Tyrosine Kinase Receptors in Oncology

 , , , and
, , , and
Abstract
:1. Introduction
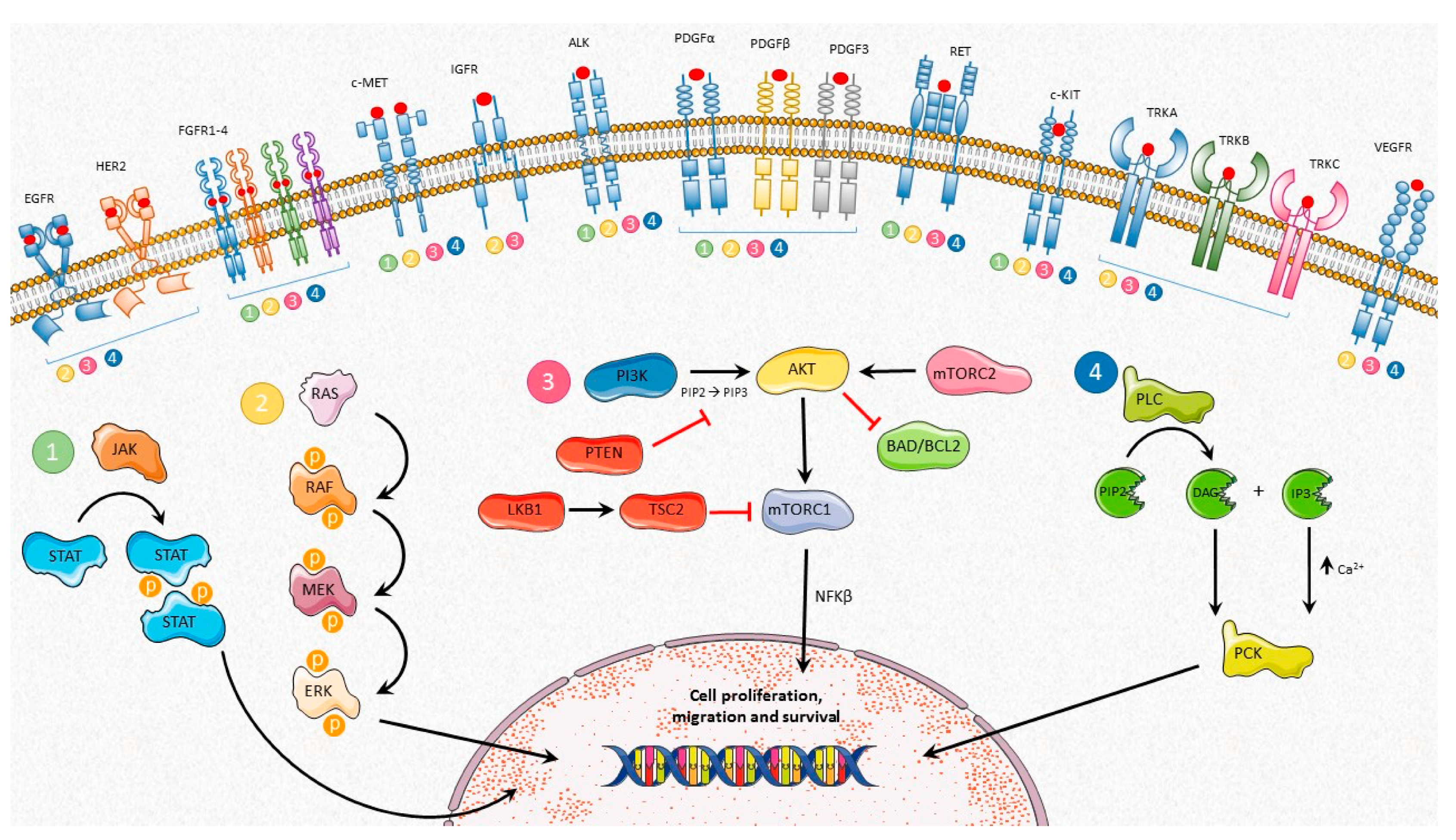
2. Roles of TKRs in Physiologic Cell Biology
3. Roles of TKRs in Tumor Biology
4. Tyrosine Kinase Receptor Inhibitors Developed for Cancer Treatment
4.1. Epidermal Growth Factor Receptor (EGFR)
4.2. HER2
4.3. Anaplastic Lymphoma Kinase (ALK)
4.4. ROS1
4.5. Neurotropic Tropomyosine Receptor Kinase (NTRK)
4.6. Fibroblast Growth Factor Receptor (FGFR)
4.7. c-KIT
4.8. Platelet-Derived Growth Factor Receptor (PDGFR)
4.9. c-MET
4.10. RET
4.11. Vascular Endothelial Growth Factor Receptor (VEGFR)
5. Discussion
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| TLA | Three letter acronym |
| LD | linear dichroism |
Appendix A

{kind=link}
| TKR Mutations | Most Representative Tumors | Other Described Alterations in Tumors |
|---|---|---|
| EGFR (primary mut.) Exon 19 (del), exon 21 (L858R, L861Q), exon 18 (G719X), exon 20 (ins) EGFR (secondary mut.) Exon 20 (T790M), D761Y, T854A, L747S | NSCLC (15–20%) NSCLC | Colorectal carcinoma (EGFR S464L, G465R, I491M, EGFR S492R) Pancreatic carcinoma Head and neck cancer (EGFR amplifications) Glioma (EGFR A289V, R108K, G598V, T263P) |
| HER2 mut. Exon 20 (ins) | NSCLC (1–3%) | Breast cancer (HER2 amplification negative; HER2 D769Y, D769H, R896C, V777L, G309E, V842I, S310F, S310Y, C311R; HER2 inframe deletion (755-759), inframe insertion (780GSP), inframe insertion (781GSP) |
| ALK rearrangement EML4-ALK, KIF5B-ALK, KLC1-ALK, HIP1-ALK ALK (secondary mut.) Exon 23 (L1196M, S1206Y), exon 25 (G1269A), exon 22 (I1171T), V1180L, F1174L | NSCLC (5–6%) NSCLC (5–6%) | Anaplastic large cell lymphoma (other not ALK L1196M; ALK F856S, A348D) Glioblastoma (ALK fusions) Inflammatory myofibroblastic tumors (ALK C1156Y) Diffuse large B-cell lymphoma (ALK G1269A) Esophageal squamous cell carcinoma (ALK L1152R) Colorectal carcinoma (ALK F1245C) |
| FGFR1-4 | NSCLC (Squamous) (FGFR1 amplif., 17%) SCLC (FGFR1 amplif., 6%) Breast cancer (FGFR1 amplif., 15%) Cholangiocarcinoma (FGFR2 fusions) Urothelial carcinoma (FGFR2-3 fusions) Gastric cancer (FGFR2 amplif., 5–10%) | Cholangiocarcinoma (other not FGFR2 fusion; FGFR2 N549H, V564F, K659M, L617V, K641R, R565A) Bladder cancer (oncogenic mutations others not FGFR3 fusions) Endometrial carcinoma (FGFR2 M536I, M538I, I548V, N550, E566G, L618M, K660E, S252W, N550K, V565I) NSCLC (Squamous) (other not FGFR1 amplification; FGFR2 W290C, S320C, K660; FGFR3 S249C, G691R) Myeloma multiple (FGFR3 K650, Y373C, V555M) |
| c-KIT mut. Exon 11, exon 9, exon 17, exon 13 | GIST (80–85%) | AML (KIT D816V, N822K) Systemic mastocytosis (KIT D816V) Thymic carcinoma (KIT H697Y) Cutaneous melanoma (KIT amplif.) |
| PDGFR mut. Exon 18, exon 12, exon 14 | GIST (6–8%) | Cutaneous melanoma (PDGFRA V658A, P577S, R841K, H845Y, G853D) Renal carcinoma (PDGFRA overexpression) Myelodisplasic syndrome (PDGFRA fusion) Dermatofibrosarcoma protuberans (translocation t(17:22)) |
| MET skipping mut. Exon 14 | NSCLC (3%) | Renal carcinoma (MET H1112R) NSCLC (MET amplif.) Colorectal, gastric carcinoma (MET amplif.) |
| ROS1 | NSCLC, cholangiocarcinoma, gastric carcinoma (ROS1 fusions, 1–2%) | NSCLC (ROS1 G2032R) Inflammatory myofibroblastic tumor |
| RET | Papillary thyroid carcinoma (RET rearrangement, 5–40%) | MEN-2 syndrome, sporadic medullary thyroid carcinoma (RET mutations) |
Appendix B
| VEGFR1 | VEGFR2 | VEGFR3 | PDFGRa | PDGFRb | c-KIT | RET | Flt-3 | MET | CSF-1R | FGFR1 | FGFR2 | FGFR2 | FGFR4 | Axl | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sunitinib | 2 | 9 | 17 | 7 | 8 | 7 | 10 | 250 | - | 890 | 830 | - | - | - | 9 |
| Pazopanib | 10 | 30 | 47 | 71 | 81 | 74 | - | - | - | - | 140 | - | 130 | 80 | - |
| Tivozanib | 30 | 6 | 15 | 40 | 49 | 78 | - | 2550 | 550 | - | 525 | - | 1250 | 1400 | - |
| Axitinib | 0.1 | 0.2 | 0.1–0.3 | 5 | 1.6 | 1.7 | >1000 | >1000 | - | 73 | 100 | - | - | - | - |
| Cabozantinib | - | 0.035 | - | - | 234 | 4.6 | 5.2 | 11.3 | 1.3 | - | 5294 | - | - | - | 7 |
| Lenvatinib | 22 | 4 | 5.2 | 25 | 39 | 0.7 | 12 | - | 1900 | - | 46 | - | - | 43 | - |
| Sorafenib | 26 | 90 | 20 | - | 57 | 68 | - | 33 | - | - | 580 | - | - | - | - |
| Regorafenib | 13 | 4.2 | 46 | - | 22 | 7 | 1.5 | - | - | - | 202 | - | - | - | - |
| Nintedanib | 34 | 21 | 13 | 59 | 65 | - | - | 26 | - | - | 69 | 37 | 100 | 610 | - |
| Vandetanib | - | 40 | 110 | - | - | - | 100 | - | - | - | - | - | - | - | - |
References
- Alberts, B. Molecular Biology of the Cell, 6th ed.; Garland Science; Taylor and Francis Group: New York, NY, USA, 2015; ISBN 978-0-8153-4432-2. [Google Scholar]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Rude Voldborg, B.; Damstrup, L.; Spang-Thomsen, M.; Skovgaard Poulsen, H. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann. Oncol. 1997, 8, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Moyes, D.L.; Tavassoli, M.; Naglik, J.R. The Role of ErbB Receptors in Infection. Trends Microbiol. 2017, 25, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef] [PubMed]
- Amatu, A.; Sartore-Bianchi, A.; Bencardino, K.; Pizzutilo, E.G.; Tosi, F.; Siena, S. Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann. Oncol. 2019, 30, viii5–viii15. [Google Scholar] [CrossRef] [Green Version]
- Farooqi, A.A.; Siddik, Z.H. Platelet-derived growth factor (PDGF) signalling in cancer: Rapidly emerging signalling landscape: PDGF-induced Signalling Cascades. Cell Biochem. Function 2015, 33, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, P.; Budnick, I.; Singh, M.; Thiruppathi, M.; Alharshawi, K.; Elshabrawy, H.; Holterman, M.J.; Prabhakar, B.S. Dual Role of GM-CSF as a Pro-Inflammatory and a Regulatory Cytokine: Implications for Immune Therapy. J. Interferon Cytokine Res. 2015, 35, 585–599. [Google Scholar] [CrossRef] [Green Version]
- Bockorny, B.; Rusan, M.; Chen, W.; Liao, R.G.; Li, Y.; Piccioni, F.; Wang, J.; Tan, L.; Thorner, A.R.; Li, T.; et al. RAS–MAPK Reactivation Facilitates Acquired Resistance in FGFR1 -Amplified Lung Cancer and Underlies a Rationale for Upfront FGFR–MEK Blockade. Mol. Cancer Ther. 2018, 17, 1526–1539. [Google Scholar] [CrossRef] [Green Version]
- Chae, Y.K.; Ranganath, K.; Hammerman, P.S.; Vaklavas, C.; Mohindra, N.; Kalyan, A.; Matsangou, M.; Costa, R.; Carneiro, B.; Villaflor, V.M.; et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: The current landscape and barriers to clinical application. Oncotarget 2017, 8, 16052–16074. [Google Scholar] [CrossRef] [Green Version]
- Kut, C.; Mac Gabhann, F.; Popel, A.S. Where is VEGF in the body? A meta-analysis of VEGF distribution in cancer. Br. J. Cancer 2007, 97, 978–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallberg, B.; Palmer, R.H. The role of the ALK receptor in cancer biology. Ann. Oncol. 2016, 27, iii4–iii15. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, D. GFRA1: A Novel Molecular Target for the Prevention of Osteosarcoma Chemoresistance. Int. J. Mol. Sci. 2018, 19, 1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miettinen, M.; Lasota, J. KIT (CD117): A Review on Expression in Normal and Neoplastic Tissues, and Mutations and Their Clinicopathologic Correlation. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Barquilla, A.; Pasquale, E.B. Eph Receptors and Ephrins: Therapeutic Opportunities. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 465–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kou, C.-T.J.; Kandpal, R.P. Differential Expression Patterns of Eph Receptors and Ephrin Ligands in Human Cancers. BioMed Res. Int. 2018, 2018, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolino, M.; Penninger, J. The Role of TAM Family Receptors in Immune Cell Function: Implications for Cancer Therapy. Cancers 2016, 8, 97. [Google Scholar] [CrossRef] [Green Version]
- Mitin, N.; Rossman, K.L.; Der, C.J. Signaling Interplay in Ras Superfamily Function. Curr. Biol. 2005, 15, R563–R574. [Google Scholar] [CrossRef] [Green Version]
- Krens, S.F.G.; Spaink, H.P.; Snaar-Jagalska, B.E. Functions of the MAPK family in vertebrate-development. FEBS Lett. 2006, 580, 4984–4990. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Ebisuya, M.; Ashida, F.; Okamoto, K.; Yonehara, S.; Nishida, E. Continuous ERK Activation Downregulates Antiproliferative Genes throughout G1 Phase to Allow Cell-Cycle Progression. Curr. Biol. 2006, 16, 1171–1182. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [Green Version]
- Morrison, D.K. MAP Kinase Pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef]
- Haga, R.B.; Ridley, A.J. Rho GTPases: Regulation and roles in cancer cell biology. Small GTPases 2016, 7, 207–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaffe, A.B.; Hall, A. RHO GTPASES: Biochemistry and Biology. Annu. Rev. Cell Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firat-Karalar, E.N.; Welch, M.D. New mechanisms and functions of actin nucleation. Curr. Opin. Cell Biol. 2011, 23, 4–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ananthakrishnan, R.; Ehrlicher, A. The Forces Behind Cell Movement. Int. J. Biol. Sci. 2007, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Pitulescu, M.E.; Adams, R.H. Eph/ephrin molecules--A hub for signaling and endocytosis. Genes Dev. 2010, 24, 2480–2492. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; He, X.; Hsu, J.-M.; Xia, W.; Chen, C.-T.; Li, L.-Y.; Lee, D.-F.; Liu, J.-C.; Zhong, Q.; Wang, X.; et al. Degradation of Mcl-1 by β-TrCP Mediates Glycogen Synthase Kinase 3-Induced Tumor Suppression and Chemosensitization. Mol. Cell. Biol. 2007, 27, 4006–4017. [Google Scholar] [CrossRef] [Green Version]
- Sears, R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Kaidanovich-Beilin, O.; Woodgett, J.R. GSK-3: Functional Insights from Cell Biology and Animal Models. Front. Mol. Neurosci. 2011, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, A.E.; Brunet, A. FOXO transcription factors: Key regulators of cellular quality control. Trends Biochem. Sci. 2014, 39, 159–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Link, W.; Fernandez-Marcos, P.J. FOXO transcription factors at the interface of metabolism and cancer: FOXO transcription factors. Int. J. Cancer 2017, 141, 2379–2391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. PtdIns(3,4,5) P 3 -Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef] [Green Version]
- Kim, L.C.; Cook, R.S.; Chen, J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2017, 36, 2191–2201. [Google Scholar] [CrossRef] [Green Version]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Puertollano, R. mTOR and lysosome regulation. F1000Prime Rep. 2014, 6, P6–P52. [Google Scholar] [CrossRef] [Green Version]
- Ballif, B.A.; Roux, P.P.; Gerber, S.A.; MacKeigan, J.P.; Blenis, J.; Gygi, S.P. Quantitative phosphorylation profiling of the ERK/p90 ribosomal S6 kinase-signaling cassette and its targets, the tuberous sclerosis tumor suppressors. Proc. Natl. Acad. Sci. USA 2005, 102, 667–672. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, B.L.J.; Hirst, S.J.; Giembycz, M.A. Protein kinase C isoenzymes: A review of their structure, regulation and role in regulating airways smooth muscle tone and mitogenesis: Protein kinase C. Br. J. Pharmacol. 2000, 130, 1433–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Caldieri, G.; Barbieri, E.; Nappo, G.; Raimondi, A.; Bonora, M.; Conte, A.; Verhoef, L.G.G.C.; Confalonieri, S.; Malabarba, M.G.; Bianchi, F.; et al. Reticulon 3–dependent ER-PM contact sites control EGFR nonclathrin endocytosis. Science 2017, 356, 617–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibach, J.; Radon, Y.; Gelléri, M.; Sonntag, M.H.; Brunsveld, L.; Bastiaens, P.I.H.; Verveer, P.J. Single Particle Tracking Reveals that EGFR Signaling Activity Is Amplified in Clathrin-Coated Pits. PLoS ONE 2015, 10, e0143162. [Google Scholar] [CrossRef] [Green Version]
- Sigismund, S.; Confalonieri, S.; Ciliberto, A.; Polo, S.; Scita, G.; Di Fiore, P.P. Endocytosis and Signaling: Cell Logistics Shape the Eukaryotic Cell Plan. Physiol. Rev. 2012, 92, 273–366. [Google Scholar] [CrossRef] [Green Version]
- Raiborg, C.; Stenmark, H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 2009, 458, 445–452. [Google Scholar] [CrossRef]
- Scharaw, S.; Iskar, M.; Ori, A.; Boncompain, G.; Laketa, V.; Poser, I.; Lundberg, E.; Perez, F.; Beck, M.; Bork, P.; et al. The endosomal transcriptional regulator RNF11 integrates degradation and transport of EGFR. J. Cell Biol. 2016, 215, 543–558. [Google Scholar] [CrossRef] [Green Version]
- Dougherty, M.K.; Müller, J.; Ritt, D.A.; Zhou, M.; Zhou, X.Z.; Copeland, T.D.; Conrads, T.P.; Veenstra, T.D.; Lu, K.P.; Morrison, D.K. Regulation of Raf-1 by Direct Feedback Phosphorylation. Mol. Cell 2005, 17, 215–224. [Google Scholar] [CrossRef]
- Chen, D.; Waters, S.B.; Holt, K.H.; Pessin, J.E. SOS Phosphorylation and Disassociation of the Grb2-SOS Complex by the ERK and JNK Signaling Pathways. J. Biol. Chem. 1996, 271, 6328–6332. [Google Scholar] [CrossRef] [Green Version]
- Keyse, S.M. Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr. Opin. Cell Biol. 2000, 12, 186–192. [Google Scholar] [CrossRef]
- Mason, J.M.; Morrison, D.J.; Albert Basson, M.; Licht, J.D. Sprouty proteins: Multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol. 2006, 16, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Mialki, R.K.; Dong, S.; Khoo, A.; Mallampalli, R.K.; Zhao, Y.; Zhao, J. A new mechanism of RhoA ubiquitination and degradation: Roles of SCF FBXL19 E3 ligase and Erk2. Biochim. Biophys. Acta BBA Mol. Cell Res. 2013, 1833, 2757–2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Haddadi, N.; Lin, Y.; Travis, G.; Simpson, A.M.; Nassif, N.T.; McGowan, E.M. PTEN/PTENP1: ‘Regulating the regulator of RTK-dependent PI3K/Akt signalling’, new targets for cancer therapy. Mol. Cancer 2018, 17, 37. [Google Scholar] [CrossRef] [PubMed]
- Planchon, S.M.; Waite, K.A.; Eng, C. The nuclear affairs of PTEN. J. Cell Sci. 2008, 121, 249–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Khouri, A.M.; Ma, Y.; Togo, S.H.; Williams, S.; Mustelin, T. Cooperative Phosphorylation of the Tumor Suppressor Phosphatase and Tensin Homologue (PTEN) by Casein Kinases and Glycogen Synthase Kinase 3β. J. Biol. Chem. 2005, 280, 35195–35202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holz, M.K.; Blenis, J. Identification of S6 Kinase 1 as a Novel Mammalian Target of Rapamycin (mTOR)-phosphorylating Kinase. J. Biol. Chem. 2005, 280, 26089–26093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Gan, W.; Inuzuka, H.; Lazorchak, A.S.; Gao, D.; Arojo, O.; Liu, D.; Wan, L.; Zhai, B.; Yu, Y.; et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat. Cell Biol. 2013, 15, 1340–1350. [Google Scholar] [CrossRef]
- Rommel, C.; Bodine, S.C.; Clarke, B.A.; Rossman, R.; Nunez, L.; Stitt, T.N.; Yancopoulos, G.D.; Glass, D.J. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat. Cell Biol. 2001, 3, 1009–1013. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, N.; Sonenberg, N. Signalling to eIF4E in cancer. Biochem. Soc. Trans. 2015, 43, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowell, P. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H.J. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.; Akita, R.; Vandlen, R.; Toomre, D.; Schlessinger, J.; Mellman, I. Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 2010, 464, 783–787. [Google Scholar] [CrossRef]
- Schneider, M.R.; Yarden, Y. The EGFR-HER2 module: A stem cell approach to understanding a prime target and driver of solid tumors. Oncogene 2016, 35, 2949–2960. [Google Scholar] [CrossRef] [Green Version]
- Grandal, M.V.; Zandi, R.; Pedersen, M.W.; Willumsen, B.M.; van Deurs, B.; Poulsen, H.S. EGFRvIII escapes down-regulation due to impaired internalization and sorting to lysosomes. Carcinogenesis 2007, 28, 1408–1417. [Google Scholar] [CrossRef] [Green Version]
- Kon, S.; Kobayashi, N.; Satake, M. Altered trafficking of mutated growth factor receptors and their associated molecules: Implication for human cancers. Cell. Logist. 2014, 4, e28461. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Lanzetti, L.; Di Fiore, P.P. Behind the Scenes: Endo/Exocytosis in the Acquisition of Metastatic Traits. Cancer Res. 2017, 77, 1813–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Chetrit, N.; Chetrit, D.; Russell, R.; Körner, C.; Mancini, M.; Abdul-Hai, A.; Itkin, T.; Carvalho, S.; Cohen-Dvashi, H.; Koestler, W.J.; et al. Synaptojanin 2 is a druggable mediator of metastasis and the gene is overexpressed and amplified in breast cancer. Sci. Signal. 2015, 8, ra7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducray, S.P.; Natarajan, K.; Garland, G.D.; Turner, S.D.; Egger, G. The Transcriptional Roles of ALK Fusion Proteins in Tumorigenesis. Cancers 2019, 11, 1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomas, A.; Vaughan, S.O.; Burgoyne, T.; Sorkin, A.; Hartley, J.A.; Hochhauser, D.; Futter, C.E. WASH and Tsg101/ALIX-dependent diversion of stress-internalized EGFR from the canonical endocytic pathway. Nat. Commun. 2015, 6, 7324. [Google Scholar] [CrossRef] [Green Version]
- Franovic, A.; Gunaratnam, L.; Smith, K.; Robert, I.; Patten, D.; Lee, S. Translational up-regulation of the EGFR by tumor hypoxia provides a nonmutational explanation for its overexpression in human cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 13092–13097. [Google Scholar] [CrossRef] [Green Version]
- Han, W.; Lo, H.-W. Landscape of EGFR signaling network in human cancers: Biology and therapeutic response in relation to receptor subcellular locations. Cancer Lett. 2012, 318, 124–134. [Google Scholar] [CrossRef] [Green Version]
- Lo, H.-W.; Hsu, S.-C.; Ali-Seyed, M.; Gunduz, M.; Xia, W.; Wei, Y.; Bartholomeusz, G.; Shih, J.-Y.; Hung, M.-C. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell 2005, 7, 575–589. [Google Scholar] [CrossRef] [Green Version]
- Lo, H.-W.; Cao, X.; Zhu, H.; Ali-Osman, F. Cyclooxygenase-2 Is a Novel Transcriptional Target of the Nuclear EGFR-STAT3 and EGFRvIII-STAT3 Signaling Axes. Mol. Cancer Res. 2010, 8, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Huo, L.; Wang, Y.-N.; Xia, W.; Hsu, S.-C.; Lai, C.-C.; Li, L.-Y.; Chang, W.-C.; Wang, Y.; Hsu, M.-C.; Yu, Y.-L.; et al. RNA helicase A is a DNA-binding partner for EGFR-mediated transcriptional activation in the nucleus. Proc. Natl. Acad. Sci. USA 2010, 107, 16125–16130. [Google Scholar] [CrossRef] [Green Version]
- Dittmann, K.; Mayer, C.; Fehrenbacher, B.; Schaller, M.; Raju, U.; Milas, L.; Chen, D.J.; Kehlbach, R.; Rodemann, H.P. Radiation-induced Epidermal Growth Factor Receptor Nuclear Import Is Linked to Activation of DNA-dependent Protein Kinase. J. Biol. Chem. 2005, 280, 31182–31189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittmann, K.; Mayer, C.; Kehlbach, R.; Rodemann, H.P. The radioprotector Bowman–Birk proteinase inhibitor stimulates DNA repair via epidermal growth factor receptor phosphorylation and nuclear transport. Radiother. Oncol. 2008, 86, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Makinoshima, H.; Takita, M.; Matsumoto, S.; Yagishita, A.; Owada, S.; Esumi, H.; Tsuchihara, K. Epidermal Growth Factor Receptor (EGFR) Signaling Regulates Global Metabolic Pathways in EGFR-mutated Lung Adenocarcinoma. J. Biol. Chem. 2014, 289, 20813–20823. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, D.; Prins, R.M.; Dang, J.; Kuga, D.; Iwanami, A.; Soto, H.; Lin, K.Y.; Huang, T.T.; Akhavan, D.; Hock, M.B.; et al. EGFR Signaling Through an Akt-SREBP-1-Dependent, Rapamycin-Resistant Pathway Sensitizes Glioblastomas to Antilipogenic Therapy. Sci. Signal. 2009, 2, ra82. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Song, F.; Zhao, X.; Jiang, H.; Wu, X.; Wang, B.; Zhou, M.; Tian, M.; Shi, B.; Wang, H.; et al. EGFR modulates monounsaturated fatty acid synthesis through phosphorylation of SCD1 in lung cancer. Mol. Cancer 2017, 16, 127. [Google Scholar] [CrossRef] [Green Version]
- Makinoshima, H.; Takita, M.; Saruwatari, K.; Umemura, S.; Obata, Y.; Ishii, G.; Matsumoto, S.; Sugiyama, E.; Ochiai, A.; Abe, R.; et al. Signaling through the Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (mTOR) Axis Is Responsible for Aerobic Glycolysis mediated by Glucose Transporter in Epidermal Growth Factor Receptor ( EGFR )-mutated Lung Adenocarcinoma. J. Biol. Chem. 2015, 290, 17495–17504. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.-O.; Li, C.-W.; Xia, W.; Lee, H.-H.; Chang, S.-S.; Shen, J.; Hsu, J.L.; Raftery, D.; Djukovic, D.; Gu, H.; et al. EGFR Signaling Enhances Aerobic Glycolysis in Triple-Negative Breast Cancer Cells to Promote Tumor Growth and Immune Escape. Cancer Res. 2016, 76, 1284–1296. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro Franco, P.I.; Rodrigues, A.P.; de Menezes, L.B.; Pacheco Miguel, M. Tumor microenvironment components: Allies of cancer progression. Pathol. Res. Pract. 2020, 216, 152729. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Melincovici, C.S.; Boşca, A.B.; Şuşman, S.; Mărginean, M.; Mihu, C.; Istrate, M.; Moldovan, I.M.; Roman, A.L.; Mihu, C.M. Vascular endothelial growth factor (VEGF)–Key factor in normal and pathological angiogenesis. Romanian J. Morphol. Embryol. Rev. Roum. Morphol. Embryol. 2018, 59, 455–467. [Google Scholar]
- Ferland-McCollough, D.; Slater, S.; Richard, J.; Reni, C.; Mangialardi, G. Pericytes, an overlooked player in vascular pathobiology. Pharmacol. Ther. 2017, 171, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of Endothelial to Mesenchymal Transition as a Source for Carcinoma-Associated Fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [Green Version]
- Petersen, O.W.; Nielsen, H.L.; Gudjonsson, T.; Villadsen, R.; Rank, F.; Niebuhr, E.; Bissell, M.J.; Rønnov-Jessen, L. Epithelial to Mesenchymal Transition in Human Breast Cancer Can Provide a Nonmalignant Stroma. Am. J. Pathol. 2003, 162, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Goswami, S.; Sahai, E.; Wyckoff, J.B.; Cammer, M.; Cox, D.; Pixley, F.J.; Stanley, E.R.; Segall, J.E.; Condeelis, J.S. Macrophages Promote the Invasion of Breast Carcinoma Cells via a Colony-Stimulating Factor-1/Epidermal Growth Factor Paracrine Loop. Cancer Res. 2005, 65, 5278–5283. [Google Scholar] [CrossRef] [Green Version]
- Schoppmann, S.F.; Birner, P.; Stöckl, J.; Kalt, R.; Ullrich, R.; Caucig, C.; Kriehuber, E.; Nagy, K.; Alitalo, K.; Kerjaschki, D. Tumor-Associated Macrophages Express Lymphatic Endothelial Growth Factors and Are Related to Peritumoral Lymphangiogenesis. Am. J. Pathol. 2002, 161, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef]
- Lindeman, N.I.; Cagle, P.T.; Beasley, M.B.; Chitale, D.A.; Dacic, S.; Giaccone, G.; Jenkins, R.B.; Kwiatkowski, D.J.; Saldivar, J.-S.; Squire, J.; et al. Molecular Testing Guideline for Selection of Lung Cancer Patients for EGFR and ALK Tyrosine Kinase Inhibitors: Guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. Arch. Pathol. Lab. Med. 2013, 137, 828–860. [Google Scholar] [CrossRef] [Green Version]
- Moran, T.; Sequist, L.V. Timing of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Therapy in Patients With Lung Cancer With EGFR Mutations. J. Clin. Oncol. 2012, 30, 3330–3336. [Google Scholar] [CrossRef]
- Shigematsu, H.; Gazdar, A.F. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int. J. Cancer 2006, 118, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Au, J.S.-K.; Thongprasert, S.; Srinivasan, S.; Tsai, C.-M.; Khoa, M.T.; Heeroma, K.; Itoh, Y.; Cornelio, G.; Yang, P.-C. A Prospective, Molecular Epidemiology Study of EGFR Mutations in Asian Patients with Advanced Non–Small-Cell Lung Cancer of Adenocarcinoma Histology (PIONEER). J. Thorac. Oncol. 2014, 9, 154–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuoka, M.; Wu, Y.-L.; Thongprasert, S.; Sunpaweravong, P.; Leong, S.-S.; Sriuranpong, V.; Chao, T.-Y.; Nakagawa, K.; Chu, D.-T.; Saijo, N.; et al. Biomarker Analyses and Final Overall Survival Results From a Phase III, Randomized, Open-Label, First-Line Study of Gefitinib Versus Carboplatin/Paclitaxel in Clinically Selected Patients With Advanced Non–Small-Cell Lung Cancer in Asia (IPASS). J. Clin. Oncol. 2011, 29, 2866–2874. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or Carboplatin–Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Inoue, A.; Kobayashi, K.; Maemondo, M.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin–paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann. Oncol. 2013, 24, 54–59. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.-Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Final overall survival results from a randomised, phase III study of erlotinib versus chemotherapy as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer (OPTIMAL, CTONG-0802). Ann. Oncol. 2015, 26, 1877–1883. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Zhou, C.; Liam, C.-K.; Wu, G.; Liu, X.; Zhong, Z.; Lu, S.; Cheng, Y.; Han, B.; Chen, L.; et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: Analyses from the phase III, randomized, open-label, ENSURE study. Ann. Oncol. 2015, 26, 1883–1889. [Google Scholar] [CrossRef]
- Cappuzzo, F.; Ciuleanu, T.; Stelmakh, L.; Cicenas, S.; Szczésna, A.; Juhász, E.; Esteban, E.; Molinier, O.; Brugger, W.; Melezínek, I.; et al. Erlotinib as maintenance treatment in advanced non-small-cell lung cancer: A multicentre, randomised, placebo-controlled phase 3 study. Lancet Oncol. 2010, 11, 521–529. [Google Scholar] [CrossRef]
- Cicènas, S.; Geater, S.L.; Petrov, P.; Hotko, Y.; Hooper, G.; Xia, F.; Mudie, N.; Wu, Y.-L. Maintenance erlotinib versus erlotinib at disease progression in patients with advanced non-small-cell lung cancer who have not progressed following platinum-based chemotherapy (IUNO study). Lung Cancer 2016, 102, 30–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, K.; Garon, E.B.; Seto, T.; Nishio, M.; Ponce Aix, S.; Paz-Ares, L.; Chiu, C.-H.; Park, K.; Novello, S.; Nadal, E.; et al. Ramucirumab plus erlotinib in patients with untreated, EGFR-mutated, advanced non-small-cell lung cancer (RELAY): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 1655–1669. [Google Scholar] [CrossRef] [Green Version]
- Hammel, P.; Huguet, F.; van Laethem, J.-L.; Goldstein, D.; Glimelius, B.; Artru, P.; Borbath, I.; Bouché, O.; Shannon, J.; André, T.; et al. Effect of Chemoradiotherapy vs Chemotherapy on Survival in Patients With Locally Advanced Pancreatic Cancer Controlled After 4 Months of Gemcitabine With or Without Erlotinib: The LAP07 Randomized Clinical Trial. JAMA 2016, 315, 1844. [Google Scholar] [CrossRef]
- Sequist, L.V.; Yang, J.C.-H.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.-M.; Boyer, M.; et al. Phase III Study of Afatinib or Cisplatin Plus Pemetrexed in Patients With Metastatic Lung Adenocarcinoma With EGFR Mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.-L.; Zhou, C.; Hu, C.-P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): An open-label, randomised phase 3 trial. Lancet Oncol. 2014, 15, 213–222. [Google Scholar] [CrossRef]
- Yang, J.C.-H.; Wu, Y.-L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.-P.; O’Byrne, K.; Feng, J.; et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Togashi, Y.; Yatabe, Y.; Mizuuchi, H.; Jangchul, P.; Kondo, C.; Shimoji, M.; Sato, K.; Suda, K.; Tomizawa, K.; et al. EGFR Exon 18 Mutations in Lung Cancer: Molecular Predictors of Augmented Sensitivity to Afatinib or Neratinib as Compared with First- or Third-Generation TKIs. Clin. Cancer Res. 2015, 21, 5305–5313. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.-H.; Yang, C.-T.; Shih, J.-Y.; Huang, M.-S.; Su, W.-C.; Lai, R.-S.; Wang, C.-C.; Hsiao, S.-H.; Lin, Y.-C.; Ho, C.-L.; et al. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Treatment Response in Advanced Lung Adenocarcinomas with G719X/L861Q/S768I Mutations. J. Thorac. Oncol. 2015, 10, 793–799. [Google Scholar] [CrossRef] [Green Version]
- Banno, E.; Togashi, Y.; Nakamura, Y.; Chiba, M.; Kobayashi, Y.; Hayashi, H.; Terashima, M.; Velasco, M.A.; Sakai, K.; Fujita, Y.; et al. Sensitivities to various epidermal growth factor receptor-tyrosine kinase inhibitors of uncommon epidermal growth factor receptor mutations L861Q and S768I: What is the optimal epidermal growth factor receptor-tyrosine kinase inhibitor? Cancer Sci. 2016, 107, 1134–1140. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.C.-H.; Sequist, L.V.; Geater, S.L.; Tsai, C.-M.; Mok, T.S.K.; Schuler, M.; Yamamoto, N.; Yu, C.-J.; Ou, S.-H.I.; Zhou, C.; et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: A combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol. 2015, 16, 830–838. [Google Scholar] [CrossRef]
- O’Kane, G.M.; Bradbury, P.A.; Feld, R.; Leighl, N.B.; Liu, G.; Pisters, K.-M.; Kamel-Reid, S.; Tsao, M.S.; Shepherd, F.A. Uncommon EGFR mutations in advanced non-small cell lung cancer. Lung Cancer 2017, 109, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Lee, M.; Linke, R.; Rosell, R.; Corral, J.; et al. Improvement in Overall Survival in a Randomized Study That Compared Dacomitinib With Gefitinib in Patients With Advanced Non–Small-Cell Lung Cancer and EGFR -Activating Mutations. J. Clin. Oncol. 2018, 36, 2244–2250. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib Is Associated with a Second Mutation in the EGFR Kinase Domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inukai, M.; Toyooka, S.; Ito, S.; Asano, H.; Ichihara, S.; Soh, J.; Suehisa, H.; Ouchida, M.; Aoe, K.; Aoe, M.; et al. Presence of Epidermal Growth Factor Receptor Gene T790M Mutation as a Minor Clone in Non–Small Cell Lung Cancer. Cancer Res. 2006, 66, 7854–7858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.L.; Tan, S.Z.; Liu, G.; Tsao, M.-S. Known and putative mechanisms of resistance to EGFR targeted therapies in NSCLC patients with EGFR mutations-a review. Transl. Lung Cancer Res. 2015, 4, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Yang, J.C.-H.; Kim, D.-W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.-J.; Kim, S.-W.; Su, W.-C.; Horn, L.; et al. AZD9291 in EGFR Inhibitor–Resistant Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Han, J.-Y.; Katakami, N.; Kim, H.R.; Hodge, R.; Kaur, P.; Brown, A.P.; Ghiorghiu, D.; et al. CNS Efficacy of Osimertinib in Patients With T790M-Positive Advanced Non–Small-Cell Lung Cancer: Data From a Randomized Phase III Trial (AURA3). J. Clin. Oncol. 2018, 36, 2702–2709. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR -Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M–Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527. [Google Scholar] [CrossRef] [Green Version]
- Piotrowska, Z.; Isozaki, H.; Lennerz, J.K.; Gainor, J.F.; Lennes, I.T.; Zhu, V.W.; Marcoux, N.; Banwait, M.K.; Digumarthy, S.R.; Su, W.; et al. Landscape of Acquired Resistance to Osimertinib in EGFR -Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition with Osimertinib and BLU-667 for Acquired RET Fusion. Cancer Discov. 2018, 8, 1529–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasako, S.; Terasaka, M.; Abe, N.; Uno, T.; Ohsawa, H.; Hashimoto, A.; Fujita, R.; Tanaka, K.; Okayama, T.; Wadhwa, R.; et al. TAS6417, A Novel EGFR Inhibitor Targeting Exon 20 Insertion Mutations. Mol. Cancer Ther. 2018, 17, 1648–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, A.; Asselin, M.-C.; Reymen, B.; Jackson, A.; Lambin, P.; West, C.M.L.; O’Connor, J.P.B.; Faivre-Finn, C. Targeting Hypoxia to Improve Non–Small Cell Lung Cancer Outcome. JNCI J. Natl. Cancer Inst. 2018, 110, 14–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piotrowska, Z.; Costa, D.B.; Oxnard, G.R.; Huberman, M.; Gainor, J.F.; Lennes, I.T.; Muzikansky, A.; Shaw, A.T.; Azzoli, C.G.; Heist, R.S.; et al. Activity of the Hsp90 inhibitor luminespib among non-small-cell lung cancers harboring EGFR exon 20 insertions. Ann. Oncol. 2018, 29, 2092–2097. [Google Scholar] [CrossRef] [PubMed]
- Singla, H.; Munshi, A.; Banipal, R.P.S.; Kumar, V. Recent Updates on the Therapeutic Potential of HER2 Tyrosine Kinase Inhibitors for the Treatment of Breast Cancer. Curr. Cancer Drug Targets 2018, 18, 306–327. [Google Scholar] [CrossRef]
- Bauerfeind, I.; Elling, D.; Heinemann, V. Lapatinib in the Treatment of Hormone Receptor-Positive/ErbB2-Positive Breast Cancer. Breast Care 2010, 5, 13–15. [Google Scholar] [CrossRef] [Green Version]
- Johnston, S.R.D.; Hegg, R.; Im, S.-A.; Park, I.H.; Burdaeva, O.; Kurteva, G.; Press, M.F.; Tjulandin, S.; Iwata, H.; Simon, S.D.; et al. Phase III, Randomized Study of Dual Human Epidermal Growth Factor Receptor 2 (HER2) Blockade With Lapatinib Plus Trastuzumab in Combination With an Aromatase Inhibitor in Postmenopausal Women With HER2-Positive, Hormone Receptor–Positive Metastatic Breast Cancer: ALTERNATIVE. J. Clin. Oncol. 2018, 36, 741–748. [Google Scholar] [CrossRef]
- Retraction. Phase III, Randomized Study of Dual Human Epidermal Growth Factor Receptor 2 (HER2) Blockade With Lapatinib Plus Trastuzumab in Combination With an Aromatase Inhibitor in Postmenopausal Women With HER2-Positive, Hormone Receptor–Positive Metastatic Breast Cancer: Updated Results of ALTERNATIVE. J. Clin. Oncol. 2020. [CrossRef]
- Blackwell, K.L.; Burstein, H.J.; Storniolo, A.M.; Rugo, H.; Sledge, G.; Koehler, M.; Ellis, C.; Casey, M.; Vukelja, S.; Bischoff, J.; et al. Randomized Study of Lapatinib Alone or in Combination With Trastuzumab in Women With ErbB2-Positive, Trastuzumab-Refractory Metastatic Breast Cancer. J. Clin. Oncol. 2010, 28, 1124–1130. [Google Scholar] [CrossRef]
- Blackwell, K.L.; Burstein, H.J.; Storniolo, A.M.; Rugo, H.S.; Sledge, G.; Aktan, G.; Ellis, C.; Florance, A.; Vukelja, S.; Bischoff, J.; et al. Overall Survival Benefit With Lapatinib in Combination With Trastuzumab for Patients With Human Epidermal Growth Factor Receptor 2–Positive Metastatic Breast Cancer: Final Results From the EGF104900 Study. J. Clin. Oncol. 2012, 30, 2585–2592. [Google Scholar] [CrossRef] [PubMed]
- Cameron, D.; Casey, M.; Press, M.; Lindquist, D.; Pienkowski, T.; Romieu, C.G.; Chan, S.; Jagiello-Gruszfeld, A.; Kaufman, B.; Crown, J.; et al. A phase III randomized comparison of lapatinib plus capecitabine versus capecitabine alone in women with advanced breast cancer that has progressed on trastuzumab: Updated efficacy and biomarker analyses. Breast Cancer Res. Treat. 2008, 112, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B.; et al. Lapatinib plus Capecitabine for HER2-Positive Advanced Breast Cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M.; Holmes, F.A.; Ejlertsen, B.; Delaloge, S.; Moy, B.; Iwata, H.; von Minckwitz, G.; Chia, S.K.L.; Mansi, J.; Barrios, C.H.; et al. Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1688–1700. [Google Scholar] [CrossRef]
- Saura, C.; Oliveira, M.; Feng, Y.-H.; Dai, M.-S.; Chen, S.-W.; Hurvitz, S.A.; Kim, S.-B.; Moy, B.; Delaloge, S.; Gradishar, W.; et al. Neratinib Plus Capecitabine Versus Lapatinib Plus Capecitabine in HER2-Positive Metastatic Breast Cancer Previously Treated With ≥ 2 HER2-Directed Regimens: Phase III NALA Trial. J. Clin. Oncol. 2020, 38, 3138–3149. [Google Scholar] [CrossRef]
- Ben-Baruch, N.E.; Bose, R.; Kavuri, S.M.; Ma, C.X.; Ellis, M.J. HER2 -Mutated Breast Cancer Responds to Treatment With Single-Agent Neratinib, a Second-Generation HER2/EGFR Tyrosine Kinase Inhibitor. J. Natl. Compr. Canc. Netw. 2015, 13, 1061–1064. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol. Res. 2019, 139, 395–411. [Google Scholar] [CrossRef]
- Duchnowska, R.; Loibl, S.; Jassem, J. Tyrosine kinase inhibitors for brain metastases in HER2-positive breast cancer. Cancer Treat. Rev. 2018, 67, 71–77. [Google Scholar] [CrossRef]
- Murthy, R.; Borges, V.F.; Conlin, A.; Chaves, J.; Chamberlain, M.; Gray, T.; Vo, A.; Hamilton, E. Tucatinib with capecitabine and trastuzumab in advanced HER2-positive metastatic breast cancer with and without brain metastases: A non-randomised, open-label, phase 1b study. Lancet Oncol. 2018, 19, 880–888. [Google Scholar] [CrossRef]
- Arcila, M.E.; Chaft, J.E.; Nafa, K.; Roy-Chowdhuri, S.; Lau, C.; Zaidinski, M.; Paik, P.K.; Zakowski, M.F.; Kris, M.G.; Ladanyi, M. Prevalence, Clinicopathologic Associations, and Molecular Spectrum of ERBB2 (HER2) Tyrosine Kinase Mutations in Lung Adenocarcinomas. Clin. Cancer Res. 2012, 18, 4910–4918. [Google Scholar] [CrossRef] [Green Version]
- Mazières, J.; Peters, S.; Lepage, B.; Cortot, A.B.; Barlesi, F.; Beau-Faller, M.; Besse, B.; Blons, H.; Mansuet-Lupo, A.; Urban, T.; et al. Lung Cancer That Harbors an HER2 Mutation: Epidemiologic Characteristics and Therapeutic Perspectives. J. Clin. Oncol. 2013, 31, 1997–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazières, J.; Barlesi, F.; Filleron, T.; Besse, B.; Monnet, I.; Beau-Faller, M.; Peters, S.; Dansin, E.; Früh, M.; Pless, M.; et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targeted drugs: Results from the European EUHER2 cohort. Ann. Oncol. 2016, 27, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Tchekmedyian, N.; Chu, D.T.; Reddy, G.; Bhat, G.; Socinski, M.A. A phase 2 study of poziotinib in patients with EGFR or HER2 exon 20 mutation-positive non-small cell lung cancer. J. Clin. Oncol. 2018, 36, TPS9106. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, T.; Qin, Z.; Jiang, J.; Wang, Q.; Yang, S.; Rivard, C.; Gao, G.; Ng, T.L.; Tu, M.M.; et al. HER2 exon 20 insertions in non-small-cell lung cancer are sensitive to the irreversible pan-HER receptor tyrosine kinase inhibitor pyrotinib. Ann. Oncol. 2019, 30, 447–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Shao, Y.; Qin, H.-F.; Tai, Y.-H.; Gao, H.-J. ALK- rearrangement in non-small-cell lung cancer (NSCLC): ALK rearrangement in lung cancer. Thorac. Cancer 2018, 9, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Yeap, B.Y.; Mino-Kenudson, M.; Digumarthy, S.R.; Costa, D.B.; Heist, R.S.; Solomon, B.; Stubbs, H.; Admane, S.; McDermott, U.; et al. Clinical Features and Outcome of Patients With Non–Small-Cell Lung Cancer Who Harbor EML4-ALK. J. Clin. Oncol. 2009, 27, 4247–4253. [Google Scholar] [CrossRef] [Green Version]
- Boland, J.M.; Erdogan, S.; Vasmatzis, G.; Yang, P.; Tillmans, L.S.; Johnson, M.R.E.; Wang, X.; Peterson, L.M.; Halling, K.C.; Oliveira, A.M.; et al. Anaplastic lymphoma kinase immunoreactivity correlates with ALK gene rearrangement and transcriptional up-regulation in non–small cell lung carcinomas. Hum. Pathol. 2009, 40, 1152–1158. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, D.-W.; Nakagawa, K.; Seto, T.; Crinó, L.; Ahn, M.-J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus Chemotherapy in Advanced ALK -Positive Lung Cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [Green Version]
- Solomon, B.J.; Mok, T.; Kim, D.-W.; Wu, Y.-L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-Line Crizotinib versus Chemotherapy in ALK -Positive Lung Cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [Green Version]
- Solomon, B.J.; Kim, D.-W.; Wu, Y.-L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; Tang, Y.; et al. Final Overall Survival Analysis From a Study Comparing First-Line Crizotinib Versus Chemotherapy in ALK-Mutation-Positive Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2251–2258. [Google Scholar] [CrossRef]
- Soria, J.-C.; Tan, D.S.W.; Chiari, R.; Wu, Y.-L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.-J.; et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK -rearranged non-small-cell lung cancer (ASCEND-4): A randomised, open-label, phase 3 study. The Lancet 2017, 389, 917–929. [Google Scholar] [CrossRef]
- Hida, T.; Nokihara, H.; Kondo, M.; Kim, Y.H.; Azuma, K.; Seto, T.; Takiguchi, Y.; Nishio, M.; Yoshioka, H.; Imamura, F.; et al. Alectinib versus crizotinib in patients with ALK -positive non-small-cell lung cancer (J-ALEX): An open-label, randomised phase 3 trial. Lancet 2017, 390, 29–39. [Google Scholar] [CrossRef]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.-W.; Ou, S.-H.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus Crizotinib in Untreated ALK -Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Dziadziuszko, R.; Peters, S.; Mok, T.; Noe, J.; Nowicka, M.; Gadgeel, S.M.; Cheema, P.; Pavlakis, N.; de Marinis, F.; et al. Updated Efficacy and Safety Data and Impact of the EML4-ALK Fusion Variant on the Efficacy of Alectinib in Untreated ALK-Positive Advanced Non–Small Cell Lung Cancer in the Global Phase III ALEX Study. J. Thorac. Oncol. 2019, 14, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Takeuchi, S.; Arai, S.; Katayama, R.; Nanjo, S.; Tanimoto, A.; Nishiyama, A.; Nakagawa, T.; Taniguchi, H.; Suzuki, T.; et al. Epithelial-to-Mesenchymal Transition Is a Mechanism of ALK Inhibitor Resistance in Lung Cancer Independent of ALK Mutation Status. Cancer Res. 2019, 79, 1658–1670. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, T.M.; Crinò, L.; Gridelli, C.; Kiura, K.; Liu, G.; Novello, S.; Bearz, A.; Gautschi, O.; Mok, T.; et al. Ceritinib versus chemotherapy in patients with ALK-rearranged non-small-cell lung cancer previously given chemotherapy and crizotinib (ASCEND-5): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 874–886. [Google Scholar] [CrossRef]
- Novello, S.; Mazières, J.; Oh, I.-J.; de Castro, J.; Migliorino, M.R.; Helland, Å.; Dziadziuszko, R.; Griesinger, F.; Kotb, A.; Zeaiter, A.; et al. Alectinib versus chemotherapy in crizotinib-pretreated anaplastic lymphoma kinase (ALK)-positive non-small-cell lung cancer: Results from the phase III ALUR study. Ann. Oncol. 2018, 29, 1409–1416. [Google Scholar] [CrossRef]
- Ou, S.-H.I.; Ahn, J.S.; De Petris, L.; Govindan, R.; Yang, J.C.-H.; Hughes, B.; Lena, H.; Moro-Sibilot, D.; Bearz, A.; Ramirez, S.V.; et al. Alectinib in Crizotinib-Refractory ALK- Rearranged Non–Small-Cell Lung Cancer: A Phase II Global Study. J. Clin. Oncol. 2016, 34, 661–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, A.T.; Gandhi, L.; Gadgeel, S.; Riely, G.J.; Cetnar, J.; West, H.; Camidge, D.R.; Socinski, M.A.; Chiappori, A.; Mekhail, T.; et al. Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer: A single-group, multicentre, phase 2 trial. Lancet Oncol. 2016, 17, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Anjum, R.; Squillace, R.; Nadworny, S.; Zhou, T.; Keats, J.; Ning, Y.; Wardwell, S.D.; Miller, D.; Song, Y.; et al. The Potent ALK Inhibitor Brigatinib (AP26113) Overcomes Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in Preclinical Models. Clin. Cancer Res. 2016, 22, 5527–5538. [Google Scholar] [CrossRef] [Green Version]
- Camidge, D.R.; Kim, H.R.; Ahn, M.-J.; Yang, J.C.-H.; Han, J.-Y.; Lee, J.-S.; Hochmair, M.J.; Li, J.Y.-C.; Chang, G.-C.; Lee, K.H.; et al. Brigatinib versus Crizotinib in ALK -Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2027–2039. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.M.; Hansen, K.H.; Paz-Ares Rodríguez, L.; West, H.L.; Reckamp, K.L.; Leighl, N.B.; Tiseo, M.; Smit, E.F.; Kim, D.-W.; Gettinger, S.N.; et al. Brigatinib in Crizotinib-Refractory ALK+ NSCLC: 2-Year Follow-up on Systemic and Intracranial Outcomes in the Phase 2 ALTA Trial. J. Thorac. Oncol. 2020, 15, 404–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK -Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baglivo, S.; Ricciuti, B.; Ludovini, V.; Metro, G.; Siggillino, A.; De Giglio, A.; Chiari, R. Dramatic Response to Lorlatinib in a Heavily Pretreated Lung Adenocarcinoma Patient Harboring G1202R Mutation and a Synchronous Novel R1192P ALK Point Mutation. J. Thorac. Oncol. 2018, 13, e145–e147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, R.; Friboulet, L.; Koike, S.; Lockerman, E.L.; Khan, T.M.; Gainor, J.F.; Iafrate, A.J.; Takeuchi, K.; Taiji, M.; Okuno, Y.; et al. Two Novel ALK Mutations Mediate Acquired Resistance to the Next-Generation ALK Inhibitor Alectinib. Clin. Cancer Res. 2014, 20, 5686–5696. [Google Scholar] [CrossRef] [Green Version]
- Solomon, B.J.; Besse, B.; Bauer, T.M.; Felip, E.; Soo, R.A.; Camidge, D.R.; Chiari, R.; Bearz, A.; Lin, C.-C.; Gadgeel, S.M.; et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: Results from a global phase 2 study. Lancet Oncol. 2018, 19, 1654–1667. [Google Scholar] [CrossRef]
- Shaw, A.T.; Solomon, B.J.; Besse, B.; Bauer, T.M.; Lin, C.-C.; Soo, R.A.; Riely, G.J.; Ou, S.-H.I.; Clancy, J.S.; Li, S.; et al. ALK Resistance Mutations and Efficacy of Lorlatinib in Advanced Anaplastic Lymphoma Kinase-Positive Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2019, 37, 1370–1379. [Google Scholar] [CrossRef]
- Pal, P.; Khan, Z. ROS1-1. J. Clin. Pathol. 2017, 70, 1001–1009. [Google Scholar] [CrossRef]
- Uguen, A.; De Braekeleer, M. ROS1 fusions in cancer: A review. Future Oncol. 2016, 12, 1911–1928. [Google Scholar] [CrossRef]
- Patil, T.; Simons, E.; Mushtaq, R.; Pacheco, J.M.; Doebele, R.C.; Bowles, D.W. Targeted therapies for ROS1-rearranged non-small cell lung cancer. Drugs Today 2019, 55, 641. [Google Scholar] [CrossRef]
- Shaw, A.T.; Ou, S.-H.I.; Bang, Y.-J.; Camidge, D.R.; Solomon, B.J.; Salgia, R.; Riely, G.J.; Varella-Garcia, M.; Shapiro, G.I.; Costa, D.B.; et al. Crizotinib in ROS1 -Rearranged Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2014, 371, 1963–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, A.T.; Riely, G.J.; Bang, Y.-J.; Kim, D.-W.; Camidge, D.R.; Solomon, B.J.; Varella-Garcia, M.; Iafrate, A.J.; Shapiro, G.I.; Usari, T.; et al. Crizotinib in ROS1-rearranged advanced non-small-cell lung cancer (NSCLC): Updated results, including overall survival, from PROFILE 1001. Ann. Oncol. 2019, 30, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Siena, S.; Dziadziuszko, R.; Barlesi, F.; Krebs, M.G.; Shaw, A.T.; de Braud, F.; Rolfo, C.; Ahn, M.-J.; Wolf, J.; et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 261–270. [Google Scholar] [CrossRef]
- Lim, S.M.; Kim, H.R.; Lee, J.-S.; Lee, K.H.; Lee, Y.-G.; Min, Y.J.; Cho, E.K.; Lee, S.S.; Kim, B.-S.; Choi, M.Y.; et al. Open-Label, Multicenter, Phase II Study of Ceritinib in Patients With Non–Small-Cell Lung Cancer Harboring ROS1 Rearrangement. J. Clin. Oncol. 2017, 35, 2613–2618. [Google Scholar] [CrossRef]
- Shaw, A.T.; Solomon, B.J.; Chiari, R.; Riely, G.J.; Besse, B.; Soo, R.A.; Kao, S.; Lin, C.-C.; Bauer, T.M.; Clancy, J.S.; et al. Lorlatinib in advanced ROS1-positive non-small-cell lung cancer: A multicentre, open-label, single-arm, phase 1–2 trial. Lancet Oncol. 2019, 20, 1691–1701. [Google Scholar] [CrossRef]
- Drilon, A.; Ou, S.-H.I.; Cho, B.C.; Kim, D.-W.; Lee, J.; Lin, J.J.; Zhu, V.W.; Ahn, M.-J.; Camidge, D.R.; Nguyen, J.; et al. Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent- Front Mutations. Cancer Discov. 2018, 8, 1227–1236. [Google Scholar] [CrossRef] [Green Version]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Solomon, J.P.; Benayed, R.; Hechtman, J.F.; Ladanyi, M. Identifying patients with NTRK fusion cancer. Ann. Oncol. 2019, 30, viii16–viii22. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Bauer, T.M.; Lee, J.J.; Dowlati, A.; Brose, M.S.; Farago, A.F.; Taylor, M.; Shaw, A.T.; Montez, S.; Meric-Bernstam, F.; et al. Larotrectinib in adult patients with solid tumours: A multi-centre, open-label, phase I dose-escalation study. Ann. Oncol. 2019, 30, 325–331. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion–Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Cho, B.C.; Drilon, A.E.; Doebele, R.C.; Kim, D.-W.; Lin, J.J.; Lee, J.; Ahn, M.-J.; Zhu, V.W.; Ejadi, S.; Camidge, D.R.; et al. Safety and preliminary clinical activity of repotrectinib in patients with advanced ROS1 fusion-positive non-small cell lung cancer (TRIDENT-1 study). J. Clin. Oncol. 2019, 37, 9011. [Google Scholar] [CrossRef]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Hirota, S. Gain-of-Function Mutations of c-kit in Human Gastrointestinal Stromal Tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Abbaspour Babaei, M.; Kamalidehghan, B.; Saleem, M.; Zaman Huri, H.; Ahmadipour, F. Receptor tyrosine kinase (c-Kit) inhibitors: A potential therapeutic target in cancer cells. Drug Des. Devel. Ther. 2016, 10, 2443–2459. [Google Scholar] [CrossRef] [Green Version]
- Demetri, G.D.; von Mehren, M.; Blanke, C.D.; Van den Abbeele, A.D.; Eisenberg, B.; Roberts, P.J.; Heinrich, M.C.; Tuveson, D.A.; Singer, S.; Janicek, M.; et al. Efficacy and Safety of Imatinib Mesylate in Advanced Gastrointestinal Stromal Tumors. N. Engl. J. Med. 2002, 347, 472–480. [Google Scholar] [CrossRef]
- von Mehren, M.; Heinrich, M.C.; Joensuu, H.; Blanke, C.D.; Wehrle, E.; Demetri, G.D. Follow-up results after 9 years (yrs) of the ongoing, phase II B2222 trial of imatinib mesylate (IM) in patients (pts) with metastatic or unresectable KIT+ gastrointestinal stromal tumors (GIST). J. Clin. Oncol. 2011, 29, 10016. [Google Scholar] [CrossRef]
- Demetri, G.D.; Rankin, C.J.; Benjamin, R.S.; Borden, E.C.; Ryan, C.W.; Priebat, D.A.; Tap, W.D.; von Mehren, M.; Blackstein, M.E.; Heinrich, M.C.; et al. Long-term disease control of advanced gastrointestinal stromal tumors (GIST) with imatinib (IM): 10-year outcomes from SWOG phase III intergroup trial S0033. J. Clin. Oncol. 2014, 32, 10508. [Google Scholar] [CrossRef]
- Verweij, J.; Casali, P.G.; Zalcberg, J.; LeCesne, A.; Reichardt, P.; Blay, J.-Y.; Issels, R.; van Oosterom, A.; Hogendoorn, P.C.; Van Glabbeke, M.; et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: Randomised trial. Lancet 2004, 364, 1127–1134. [Google Scholar] [CrossRef]
- Blanke, C.D.; Rankin, C.; Demetri, G.D.; Ryan, C.W.; von Mehren, M.; Benjamin, R.S.; Raymond, A.K.; Bramwell, V.H.C.; Baker, L.H.; Maki, R.G.; et al. Phase III Randomized, Intergroup Trial Assessing Imatinib Mesylate At Two Dose Levels in Patients With Unresectable or Metastatic Gastrointestinal Stromal Tumors Expressing the Kit Receptor Tyrosine Kinase: S0033. J. Clin. Oncol. 2008, 26, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST) Comparison of Two Doses of Imatinib for the Treatment of Unresectable or Metastatic Gastrointestinal Stromal Tumors: A Meta-Analysis of 1,640 Patients. J. Clin. Oncol. 2010, 28, 1247–1253. [CrossRef] [Green Version]
- DeMatteo, R.P.; Ballman, K.V.; Antonescu, C.R.; Maki, R.G.; Pisters, P.W.; Demetri, G.D.; Blackstein, M.E.; Blanke, C.D.; von Mehren, M.; Brennan, M.F.; et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: A randomised, double-blind, placebo-controlled trial. Lancet 2009, 373, 1097–1104. [Google Scholar] [CrossRef] [Green Version]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.-K.; Blay, J.-Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Serrano, C.; Mariño-Enríquez, A.; Tao, D.L.; Ketzer, J.; Eilers, G.; Zhu, M.; Yu, C.; Mannan, A.M.; Rubin, B.P.; Demetri, G.D.; et al. Complementary activity of tyrosine kinase inhibitors against secondary kit mutations in imatinib-resistant gastrointestinal stromal tumours. Br. J. Cancer 2019, 120, 612–620. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. The role of small molecule Kit protein-tyrosine kinase inhibitors in the treatment of neoplastic disorders. Pharmacol. Res. 2018, 133, 35–52. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Lennartsson, J. The PDGF/PDGFR pathway as a drug target. Mol. Aspects Med. 2018, 62, 75–88. [Google Scholar] [CrossRef]
- Medeiros, F.; Corless, C.L.; Duensing, A.; Hornick, J.L.; Oliveira, A.M.; Heinrich, M.C.; Fletcher, J.A.; Fletcher, C.D.M. KIT-Negative Gastrointestinal Stromal Tumors: Proof of Concept and Therapeutic Implications. Am. J. Surg. Pathol. 2004, 28, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Corless, C.L.; Schroeder, A.; Griffith, D.; Town, A.; McGreevey, L.; Harrell, P.; Shiraga, S.; Bainbridge, T.; Morich, J.; Heinrich, M.C. PDGFRA Mutations in Gastrointestinal Stromal Tumors: Frequency, Spectrum and In Vitro Sensitivity to Imatinib. J. Clin. Oncol. 2005, 23, 5357–5364. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Blanke, C.D.; von Mehren, M.; Joensuu, H.; McGreevey, L.S.; Chen, C.-J.; Van den Abbeele, A.D.; Druker, B.J.; et al. Kinase Mutations and Imatinib Response in Patients With Metastatic Gastrointestinal Stromal Tumor. J. Clin. Oncol. 2003, 21, 4342–4349. [Google Scholar] [CrossRef] [PubMed]
- Cassier, P.A.; Fumagalli, E.; Rutkowski, P.; Schoffski, P.; Van Glabbeke, M.; Debiec-Rychter, M.; Emile, J.-F.; Duffaud, F.; Martin-Broto, J.; Landi, B.; et al. Outcome of Patients with Platelet-Derived Growth Factor Receptor Alpha-Mutated Gastrointestinal Stromal Tumors in the Tyrosine Kinase Inhibitor Era. Clin. Cancer Res. 2012, 18, 4458–4464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Morimitsu, Y.; Okamoto, S.; Hisaoka, M.; Ishida, T.; Sheng, W.; Hashimoto, H. COL1A1-PDGFB Fusion Transcripts in Fibrosarcomatous Areas of Six Dermatofibrosarcomas Protuberans. J. Mol. Diagn. 2000, 2, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Abbott, J.J.; Erickson-Johnson, M.; Wang, X.; Nascimento, A.G.; Oliveira, A.M. Gains of COL1A1-PDGFB genomic copies occur in fibrosarcomatous transformation of dermatofibrosarcoma protuberans. Mod. Pathol. 2006, 19, 1512–1518. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.P.; Schuetze, S.M.; Eary, J.F.; Norwood, T.H.; Mirza, S.; Conrad, E.U.; Bruckner, J.D. Molecular Targeting of Platelet-Derived Growth Factor B by Imatinib Mesylate in a Patient With Metastatic Dermatofibrosarcoma Protuberans. J. Clin. Oncol. 2002, 20, 3586–3591. [Google Scholar] [CrossRef] [PubMed]
- McArthur, G.A.; Demetri, G.D.; van Oosterom, A.; Heinrich, M.C.; Debiec-Rychter, M.; Corless, C.L.; Nikolova, Z.; Dimitrijevic, S.; Fletcher, J.A. Molecular and Clinical Analysis of Locally Advanced Dermatofibrosarcoma Protuberans Treated With Imatinib: Imatinib Target Exploration Consortium Study B2225. J. Clin. Oncol. 2005, 23, 866–873. [Google Scholar] [CrossRef]
- Rutkowski, P.; Van Glabbeke, M.; Rankin, C.J.; Ruka, W.; Rubin, B.P.; Debiec-Rychter, M.; Lazar, A.; Gelderblom, H.; Sciot, R.; Lopez-Terrada, D.; et al. Imatinib Mesylate in Advanced Dermatofibrosarcoma Protuberans: Pooled Analysis of Two Phase II Clinical Trials. J. Clin. Oncol. 2010, 28, 1772–1779. [Google Scholar] [CrossRef]
- Ugurel, S.; Mentzel, T.; Utikal, J.; Helmbold, P.; Mohr, P.; Pfohler, C.; Schiller, M.; Hauschild, A.; Hein, R.; Kampgen, E.; et al. Neoadjuvant Imatinib in Advanced Primary or Locally Recurrent Dermatofibrosarcoma Protuberans: A Multicenter Phase II DeCOG Trial with Long-term Follow-up. Clin. Cancer Res. 2014, 20, 499–510. [Google Scholar] [CrossRef] [Green Version]
- Navarrete-Dechent, C.; Mori, S.; Barker, C.A.; Dickson, M.A.; Nehal, K.S. Imatinib Treatment for Locally Advanced or Metastatic Dermatofibrosarcoma Protuberans: A Systematic Review. JAMA Dermatol. 2019, 155, 361. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Jones, R.L.; von Mehren, M.; Schöffski, P.; Serrano, C.; Kang, Y.-K.; Cassier, P.A.; Mir, O.; Eskens, F.; Tap, W.D.; et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): A multicentre, open-label, phase 1 trial. Lancet Oncol. 2020, 21, 935–946. [Google Scholar] [CrossRef]
- Blay, J.-Y.; Serrano, C.; Heinrich, M.C.; Zalcberg, J.; Bauer, S.; Gelderblom, H.; Schöffski, P.; Jones, R.L.; Attia, S.; D’Amato, G.; et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 923–934. [Google Scholar] [CrossRef]
- Fernandes, M.; Duplaquet, L.; Tulasne, D. Proteolytic cleavages of MET: The divide-and-conquer strategy of a receptor tyrosine kinase. BMB Rep. 2019, 52, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Leonardi, G.C.; Kravets, S.; Dahlberg, S.E.; Drilon, A.; Noonan, S.A.; Camidge, D.R.; Ou, S.-H.I.; Costa, D.B.; Gadgeel, S.M.; et al. Impact of MET inhibitors on survival among patients with non-small cell lung cancer harboring MET exon 14 mutations: A retrospective analysis. Lung Cancer 2019, 133, 96–102. [Google Scholar] [CrossRef]
- Zhang, H.; Feng, Q.; Chen, W.-D.; Wang, Y.-D. HGF/c-MET: A Promising Therapeutic Target in the Digestive System Cancers. Int. J. Mol. Sci. 2018, 19, 3295. [Google Scholar] [CrossRef] [Green Version]
- Wolf, J.; Seto, T.; Han, J.-Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.-W.; Hida, T.; De Jonge, M.J.; Orlov, S.V.; et al. Capmatinib (INC280) in METΔex14 -mutated advanced non-small cell lung cancer (NSCLC): Efficacy data from the phase II GEOMETRY mono-1 study. J. Clin. Oncol. 2019, 37, 9004. [Google Scholar] [CrossRef]
- Schuler, M.; Berardi, R.; Lim, W.-T.; de Jonge, M.; Bauer, T.M.; Azaro, A.; Gottfried, M.; Han, J.-Y.; Lee, D.H.; Wollner, M.; et al. Molecular correlates of response to capmatinib in advanced non-small-cell lung cancer: Clinical and biomarker results from a phase I trial. Ann. Oncol. 2020, 31, 789–797. [Google Scholar] [CrossRef]
- Camidge, D.R.; Ou, S.-H.I.; Shapiro, G.; Otterson, G.A.; Villaruz, L.C.; Villalona-Calero, M.A.; Iafrate, A.J.; Varella-Garcia, M.; Dacic, S.; Cardarella, S.; et al. Efficacy and safety of crizotinib in patients with advanced c-MET -amplified non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2014, 32, 8001. [Google Scholar] [CrossRef]
- Drilon, A.E.; Camidge, D.R.; Ou, S.-H.I.; Clark, J.W.; Socinski, M.A.; Weiss, J.; Riely, G.J.; Winter, M.; Wang, S.C.; Monti, K.; et al. Efficacy and safety of crizotinib in patients (pts) with advanced MET exon 14-altered non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2016, 34, 108. [Google Scholar] [CrossRef]
- Drilon, A.; Clark, J.W.; Weiss, J.; Ou, S.-H.I.; Camidge, D.R.; Solomon, B.J.; Otterson, G.A.; Villaruz, L.C.; Riely, G.J.; Heist, R.S.; et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat. Med. 2020, 26, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Drilon, A.; Fan, P.-D.; Yu, H.; Rekhtman, N.; Ginsberg, M.S.; Borsu, L.; Schultz, N.; Berger, M.F.; Rudin, C.M.; et al. Response to MET Inhibitors in Patients with Stage IV Lung Adenocarcinomas Harboring MET Mutations Causing Exon 14 Skipping. Cancer Discov. 2015, 5, 842–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in Non–Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N. Engl. J. Med. 2020, 383, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Jhiang, S.M. The RET proto-oncogene in human cancers. Oncogene 2000, 19, 5590–5597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manié, S.; Santoro, M.; Fusco, A.; Billaud, M. The RET receptor: Function in development and dysfunction in congenital malformation. Trends Genet. 2001, 17, 580–589. [Google Scholar] [CrossRef]
- Li, A.Y.; McCusker, M.G.; Russo, A.; Scilla, K.A.; Gittens, A.; Arensmeyer, K.; Mehra, R.; Adamo, V.; Rolfo, C. RET fusions in solid tumors. Cancer Treat. Rev. 2019, 81, 101911. [Google Scholar] [CrossRef]
- Subbiah, V.; Yang, D.; Velcheti, V.; Drilon, A.; Meric-Bernstam, F. State-of-the-Art Strategies for Targeting RET -Dependent Cancers. J. Clin. Oncol. 2020, 38, 1209–1221. [Google Scholar] [CrossRef]
- Drilon, A.; Oxnard, G.; Wirth, L.; Besse, B.; Gautschi, O.; Tan, S.W.D.; Loong, H.; Bauer, T.; Kim, Y.J.; Horiike, A.; et al. PL02.08 Registrational Results of LIBRETTO-001: A Phase 1/2 Trial of LOXO-292 in Patients with RET Fusion-Positive Lung Cancers. J. Thorac. Oncol. 2019, 14, S6–S7. [Google Scholar] [CrossRef]
- Subbiah, V.; Gainor, J.F.; Oxnard, G.R.; Tan, D.S.-W.; Owen, D.H.; Cho, B.C.; Loong, H.H.F.; McCoach, C.E.; Weiss, J.; Kim, Y.; et al. Intracranial activity of selpercatinib (LOXO-292) in RET fusion-positive non-small cell lung cancer (NSCLC) patients on the LIBRETTO-001 trial. J. Clin. Oncol. 2020, 38, 9516. [Google Scholar] [CrossRef]
- Shah, M.H.; Sherman, E.J.; Robinson, B.; Solomon, B.J.; Kang, H.; Lorch, J.H.; Worden, F.P.; Brose, M.S.; Leboulleux, S.; Godbert, Y.; et al. Selpercatinib (LOXO-292) in patients with RET -mutant medullary thyroid cancer. J. Clin. Oncol. 2020, 38, 3594. [Google Scholar] [CrossRef]
- Markham, A. Selpercatinib: First Approval. Drugs 2020, 80, 1119–1124. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Hu, M.I.-N.; Gainor, J.F.; Mansfield, A.S.; Alonso, G.; Taylor, M.H.; Zhu, V.W.; Garrido Lopez, P.; Amatu, A.; Doebele, R.C.; et al. Clinical activity of the RET inhibitor pralsetinib (BLU-667) in patients with RET fusion+ solid tumors. J. Clin. Oncol. 2020, 38, 109. [Google Scholar] [CrossRef]
- Subbiah, V.; Gainor, J.F.; Rahal, R.; Brubaker, J.D.; Kim, J.L.; Maynard, M.; Hu, W.; Cao, Q.; Sheets, M.P.; Wilson, D.; et al. Precision Targeted Therapy with BLU-667 for RET -Driven Cancers. Cancer Discov. 2018, 8, 836–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drilon, A.E.; Zhai, D.; Rogers, E.; Deng, W.; Zhang, X.; Ung, J.; Lee, D.; Rodon, L.; Graber, A.; Zimmerman, Z.F.; et al. The next-generation RET inhibitor TPX-0046 is active in drug-resistant and naïve RET-driven cancer models. J. Clin. Oncol. 2020, 38, 3616. [Google Scholar] [CrossRef]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus Placebo in Radioiodine-Refractory Thyroid Cancer. N. Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Schlumberger, M.; Elisei, R.; Müller, S.; Schöffski, P.; Brose, M.; Shah, M.; Licitra, L.; Krajewska, J.; Kreissl, M.C.; Niederle, B.; et al. Overall survival analysis of EXAM, a phase III trial of cabozantinib in patients with radiographically progressive medullary thyroid carcinoma. Ann. Oncol. 2017, 28, 2813–2819. [Google Scholar] [CrossRef]
- Wells, S.A.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in Patients With Locally Advanced or Metastatic Medullary Thyroid Cancer: A Randomized, Double-Blind Phase III Trial. J. Clin. Oncol. 2012, 30, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Parveen, A.; Subedi, L.; Kim, H.; Khan, Z.; Zahra, Z.; Farooqi, M.; Kim, S. Phytochemicals Targeting VEGF and VEGF-Related Multifactors as Anticancer Therapy. J. Clin. Med. 2019, 8, 350. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Iring, A.; Strilic, B.; Albarrán Juárez, J.; Kaur, H.; Troidl, K.; Tonack, S.; Burbiel, J.C.; Müller, C.E.; Fleming, I.; et al. P2Y2 and Gq/G11 control blood pressure by mediating endothelial mechanotransduction. J. Clin. Investig. 2015, 125, 3077–3086. [Google Scholar] [CrossRef] [Green Version]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus Interferon Alfa in Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Raymond, E.; Dahan, L.; Raoul, J.-L.; Bang, Y.-J.; Borbath, I.; Lombard-Bohas, C.; Valle, J.; Metrakos, P.; Smith, D.; Vinik, A.; et al. Sunitinib Malate for the Treatment of Pancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2011, 364, 501–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternberg, C.N.; Hawkins, R.E.; Wagstaff, J.; Salman, P.; Mardiak, J.; Barrios, C.H.; Zarba, J.J.; Gladkov, O.A.; Lee, E.; Szczylik, C.; et al. A randomised, double-blind phase III study of pazopanib in patients with advanced and/or metastatic renal cell carcinoma: Final overall survival results and safety update. Eur. J. Cancer 2013, 49, 1287–1296. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Cella, D.; Reeves, J.; Hawkins, R.; Guo, J.; Nathan, P.; Staehler, M.; de Souza, P.; Merchan, J.R.; et al. Pazopanib versus Sunitinib in Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2013, 369, 722–731. [Google Scholar] [CrossRef] [Green Version]
- van der Graaf, W.T.; Blay, J.-Y.; Chawla, S.P.; Kim, D.-W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Motzer, R.J.; Nosov, D.; Eisen, T.; Bondarenko, I.; Lesovoy, V.; Lipatov, O.; Tomczak, P.; Lyulko, O.; Alyasova, A.; Harza, M.; et al. Tivozanib Versus Sorafenib As Initial Targeted Therapy for Patients With Metastatic Renal Cell Carcinoma: Results From a Phase III Trial. J. Clin. Oncol. 2013, 31, 3791–3799. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Tomczak, P.; Hutson, T.E.; Michaelson, M.D.; Negrier, S.; Oudard, S.; Gore, M.E.; Tarazi, J.; Hariharan, S.; et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: Overall survival analysis and updated results from a randomised phase 3 trial. Lancet Oncol. 2013, 14, 552–562. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Halabi, S.; Sanford, B.L.; Hahn, O.; Michaelson, M.D.; Walsh, M.K.; Feldman, D.R.; Olencki, T.; Picus, J.; Small, E.J.; et al. Cabozantinib Versus Sunitinib As Initial Targeted Therapy for Patients With Metastatic Renal Cell Carcinoma of Poor or Intermediate Risk: The Alliance A031203 CABOSUN Trial. J. Clin. Oncol. 2017, 35, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Tannir, N.M.; Mainwaring, P.N.; Rini, B.I.; Hammers, H.J.; Donskov, F.; Roth, B.J.; Peltola, K.; et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): Final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2016, 17, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Wirth, L.J.; Tahara, M.; Robinson, B.; Francis, S.; Brose, M.S.; Habra, M.A.; Newbold, K.; Kiyota, N.; Dutcus, C.E.; Mathias, E.; et al. Treatment-emergent hypertension and efficacy in the phase 3 Study of (E7080) lenvatinib in differentiated cancer of the thyroid (SELECT): Treatment-Emergent HTN With Lenvatinib in RR-DTC. Cancer 2018, 124, 2365–2372. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Glen, H.; Michaelson, M.D.; Molina, A.; Eisen, T.; Jassem, J.; Zolnierek, J.; Maroto, J.P.; Mellado, B.; et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015, 16, 1473–1482. [Google Scholar] [CrossRef] [Green Version]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Ulivi, P.; Arienti, C.; Amadori, D.; Fabbri, F.; Carloni, S.; Tesei, A.; Vannini, I.; Silvestrini, R.; Zoli, W. Role of RAF/MEK/ERK pathway, p-STAT-3 and Mcl-1 in sorafenib activity in human pancreatic cancer cell lines. J. Cell. Physiol. 2009, 220, 214–221. [Google Scholar] [CrossRef]
- Rahmani, M.; Davis, E.M.; Crabtree, T.R.; Habibi, J.R.; Nguyen, T.K.; Dent, P.; Grant, S. The Kinase Inhibitor Sorafenib Induces Cell Death through a Process Involving Induction of Endoplasmic Reticulum Stress. Mol. Cell. Biol. 2007, 27, 5499–5513. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in Advanced Clear-Cell Renal-Cell Carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schütz, G.; Thierauch, K.-H.; Zopf, D. Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Battaglin, F.; Wang, J.; Lo, J.H.; Soni, S.; Zhang, W.; Lenz, H.-J. Molecular insight of regorafenib treatment for colorectal cancer. Cancer Treat. Rev. 2019, 81, 101912. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Cutsem, E.V.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Reck, M.; Kaiser, R.; Mellemgaard, A.; Douillard, J.-Y.; Orlov, S.; Krzakowski, M.; von Pawel, J.; Gottfried, M.; Bondarenko, I.; Liao, M.; et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): A phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014, 15, 143–155. [Google Scholar] [CrossRef]
- Li, J.; Qin, S.; Xu, J.; Guo, W.; Xiong, J.; Bai, Y.; Sun, G.; Yang, Y.; Wang, L.; Xu, N.; et al. Apatinib for Chemotherapy-Refractory Advanced Metastatic Gastric Cancer: Results From a Randomized, Placebo-Controlled, Parallel-Arm, Phase II Trial. J. Clin. Oncol. 2013, 31, 3219–3225. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.-K.; Kang, W.K.; Di Bartolomeo, M.; Chau, I.; Yoon, H.H.; Cascinu, S.; Ryu, M.-H.; Kim, J.G.; Lee, K.-W.; Oh, S.C.; et al. Randomized phase III ANGEL study of rivoceranib (apatinib) + best supportive care (BSC) vs placebo + BSC in patients with advanced/metastatic gastric cancer who failed ≥2 prior chemotherapy regimens. Ann. Oncol. 2019, 30, v877–v878. [Google Scholar] [CrossRef]
- Zhang, Y.; Zou, J.-Y.; Wang, Z.; Wang, Y. Fruquintinib: A novel antivascular endothelial growth factor receptor tyrosine kinase inhibitor for the treatment of metastatic colorectal cancer. Cancer Manag. Res. 2019, 11, 7787–7803. [Google Scholar] [CrossRef] [Green Version]
- Shen, G.; Zheng, F.; Ren, D.; Du, F.; Dong, Q.; Wang, Z.; Zhao, F.; Ahmad, R.; Zhao, J. Anlotinib: A novel multi-targeting tyrosine kinase inhibitor in clinical development. J. Hematol. Oncol.J Hematol Oncol 2018, 11, 120. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.A.; McKee, A.E.; Kibbe, W.A.; Villaflor, V.M. Complexity of Delivering Precision Medicine: Opportunities and Challenges. Am. Soc. Clin. Oncol. Educ. Book 2018, 998–1007. [Google Scholar] [CrossRef]
- Offin, M.; Liu, D.; Drilon, A. Tumor-Agnostic Drug Development. Am. Soc. Clin. Oncol. Educ. Book 2018, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Lavacchi, D.; Roviello, G.; D’Angelo, A. Tumor-Agnostic Treatment for Cancer: When How is Better than Where. Clin. Drug Investig. 2020, 40, 519–527. [Google Scholar] [CrossRef] [PubMed]
- McNeil, C. NCI-MATCH Launch Highlights New Trial Design in Precision-Medicine Era. J. Natl. Cancer Inst. 2015, 107, djv193. [Google Scholar] [CrossRef] [PubMed]
- Park, J.J.H.; Hsu, G.; Siden, E.G.; Thorlund, K.; Mills, E.J. An overview of precision oncology basket and umbrella trials for clinicians. CA. Cancer J. Clin. 2020, 70, 125–137. [Google Scholar] [CrossRef]
- Horak, P.; Fröhling, S.; Glimm, H. Integrating next-generation sequencing into clinical oncology: Strategies, promises and pitfalls. ESMO Open 2016, 1, e000094. [Google Scholar] [CrossRef] [Green Version]
- Kamps, R.; Brandão, R.; Bosch, B.; Paulussen, A.; Xanthoulea, S.; Blok, M.; Romano, A. Next-Generation Sequencing in Oncology: Genetic Diagnosis, Risk Prediction and Cancer Classification. Int. J. Mol. Sci. 2017, 18, 308. [Google Scholar] [CrossRef]
- Blueprint Medicines Announces the Achievement of Key Portfolio Milestones. Available online: https://bit.ly/2R3cW9R (accessed on 15 July 2020).
- Dirkx, A.E.M.; Egbrink, M.G.O.; Castermans, K.; Schaft, D.W.J.; Thijssen, V.L.J.L.; Dings, R.P.M.; Kwee, L.; Mayo, K.H.; Wagstaff, J.; Steege, J.C.A.B.; et al. Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte-endothelium interactions and infiltration in tumors. FASEB J. 2006, 20, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, S.; Sho, M.; Yamato, I.; Yoshiji, H.; Wakatsuki, K.; Nishiwada, S.; Yagita, H.; Nakajima, Y. Simultaneous blockade of programmed death 1 and vascular endothelial growth factor receptor 2 (VEGFR2) induces synergistic anti-tumour effect in vivo: Synergistic anti-tumour effect by dual blockade of PD-1 and VEGFR2. Clin. Exp. Immunol. 2013, 172, 500–506. [Google Scholar] [CrossRef]
- Amin, A.; Plimack, E.R.; Infante, J.R.; Ernstoff, M.S.; Rini, B.I.; McDermott, D.F.; Knox, J.J.; Pal, S.K.; Voss, M.H.; Sharma, P.; et al. Nivolumab (anti-PD-1; BMS-936558, ONO-4538) in combination with sunitinib or pazopanib in patients (pts) with metastatic renal cell carcinoma (mRCC). J. Clin. Oncol. 2014, 32, 5010. [Google Scholar] [CrossRef]
- Chowdhury, S.; McDermott, D.F.; Voss, M.H.; Hawkins, R.E.; Aimone, P.; Voi, M.; Isabelle, N.; Wu, Y.; Infante, J.R. A phase I/II study to assess the safety and efficacy of pazopanib (PAZ) and pembrolizumab (PEM) in patients (pts) with advanced renal cell carcinoma (aRCC). J. Clin. Oncol. 2017, 35, 4506. [Google Scholar] [CrossRef]
- Rini, B.I.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.L.; et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): A multicentre, open-label, phase 3, randomised controlled trial. Lancet 2019, 393, 2404–2415. [Google Scholar] [CrossRef]
- Jänne, P.A.; Wang, X.; Socinski, M.A.; Crawford, J.; Stinchcombe, T.E.; Gu, L.; Capelletti, M.; Edelman, M.J.; Villalona-Calero, M.A.; Kratzke, R.; et al. Randomized Phase II Trial of Erlotinib Alone or With Carboplatin and Paclitaxel in Patients Who Were Never or Light Former Smokers With Advanced Lung Adenocarcinoma: CALGB 30406 Trial. J. Clin. Oncol. 2012, 30, 2063–2069. [Google Scholar] [CrossRef]
- D’Cunha, R.; Bae, S.; Murry, D.J.; An, G. TKI combination therapy: Strategy to enhance dasatinib uptake by inhibiting Pgp- and BCRP-mediated efflux: TKI combination therapy overcomes efflux transporter-mediated MDR. Biopharm. Drug Dispos. 2016, 37, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Rotow, J.K.; Costa, D.B.; Paweletz, C.P.; Awad, M.M.; Marcoux, P.; Rangachari, D.; Barbie, D.A.; Sands, J.; Cheng, M.L.; Johnson, B.E.; et al. Concurrent osimertinib plus gefitinib for first-line treatment of EGFR-mutated non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2020, 38, 9507. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Yang, J.C.-H.; Yu, H.; Kim, S.-W.; Saka, H.; Horn, L.; Goto, K.; Ohe, Y.; Mann, H.; Thress, K.S.; et al. TATTON: A multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann. Oncol. 2020, 31, 507–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendel, D.B.; Laird, A.D.; Xin, X.; Louie, S.G.; Christensen, J.G.; Li, G.; Schreck, R.E.; Abrams, T.J.; Ngai, T.J.; Lee, L.B.; et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin. Cancer Res. 2003, 9, 327–337. [Google Scholar]
- Paez, J.G. EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [Green Version]
- Bose, R.; Kavuri, S.M.; Searleman, A.C.; Shen, W.; Shen, D.; Koboldt, D.C.; Monsey, J.; Goel, N.; Aronson, A.B.; Li, S.; et al. Activating HER2 Mutations in HER2 Gene Amplification Negative Breast Cancer. Cancer Discov. 2013, 3, 224–237. [Google Scholar] [CrossRef] [Green Version]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Choi, Y.L.; Soda, M.; Yamashita, Y.; Ueno, T.; Takashima, J.; Nakajima, T.; Yatabe, Y.; Takeuchi, K.; Hamada, T.; Haruta, H.; et al. EML4-ALK Mutations in Lung Cancer That Confer Resistance to ALK Inhibitors. N. Engl. J. Med. 2010, 363, 1734–1739. [Google Scholar] [CrossRef]
- European Medicines Agency (EMA). Assessment Report of Tivozanib. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/fotivda (accessed on 15 June 2020).
- Gross-Goupil, M.; Françlois, L.; Quivy, A.; Ravaud, A. Axitinib: A Review of its Safety and Efficacy in the Treatment of Adults with Advanced Renal Cell Carcinoma. Clin. Med. Insights Oncol. 2013, 7, CMO.S10594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu-Lowe, D.D.; Zou, H.Y.; Grazzini, M.L.; Hallin, M.E.; Wickman, G.R.; Amundson, K.; Chen, J.H.; Rewolinski, D.A.; Yamazaki, S.; Wu, E.Y.; et al. Nonclinical Antiangiogenesis and Antitumor Activities of Axitinib (AG-013736), an Oral, Potent, and Selective Inhibitor of Vascular Endothelial Growth Factor Receptor Tyrosine Kinases 1, 2, 3. Clin. Cancer Res. 2008, 14, 7272–7283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a Novel MET and VEGFR2 Inhibitor, Simultaneously Suppresses Metastasis, Angiogenesis, and Tumor Growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, W.-K.; Sennino, B.; Williamson, C.W.; Falcon, B.; Hashizume, H.; Yao, L.-C.; Aftab, D.T.; McDonald, D.M. VEGF and c-Met Blockade Amplify Angiogenesis Inhibition in Pancreatic Islet Cancer. Cancer Res. 2011, 71, 4758–4768. [Google Scholar] [CrossRef] [Green Version]
- Cabanillas, M.E.; Habra, M.A. Lenvatinib: Role in thyroid cancer and other solid tumors. Cancer Treat. Rev. 2016, 42, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Fox, E.; Aungst, S. Pharmacology/toxicology NDA review and evaluation for LENVIMA (Lenvatinib 2014). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/206947Orig1s000PharmR.pdf (accessed on 9 November 2020).
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef]
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. BIBF 1120: Triple Angiokinase Inhibitor with Sustained Receptor Blockade and Good Antitumor Efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, H.; Roman, S.; Thumar, J.; Sosa, J.A. Vandetanib (ZD6474) in the Treatment of Medullary Thyroid Cancer. Clin. Med. Insights Oncol. 2011, 5, CMO.S6197. [Google Scholar] [CrossRef]
- Frampton, J.E. Vandetanib: In Medullary Thyroid Cancer. Drugs 2012, 72, 1423–1436. [Google Scholar] [CrossRef]

| Ligand | Receptors and Representative Examples | Most Representative Functions | Cellular and Tissue Distribution |
|---|---|---|---|
| EGF (Epidermal Growth Factor) | EGFR (HER1), HER2, HER3, HER4 | Stimulates survival, growth, proliferation, and differentiation of various cell lines |
|
| Insulin | Insulin receptor | Regulates carbohydrate metabolism and protein synthesis |
|
| IGF (Insulin-like Growth Factor) | IGF-1R | Stimulates cell growth and survival. Regulation of growth in young people and has anabolic effects in adults. |
|
| NGF (Nerve Growth Factor) | NTRK1 (TrkA, TrkB, TrkC) | Stimulates neural growth and differentiation |
|
| PDGF (Platelet-Derived Growth Factor) | PDGFRα, PDGFRβ | It stimulates survival, growth, proliferation and migration of various cellular subtypes. |
|
| GM-CSF (Granulocyte Macrophage-colony stimulating factor) | GM-CSFR or GMRα/β | It stimulates the proliferation of monocytes and their differentiation to macrophages. |
|
| FGF (Fibroblast Growth Factor) | FGFR1, FGFR2, FGFR3, FGFR4 | It stimulates the proliferation of various cell types and inhibits the differentiation of other types. |
|
| VEGF (Vascular Endotelial Growth Factor) | VEGFR-1 (FLT-1), VEGFR-2 (KDR), VEGFR-3 (FLT-4) | Stimulates angiogenesis. |
|
| ALK (Anaplastic Lymphoma Kinase) | ALK (CD246) | Involved in the development and function of the central nervous system. |
|
| GDNF (Glial cell-line derived neurotrophic factor) | GFRα1, GFRα2, GFRα3, GFRα4 | Regulation of survival (dopaminergic neurons), growth of neurites, cell differentiation and migration. |
|
| SCF (Mast/Stem cell growth factor) | KIT (CD117) | It intervenes in processes such as the survival of melanocytes, hematopoiesis and gametogenesis. |
|
| Ephrin | Eph receptors | Guides cell and axon migration; angiogenesis. |
|
| Gas6, Protein S. | TAM family (Tyro3, MerTK, Axl) | Cell growth, survival, differentiation. Regulation of systemic immunity |
|
| Name (Code) Trade Name | Targets | Approved Clinical Indications or Clinical Trial Study |
|---|---|---|
| Gefitinib (ZD1389) Iressa | EGFR | NSCLC |
| Erlotinib (OSI-774) Tarceva | EGFR | NSCLC |
| Afatinib (BIBW2992) Tovok | Erb1/2/4 | NSCLC |
| Osimertinib (AZD-9291) Tagrisso | EGFR | NSCLC |
| Dacomitinib (PF299804) Visimpro | Pan-HER | NSCLC |
| Saracatinib (AZD0530) | EGFR, Src | Phase II trial (NCT00752206, NCT00607594, NCT01267266) for osteosarcoma, gastric, prostate and other solid tumors |
| Lapatinib (GW572016) Tykerb | EGFR/HER2 | Breast cancer |
| Neratinib (HKI-272) Nerlynx | HER2 | Breast cancer |
| Icotinib | EGFR | NSCLC (China) |
| Poziotinib (HM781-36B) | EGFR/HER2/HER4 | Phase II trial (NCT02979821) for NSCLC |
| Tarloxotinib (TH4000) | EGFR/HER2 | Phase II trial (NCT03805841) for NSCLC and solid tumors harboring ERBB/NRG1 gene fusions |
| Lazertinib (YH25448) | EGFR | Phase III (NCT04248829) trial for NSCLC |
| AZD3759 | EGFR | Phase II/III (NCT03653546) trial for NSCLC. |
| Pyrotinib (SHR-1258) | EGFR/HER2 | Phase I trial (NCT02500199) for HER2 positive solid tumors. |
| Avitinib (AC0010MA) | EGFR | Phase I/II (NCT02330367) clinical trial for NSCLC |
| Sapitinib (AZD8931) | EGFR/HER2/HER3 | Phase I/II trial (NCT01862003) for colorectal cancer. |
| Rociletinib (CO-1686) | EGFR | Phase III (NCT02322281) for NSCLC |
| TAS6417 | EGFR/HER2/HER3 | Phase I/IIa trial (NCT04036682) for NSCLC |
| Varlitinib (ASLAN001) | EGFR/HER2/HER4 | Phase II/III (NCT03093870, NCT03130790) for billiard tract cancer, gastric cancer and hepatocarcinoma (Phase Ib; NCT03499626) |
| Olmutinib (HM61713) | EGFR | Phase Ib (NCT04510415) and phase II (NCT03228277) clinical trials for NSCLC |
| Nazartinib (EGF816) | EGFR | Phase I/II (NCT02108964) clinical trial for NSCLC |
| Mavelertinib (PF-06747775) | EGFR | Phase II trial (NCT02349633) for NSCLC |
| Naquonitinib (ASP8273) | EGFR | Phase I (NCT02113813) trial for NSCLC |
| Ibrutinib (PCI-32765) Imbruvica | EGFR, BTK | Phase I/II (NCT02321540) trial for NSCLC, MCL, CLL. |
| EAI001 | EGFR | Preclinical |
| EAI045 | EGFR | Preclinical |
| Name (Code) Trade Name | Targets | Approved Clinical Indications or Clinical Trial Study |
|---|---|---|
| Crizotinib (PF 2341066) Xalkori | ALK, MET, ROS1 | NSCLC |
| Ceritinib (LDK378) Zykadia | ALK, IGF-1R, ROS1 | NSCLC |
| Alectinib (CH5424802)Alecensa | ALK, RET | NSCLC |
| Brigatinib (AP 26113) Alunbrig | ALK, ROS1, EGFR | NSCLC |
| Lorlatinib (PF-06463922) Lorbrena | ALK, ROS1 | NSCLC |
| Ensartinib (X-396) | ALK, MET, Axl, ABL, EPHA2 LTK, ROS1, SLK | Phase II (NCT01625234) and phase III (NCT02767804) trials for NSCLC |
| Merestinib (LY2801653) | NTRK, MET, ROS1, FLT3, Axl | Phase II trial (NCT02920996) for NSCLC and solid tumors with NTRK fusion proteins. |
| Belizartinib (TSR-011) | ALK, TRKA/B/C | Phase I/II trial for solid tumors and lymphomas with NTRK fusion proteins |
| Entrectinib (RXDX-101) Rozlytrek | TRKA/B/C, ROS1 | Solid tumors with NTRK fusion proteins, ROS1-positive NSCLC |
| Larotrectinib (LOXO-101) Vitrakvi | TRKA/B/C | Solid tumors with NTRK fusion proteins |
| Repotrectinib (TPX-0005) | ROS1, TRKA/B/C, ALK | Phase I/II trial (NCT03093116) for solid tumors with NTRK fusion proteins and ROS1-positive NSCLC |
| Taletrectinib (DS-6051b) | ROS1, TRKA/B/C | Phase I trial (NCT02675491) for solid tumors with NTRK and ROS1 fusion proteins. |
| Selitrectinib (BAY2731954) | TRKA/B/C | Phase I/II trial (NCT03215511) for solid tumors with NTRK fusion proteins. |
| BMS-754807 | TRKA/B, Insulin receptor, MET | Phase II trial (NCT01225172) for breast cancer. |
| Name (Code) Trade Name | Targets | Approved Clinical Indications or Clinical Trial Study |
|---|---|---|
| Erdafitinib (JNJ-42756493) Balversa | FGFR1/2/3/4 | Urothelial bladder cancer |
| Nintedanib (BIBF-1120) Vargatef | FGFR1/2/3 | Idiopathic pulmonary fibrosis, NSCLC |
| Pemigatinib (INCB054828) Pemazyre | FGFR1/2/3 | Cholangiocarcinoma |
| Rogaratinib (BAY 1163877) | FGFR1/2/3 | Under development in SQCLC (NCT03762122), breast cancer (NCT04483505), urothelial carcinoma (NCT03473756), sarcoma GIST (NCT04595747), and gastric cancer (NCT04077255) |
| Vofatamab (B-701) | FGFR 3 | Under development in urothelial carcinoma (NCT03123055, NCT02401542) |
| Infigratinib (BGJ398) | FGFR1/2/3 | Under development in urothelial carcinoma (NCT04197986), breast cancer (NCT04504331), cholangiocarcinoma (NCT03773302), and glioblastoma (NCT04424966) |
| Derazantinib (ARQ 087) | FGFR1/2/3 | Under development in urothelial carcinoma (NCT04045613), gastric cancer (NCT04604132) and cholangiocarcinoma (NCT03230318) |
| AZD4547 | FGFR1/2/3 | Under development in NSCLC (NCT01824901), breast cancer (NCT01202591), gliomas (NCT02824133), urothelial carcinomas (NCT02546661) and gastro-esophageal cancer (NCT01457846) |
| Debio 1347 | FGFR1/2/3 | Under development in breast cancer (NCT03344536) and other solid tumors (NCT03834220) |
| Name (Code) Trade Name | Targets | Approved Clinical Indications or Clinical Trial Study |
|---|---|---|
| Imatinib (STI-571) Glivec | PDGFR, c-Kit, v-Abl | GIST, CML (Chronic Myeloid Leukemia), ALL (Acute lymphocytic leukemia), dermatofibrosarcoma protuberans, myelodisplasic síndrome, leukemias. |
| Avapritinib (BLU-285) Ayvakit | PDGFR, c-Kit | GIST |
| Ripretinib (DCC-2618) Qinlock | c-Kit, PDGFR | GIST |
| Amuvatinib (MP-470) | c-Kit, PDGFR, Flt3 | Under development in NSCLC (NCT01357395) and other solid tumors (NCT00894894) |
| Dasatinib (BMS-354825) Sprycel | c-Kit, Abl, Src | CML |
| Name (Code) Trade Name | Targets | Approved Clinical Indications or Clinical Trial Study |
|---|---|---|
| Capmatinib (INC280) Tabrecta | MET | NSCLC, under development in solid tumors harboring MET mutations. |
| Tepotinib (EMD 1214063) | MET | Under development in NSCLC (NCT02864992) and colorectal cancer (NCT04515394) |
| Glumetinib (SCC244) | MET | Under development in NSCLC (NCT04270591) and other solid tumors (NCT03457532) |
| Savolitinib (AZD6094) | MET | Under development in solid tumors harboring MET mutations. |
| AMG 337 | MET | Under development in clear cell sarcoma (NCT03132155) and other solid tumors (NCT01253707) |
| Bozitinib (PLB-1001) | MET | Under development in gliomas (NCT02978261), NSCLC (NCT04258033), renal cell carcinoma and hepatocellular carcinoma (NCT03655613), and other solid tumors (NCT03175224) |
| Merestinib (LY2801653) | MET, AXL, DDR1, DDR2 | Under development in NSCLC (NCT02920996), biliary tract cancer (NCT02711553) and other solid tumors (NCT03027284) |
| Tivantinib (ARQ197) | MET | Under development in multiple solid tumors harboring MET mutations. |
| Foretinib (GSK1363089) | MET, Tie-2, VEGFR3 | Under development in several solid tumors. |
| Crenolanib (CP-868596) | PDGFR, FLT3 | Under development in GIST (NCT02847429), glioma (NCT01393912), and esophagogastric carcinoma (NCT03193918) |
| Name (Code) Trade Name | Targets | Approved Clinical Indications or Clinical Trial Study |
|---|---|---|
| Selpercatinib (LOXO-292) Retevmo | RET | Thyroid cancer and NSCLC |
| Pralsetinib (BLU-667) Gavreto | RET | NSCLC |
| BOS172738 | RET | Under development in solid RET-mutated tumors (NCT03780517) |
| TAS0953/HM06 | RET | Under development in solid RET-mutated tumors |
| TPX-0046 | RET, SRC | Under development in solid RET-mutated tumors (NCT04161391) |
| Name (Code) Trade Name | Targets | Approved Clinical Indications or Clinical Trial Study |
|---|---|---|
| Sunitinib (SU011248) Sutent | VEGFR1/2/3, FLT3, Kit, PDFGRβ | GIST, RCC, pNET |
| Pazopanib (GW786034) Votrient | VEGFR1/2/3, PDGFRα/β, FGFR cKIT. | RCC, soft-tissue sarcoma. |
| Tivozanib (AV-951) Fotivda | VEGFR1/2/3 | RCC |
| Axitinib (AG013736) Inlyta | VEGFR1/2/3, c-KIT | RCC |
| Cabozantinib (BMS-907351) Cabometyx, cometriq | c-MET, VEGFR2, RET, TRKA, TRKB and Axl. | RCC, HCC, MTC |
| Lenvatinib (E7080) Lenvima | VEGFR1/2/3, FGFR1/2/3/4, PDGFRα, c-KIT, RET | RCC, DTC, HCC |
| Sorafenib (BAY43-9006), Nexavar | VEGFR1/2/3, PDGFRβ, Flt-3, c-KIT, RET, Raf | HCC, DTC, RCC |
| Regorafenib (BAY73-4506) Stivarga | VEGFR1/2/3, Raf, TIE-2, PDGFR, RET, CSF-1 | CRC, GIST, HCC |
| Nintedanib (BIBF1120) Vargatef | VEGFR1/2/3, PDGFRα/β), FGFR1/2/3, TRK, Flt-3 RET | NSCLC |
| Vandetanib (ZD6474) Caprelsa | VEGFR, EGFR and RET. | MTC |
| Apatinib (YN968D1) | VEGFR2 | Gastric cancer (China) |
| Fruquintinib (HMPL-013) | VEGFR1/2/3 | CRC (China) |
| Anlotinib (AL3818) | VEGFR2/3, c-Kit | NSCLC (China) |
| Motesanib (AMG-706) | VEGFR1/2/3, c-Kit, RET, PDGFR | Under development in several solid tumors |
| Linifanib (ABT-869) | VEGFR1/2, CSF-1R, FLT3, c-Kit | Under development in several solid tumors |
| Glesatinib (MGCD-265) | VEGFR1/2/3, MET, RON, Tie-2 | Under development in NSCLC (NCT02544633) and other solid tumors |
| Cediranib (AZD2171) | VEGFR1/2/3, c-Kit, PDGFRβ, FGFR1 | Under development in several solid tumors |
| Dovitinib (CHIR-258) | FGFR1/2/3, VEGF, c-Kit, FLT3 | Multiple clinical trials mainly in renal cell carcinoma (phase III), breast cancer, hepatocellular cancer, endometrial cancer and GIST |
| Tivozanib (AV-951) | VEGFR1/2/3, EphB2, PDGFR | Under development in several solid tumors |
| Vatalanib (PTK787) | VEGFR1/2/3, PDGFR, c-Kit | Under development in several solid tumors |
| Brivanib (BMS-540215) | VEGFR1/2, FGFR1 | Under development in several solid tumors |
| Apatinib (YN968D1) | VEGFR2, RET | Under development in several solid tumors |
| Ponatinib (AP24534) | VEGFR2, PDGFR, FGFR1 | Under development in several solid tumors |
| LY2874455 | VEGFR2, FGFR1/2/4 | Under development in solid tumors (NCT01212107) |
| Golvatinib (E7050) | VEGFR2, MET | Under development in several solid tumors |
| Telatinib (BAY 57-9352) | VEGFR2/3, c-Kit, PDGFR | Under development in gastric cancer (NCT00952497, NCT03817411) and other solid tumors (NCT03175497) |
| Lucitanib (E-3810) | VEGFR1/2/3, FGFR1/2 | Under development in several solid tumors |
| Sulfatinib | VEGFR1/2/3, FGFR1, CSF1R | Under development in neuroendocrine tumors (NCT04579679), thyroid cancer (NCT04524884), biliary tract carcinoma (NCT03873532) and other solid tumors (NCT04579757, NCT02549937) |
| Name (Code) Trade Name | Targets | Approved Clinical Indications or Clinical Trial Study |
|---|---|---|
| Bosutinib (SKI-606) | SRC, STAT3 | Under development in breast cancer (NCT03854903) and other solid tumors (NCT03023319) |
| Pexmetinib (ARRY-614) | MAPK, Tie-2 | Under development in solid tumors (NCT04074967) |
| Dubermatinib (TP-0903) | Axl | Under development in solid tumors (NCT02729298) |
| Bemcentinib (BGB324) | Axl | Under development in breast cancer (NCT03184558), pancreatic cancer (NCT03649321), and SNCLC (NCT03184571) |
| Sitravatinib (MGCD516) | Axl, VEGFR3 | Under development in several solid tumors |
| Ningetinib (C31H29FN4O5) | Axl, c-MET, VEGFR2 | Under development in NSCLC (NCT03758287) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esteban-Villarrubia, J.; Soto-Castillo, J.J.; Pozas, J.; San Román-Gil, M.; Orejana-Martín, I.; Torres-Jiménez, J.; Carrato, A.; Alonso-Gordoa, T.; Molina-Cerrillo, J. Tyrosine Kinase Receptors in Oncology. Int. J. Mol. Sci. 2020, 21, 8529. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228529
Esteban-Villarrubia J, Soto-Castillo JJ, Pozas J, San Román-Gil M, Orejana-Martín I, Torres-Jiménez J, Carrato A, Alonso-Gordoa T, Molina-Cerrillo J. Tyrosine Kinase Receptors in Oncology. International Journal of Molecular Sciences. 2020; 21(22):8529. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228529
Chicago/Turabian StyleEsteban-Villarrubia, Jorge, Juan José Soto-Castillo, Javier Pozas, María San Román-Gil, Inmaculada Orejana-Martín, Javier Torres-Jiménez, Alfredo Carrato, Teresa Alonso-Gordoa, and Javier Molina-Cerrillo. 2020. "Tyrosine Kinase Receptors in Oncology" International Journal of Molecular Sciences 21, no. 22: 8529. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228529