, A.M.; Travers, A.R.; Lewis, D.A.; Travers, J.B. Involvement of P38 MAP kinase in the augmentation of UVB-mediated apoptosis via the epidermal platelet-activating factor receptor. Arch. Dermatol. Res. 2007, 299, 263–266. [Google Scholar] [CrossRef]
- Chao, W.; Deng, J.S.; Li, P.Y.; Kuo, Y.H.; Huang, G.J. Inotilone from Inonotus linteus suppresses lung cancer metastasis in vitro and in vivo through ROS-mediated PI3K/AKT/MAPK signaling pathways. Sci. Rep. 2019, 9, 2344. [Google Scholar] [CrossRef]
- Tang, H.; Xue, G. Major Physiological Signaling Pathways in the Regulation of Cell Proliferation and Survival. Handb. Exp. Pharmacol. 2018, 249, 13–30. [Google Scholar] [CrossRef]
- Braquet, P.; Touqui, L.; Shen, T.Y.; Vargaftig, B.B. Perspectives in platelet-activating factor research. Pharmacol. Rev. 1987, 39, 97–145. [Google Scholar]
- Walterscheid, J.P.; Ullrich, S.E.; Nghiem, D.X. Platelet-activating factor, a molecular sensor for cellular damage, activates systemic immune suppression. J. Exp. Med. 2002, 195, 171–179. [Google Scholar] [CrossRef]
- Konger, R.L.; Marathe, G.K.; Yao, Y.; Zhang, Q.; Travers, J.B. Oxidized glycerophosphocholines as biologically active mediators for ultraviolet radiation-mediated effects. Prostaglandins Other Lipid Mediat. 2008, 87, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Sahu, R.P. Expression of the platelet-activating factor receptor enhances benzyl isothiocyanate-induced apoptosis in murine and human melanoma cells. Mol. Med. Rep. 2015, 12, 394–400. [Google Scholar] [CrossRef] [Green Version]
- Onuchic, A.C.; Machado, C.M.L.; Saito, R.F.; Rios, F.J.; Jancar, S.; Chammas, R. Expression of PAFR as part of a prosurvival response to chemotherapy: A novel target for combination therapy in melanoma. Mediat. Inflamm. 2012, 2012, 175408. [Google Scholar] [CrossRef] [Green Version]
- Sahu, R.P.; Turner, M.J.; DaSilva, S.C.; Rashid, B.M.; Ocana, J.A.; Perkins, S.M.; Konger, R.L.; Touloukian, C.E.; Kaplan, M.H.; Travers, J.B. The environmental stressor ultraviolet B radiation inhibits murine antitumor immunity through its ability to generate platelet-activating factor agonists. Carcinogenesis 2012, 33, 1360–1367. [Google Scholar] [CrossRef] [Green Version]
- Perry, S.W.; Norman, J.P.; Litzburg, A.; Zhang, D.; Dewhurst, S.; Gelbard, H.A. HIV-1 transactivator of transcription protein induces mitochondrial hyperpolarization and synaptic stress leading to apoptosis. J. Immunol. 2005, 74, 4333–4344. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.B.; Yu, M.R.; Song, J.S.; Ha, H. Reactive oxygen species amplify protein kinase C signaling in high glucose-induced fibronectin expression by human peritoneal mesothelial cells. Kidney Int. 2004, 65, 1170–1179. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.; Im, M.; Yim, N.-H.; Jung, Y.P.; Ma, J.Y. Aqueous extract of Bambusae Caulis in Taeniam inhibits PMA-induced tumor cell invasion and pulmonary metastasis: Suppression of NF-κB activation through ROS signaling. PLoS ONE 2013, 8, e78061. [Google Scholar] [CrossRef]
- Ponath, V.; Kaina, B. Death of Monocytes through Oxidative Burst of Macrophages and Neutrophils: Killing in Trans. PLoS ONE 2017, 12, e0170347. [Google Scholar] [CrossRef]
- Bodega, G.; Alique, M.; Puebla, L.; Carracedo, J.; Ramírez, R.M. Microvesicles: ROS scavengers and ROS producers. J. Extracell. Vesicles 2019, 8, 1626654. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-Q.; Liu, J.-T.; Fan, L.-L.; Liu, Y.; Cheng, L.; Wang, F.; Yu, H.-Q.; Gao, J.; Wei, W.; Wang, H.; et al. Exosomes derived from gefitinib-treated EGFR-mutant lung cancer cells alter cisplatin sensitivity via up-regulating autophagy. Oncotarget 2016, 7, 24585–24595. [Google Scholar] [CrossRef] [Green Version]
- Kosgodage, U.S.; Trindade, R.P.; Thompson, P.R.; Inal, J.M.; Lange, S. Chloramidine/Bisindolylmaleimide-I-Mediated Inhibition of Exosome and Microvesicle Release and Enhanced Efficacy of Cancer Chemotherapy. Int. J. Mol. Sci. 2017, 18, 1007. [Google Scholar] [CrossRef]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef] [Green Version]
- Willms, E.; Cabañas, C.; Mäger, I.; Wood, M.; Vader, P. Extracellular Vesicle Heterogeneity: Subpopulations, Isolation Techniques, and Diverse Functions in Cancer Progression. Front. Immunol. 2018, 9, 738. [Google Scholar] [CrossRef] [Green Version]
- D’Souza-Schorey, C.; Clancy, J.W. Tumor-derived microvesicles: Shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev. 2012, 26, 1287–1299. [Google Scholar] [CrossRef] [Green Version]
- Barreiro, K.; Holthofer, H. Urinary extracellular vesicles. A promising shortcut to novel biomarker discoveries. Cell Tissue Res. 2017, 369, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Moore, C.; Kosgodage, U.; Lange, S.; Inal, S.M. The emerging role of exosome and microvesicle- (EMV-) based cancer therapeutics and immunotherapy. Int. J. Cancer 2017, 141, 428–436. [Google Scholar] [CrossRef]
- Balachandran, B.; Yuana, Y. Extracellular vesicles-based drug delivery system for cancer treatment. Cogent Med. 2019, 6, 1635806. [Google Scholar] [CrossRef]
- Mentkowski, K.I.; Snitzer, J.D.; Rusnak, S.; Lang, J.K. Therapeutic Potential of Engineered Extracellular Vesicles. AAPS J. 2018, 20, 50. [Google Scholar] [CrossRef] [Green Version]
- Sheu, J.; Lee, F.; Wallace, C.G.; Tsai, T.H.; Leu, S.; Chen, Y.L.; Chai, H.T.; Lu, H.I.; Sun, C.K.; Yip, H.K. Administered circulating microparticles derived from lung cancer patients markedly improved angiogenesis, blood flow and ischemic recovery in rat critical limb ischemia. J. Transl. Med. 2015, 13, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Schubert, P.; Bakkour, S.; Culibrk, B.; Busch, M.P.; Devine, D.V. p38 mitogen-activated protein kinase regulates mitochondrial function and microvesicle release in riboflavin- and ultraviolet light–treated apheresis platelet concentrates. Transfusion 2017, 57, 1199–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thyagarajan, A.; Saylae, J.; Sahu, R.P. Acetylsalicylic acid inhibits the growth of melanoma tumors via SOX2-dependent-PAF-R-independent signaling pathway. Oncotarget 2017, 8, 49959–49972. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chauhan, S.J.; Thyagarajan, A.; Chen, Y.; Travers, J.B.; Sahu, R.P. Platelet-Activating Factor-Receptor Signaling Mediates Targeted Therapies-Induced Microvesicle Particles Release in Lung Cancer Cells. Int. J. Mol. Sci. 2020, 21, 8517. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228517
Chauhan SJ, Thyagarajan A, Chen Y, Travers JB, Sahu RP. Platelet-Activating Factor-Receptor Signaling Mediates Targeted Therapies-Induced Microvesicle Particles Release in Lung Cancer Cells. International Journal of Molecular Sciences. 2020; 21(22):8517. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228517
Chicago/Turabian StyleChauhan, Shreepa J., Anita Thyagarajan, Yanfang Chen, Jeffrey B. Travers, and Ravi P. Sahu. 2020. "Platelet-Activating Factor-Receptor Signaling Mediates Targeted Therapies-Induced Microvesicle Particles Release in Lung Cancer Cells" International Journal of Molecular Sciences 21, no. 22: 8517. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228517