Assessing the Thiamine Diphosphate Dependent Pyruvate Dehydrogenase E1 Subunit for Carboligation Reactions with Aliphatic Ketoacids



Abstract
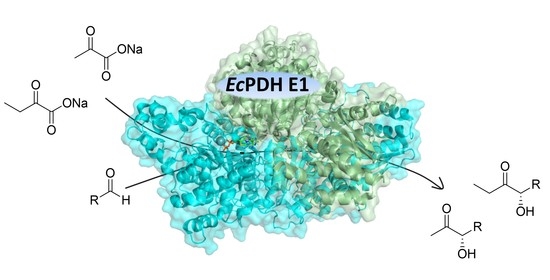
:
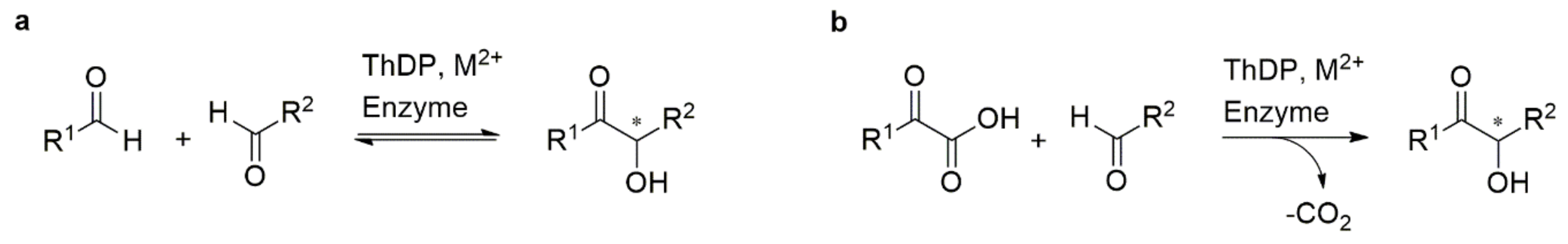
1. Introduction
2. Results and Discussion
2.1. Characterisation of WT EcPDH E1
2.1.1. Expression and Purification
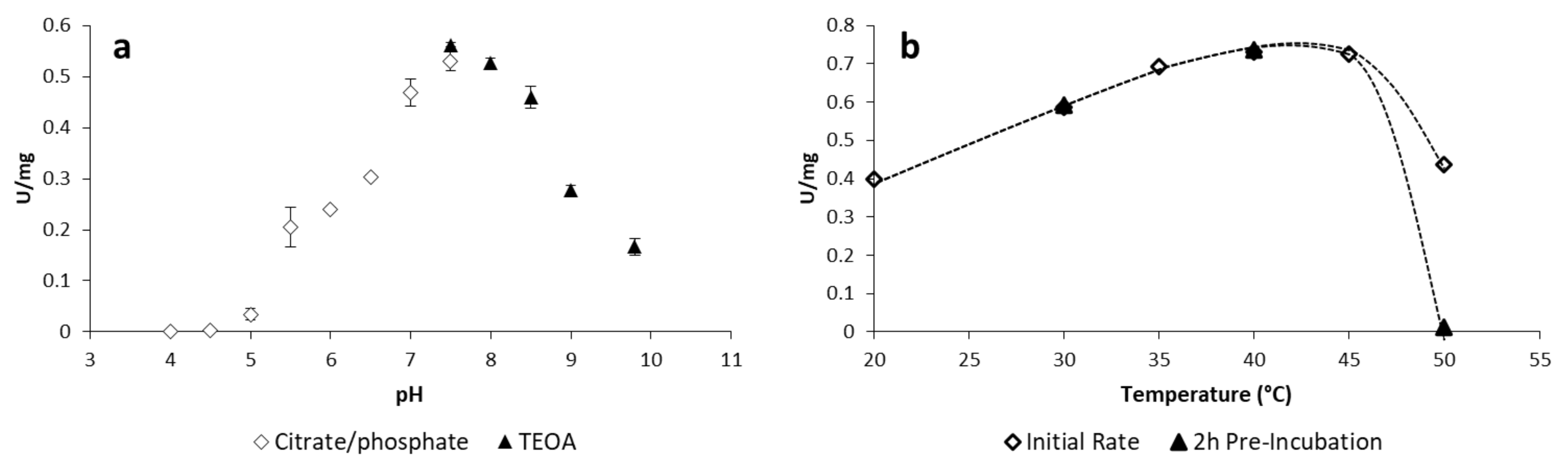
2.1.2. Optimisation of Reaction Conditions
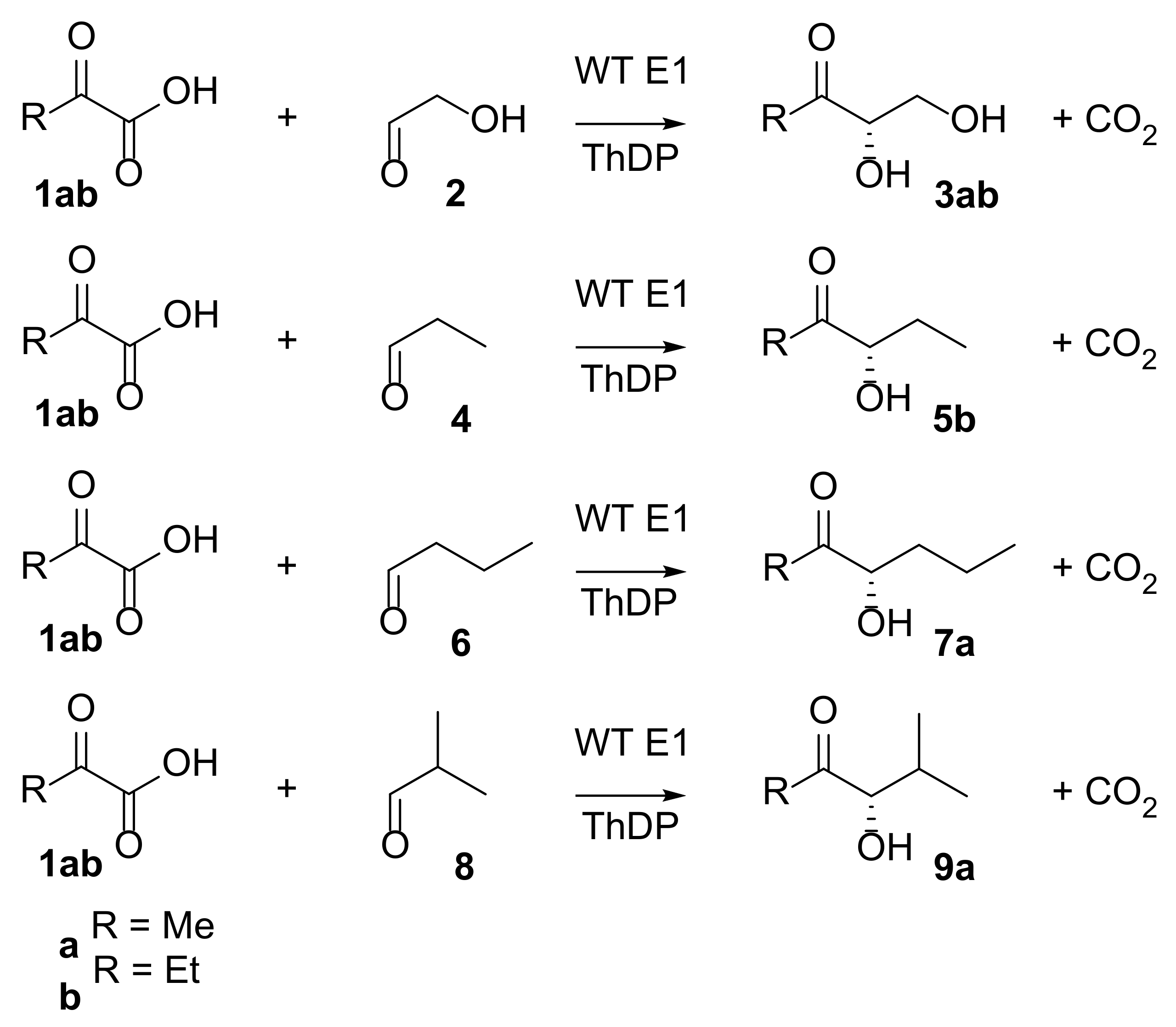
2.1.3. Preparative Scale Reactions
2.1.4. Conversion of (R)- and (S)-Configured α-hydroxyaldehyde Acceptor Substrates
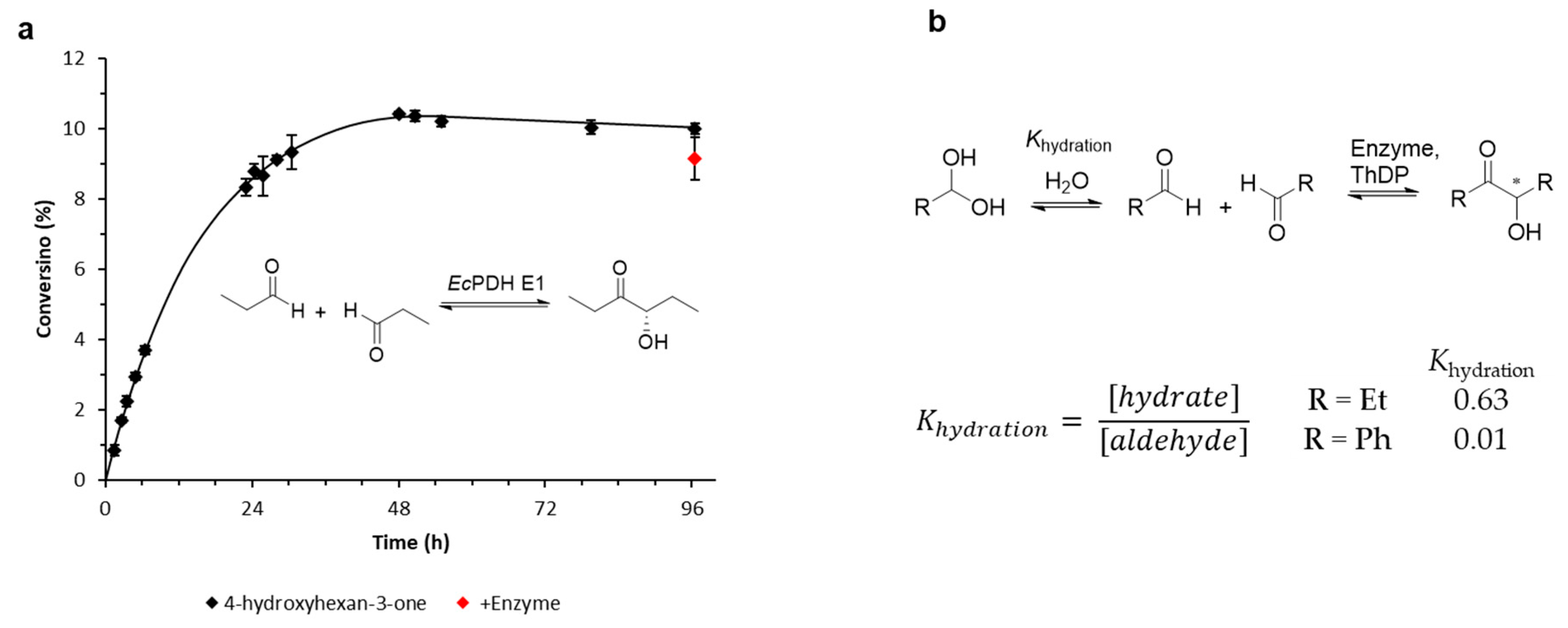
2.1.5. Thermodynamically Controlled One-Substrate Reactions
2.1.6. Determination of Kinetic Parameters
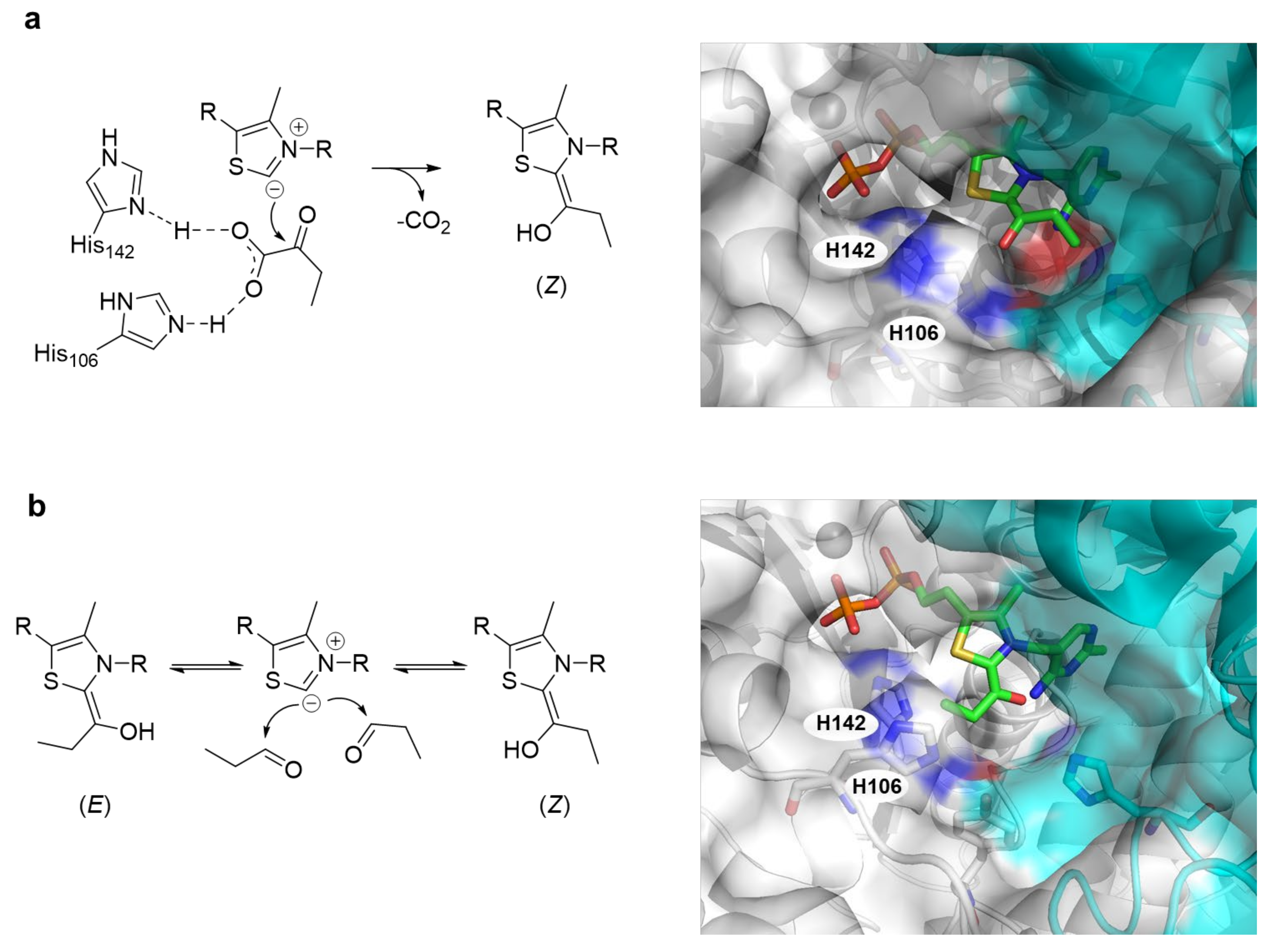
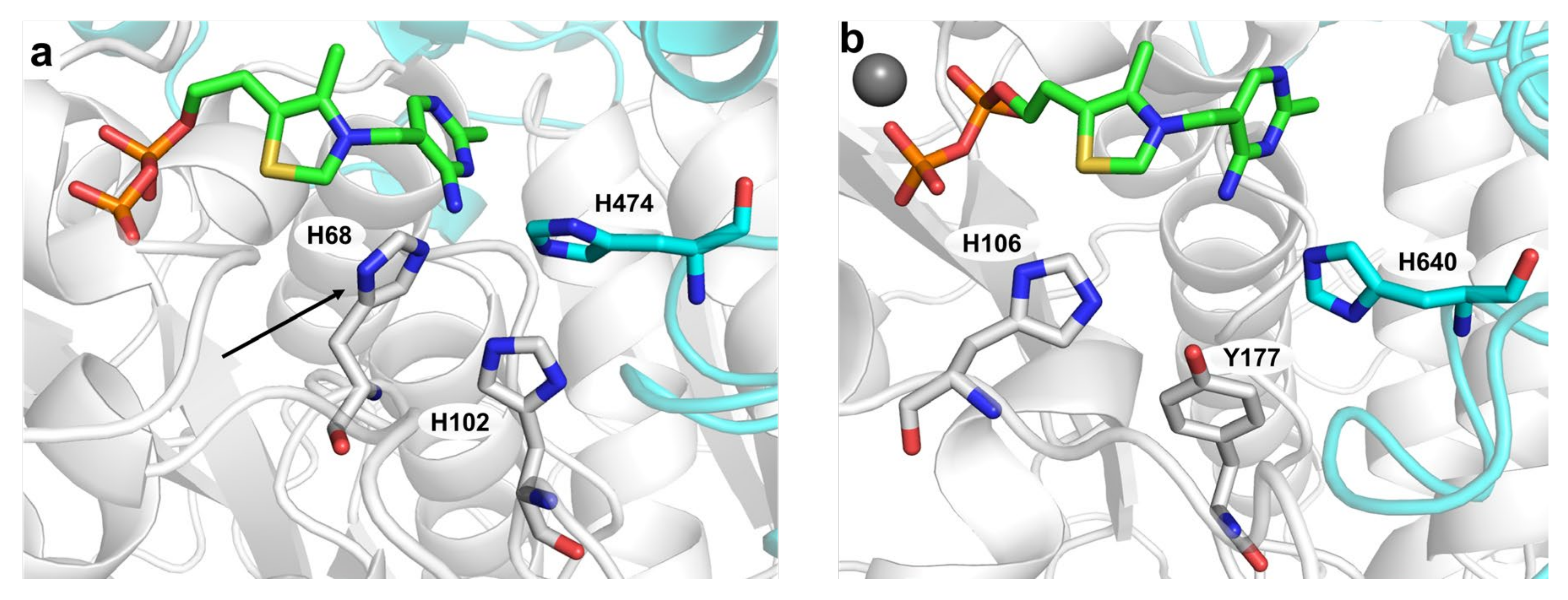
2.2. Mutagenesis
3. Materials and Methods
3.1. pH Depend. Activity
3.2. Temperature Dependent Activity
3.3. Preparative Scale Reactions
3.4. General RP-HPLC Method
3.5. Chiral GC Methods
3.6. Michaelis-Menten Analysis
3.7. Time Course for the Conversion of Racemic DL-Glyceraldehyde with Pyruvate
3.8. Homology Modelling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ThDP | Thiamine diphosphate |
| TK | Transketolase |
| EcPDH E1 | Pyruvate dehydrogenase E1 subunit from Escherichia coli |
| PDHc | Pyruvate dehydrogenase complex |
| DXS | 1-deoxy-D-xylulose-5-phosphate synthase |
| MenD | 2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexene-1-carboxylate synthase |
| BAL | Benzaldehyde lyase |
| DCPIP | 2,6-dichloroindophenol |
| HPA | Hydroxypyruvate |
| TEOA | Triethanolamine |
| TEED | Thiamine Enzyme Engineering Database |
References
- Pohl, M.; Wechsler, C.; Müller, M. Acyloin, benzoin, and related reactions. In Science of Synthesis: Biocatalysis in Organic Synthesis 2; Faber, K., Fessner, W.-D., Turner, N.J., Eds.; Georg Thieme Verlag: Stuttgart, Germany, 2014; pp. 93–127. ISBN 9783131741714. [Google Scholar]
- Kluger, R.; Tittmann, K. Thiamin Diphosphate Catalysis: Enzymic and Nonenzymic Covalent Intermediates. Chem. Rev. 2008, 108, 1797–1833. [Google Scholar] [CrossRef] [PubMed]
- Tittmann, K.; Golbik, R.; Uhlemann, K.; Khailova, L.; Schneider, G.; Patel, M.; Jordan, F.; Chipman, D.M.; Duggleby, R.G.; Hübner, G. NMR Analysis of Covalent Intermediates in Thiamin Diphosphate Enzymes. Biochemistry 2003, 42, 7885–7891. [Google Scholar] [CrossRef] [PubMed]
- Breslow, R. On the Mechanism of Thiamine Action. IV.1Evidence from Studies on Model Systems. J. Am. Chem. Soc. 1958, 80, 3719–3726. [Google Scholar] [CrossRef]
- Marsden, S.R.; Gjonaj, L.; Eustace, S.J.; Hanefeld, U. Separating Thermodynamics from Kinetics-A New Understanding of the Transketolase Reaction. ChemCatChem 2017, 9, 1808–1814. [Google Scholar] [CrossRef] [Green Version]
- Marsden, S.R.; Mestrom, L.; McMillan, D.G.G.; Hanefeld, U. Thermodynamically and Kinetically Controlled Reactions in Biocatalysis – from Concepts to Perspectives. ChemCatChem 2019, 12, 426–437. [Google Scholar] [CrossRef]
- Wohlgemuth, R. C2-Ketol elongation by transketolase-catalyzed asymmetric synthesis. J. Mol. Catal. B Enzym. 2009, 61, 23–29. [Google Scholar] [CrossRef]
- Cázares, A.; Galman, J.L.; Crago, L.G.; Smith, M.E.B.; Strafford, J.; Ríos-Solís, L.; Lye, G.J.; Dalby, P.A.; Hailes, H.C. Non-α-hydroxylated aldehydes with evolved transketolase enzymes. Org. Biomol. Chem. 2010, 8, 1301–1309. [Google Scholar] [CrossRef]
- Yi, D.; Saravanan, T.; Devamani, T.; Charmantray, F.; Hecquet, L.; Fessner, W.-D. A thermostable transketolase evolved for aliphatic aldehyde acceptors. Chem. Commun. 2015, 51, 480–483. [Google Scholar] [CrossRef] [Green Version]
- Saravanan, T.; Reif, M.-L.; Yi, D.; Lorillière, M.; Charmantray, F.; Hecquet, L.; Fessner, W.-D. Engineering a thermostable transketolase for arylated substrates. Green Chem. 2017, 19, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Hibbert, E.G.; Senussi, T.; Costelloe, S.J.; Lei, W.; Smith, M.E.; Ward, J.M.; Hailes, H.C.; Dalby, P.A. Directed evolution of transketolase activity on non-phosphorylated substrates. J. Biotechnol. 2007, 131, 425–432. [Google Scholar] [CrossRef]
- Ranoux, A.; Karmee, S.K.; Jin, J.; Bhaduri, A.; Caiazzo, A.; Arends, I.W.C.E.; Hanefeld, U. Enhancement of the Substrate Scope of Transketolase. ChemBioChem 2012, 13, 1921–1931. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.E.; Hibbert, E.G.; Jones, A.B.; Dalby, P.A.; Hailes, H.C. Enhancing and reversing the stereoselectivity of Escherichia coli transketolase via single-point mutations. Adv. Synth. Catal. 2008, 350, 2631–2638. [Google Scholar] [CrossRef]
- Westphal, R.; Vogel, D.C.; Schmitz, C.; Pleiss, J.; Müller, M.; Pohl, M.; Rother, D. A Tailor-Made Chimeric Thiamine Diphosphate Dependent Enzyme for the Direct Asymmetric Synthesis of (S)-Benzoins. Angew. Chem. Int. Ed. 2014, 53, 9376–9379. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Saravanan, T.; Lorillière, M.; Wei, D.; Charmantray, F.; Hecquet, L.; Fessner, W.-D.; Yi, D. Second-Generation Engineering of a Thermostable Transketolase (TKGst) for Aliphatic Aldehyde Acceptors with Either Improved or Reversed Stereoselectivity. ChemBioChem 2017, 18, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Pohl, M.; Sprenger, G.A.; Müller, M. A new perspective on thiamine catalysis. Curr. Opin. Biotechnol. 2004, 15, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Hailes, H.C.; Rother, D.; Muller, M.; Westphal, R.; Ward, J.M.; Pleiss, J.; Vogel, C.; Pohl, M. Engineering stereoselectivity of ThDP-dependent enzymes. FEBS J. 2013, 280, 6374–6394. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, P.P.; Bortolini, O.; Massi, A. Thiamine-Diphosphate-Dependent Enzymes as Catalytic Tools for the Asymmetric Benzoin-Type Reaction. Eur. J. Org. Chem. 2016, 2016, 4441–4459. [Google Scholar] [CrossRef]
- Wikner, C.; Nilsson, U.; Meshalkina, L.; Udekwu, C.; Lindqvist, Y.; Schneider, G. Identification of Catalytically Important Residues in Yeast Transketolase. Biochemistry 1997, 36, 15643–15649. [Google Scholar] [CrossRef]
- Wikner, C.; Meshalkina, L.; Nilsson, U.; Nikkola, M.; Lindqvist, Y.; Sundström, M.; Schneider, G. Analysis of an invariant cofactor-protein interaction in thiamin diphosphate-dependent enzymes by site-directed mutagenesis. Glutamic acid 418 in transketolase is essential for catalysis. J. Biol. Chem. 1994, 269, 32144–32150. [Google Scholar]
- Kern, D.; Kern, G.; Neef, H.; Tittmann, K.; Killenberg-Jabs, M.; Wikner, C.; Schneider, G.; Hübner, G. How Thiamin Diphosphate Is Activated in Enzymes. Science 1997, 275, 67–70. [Google Scholar] [CrossRef]
- Shin, W.; Pletcher, J.; Blank, G.; Sax, M. Ring stacking interactions between thiamin and planar molecules as seen in the crystal structure of a thiamin picrolonate dihydrate complex. J. Am. Chem. Soc. 1977, 99, 3491–3499. [Google Scholar] [CrossRef] [PubMed]
- Schellenberger, A. Sixty years of thiamin diphosphate biochemistry. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. Enzym. 1998, 1385, 177–186. [Google Scholar] [CrossRef]
- Asztalos, P.; Parthier, C.; Golbik, R.; Kleinschmidt, M.; Hübner, G.; Weiss, M.S.; Friedemann, R.; Wille, G.; Tittmann, K. Strain and Near Attack Conformers in Enzymic Thiamin Catalysis: X-ray Crystallographic Snapshots of Bacterial Transketolase in Covalent Complex with Donor Ketoses Xylulose 5-phosphate and Fructose 6-phosphate, and in Noncovalent Complex with Acceptor Aldose Ribose 5-phosphate. Biochemistry 2007, 46, 12037–12052. [Google Scholar] [CrossRef]
- Lüdtke, S.; Neumann, P.; Erixon, K.M.; Leeper, F.J.; Kluger, R.; Ficner, R.; Tittmann, K. Sub-ångström-resolution crystallography reveals physical distortions that enhance reactivity of a covalent enzymatic intermediate. Nat. Chem. 2013, 5, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Kochetov, G.; Philippov, P.; Razjivin, A.; Tikhomirova, N. Kinetics of reconstruction of holo-transketolase. FEBS Lett. 1975, 53, 211–212. [Google Scholar] [CrossRef] [Green Version]
- Kovina, M.V.; Kochetov, G.A. Cooperativity and flexibility of active sites in homodimeric transketolase. FEBS Lett. 1998, 440, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Ga, K.; Ia, S.; Kochetov, G.; Sevostyanova, I. Binding of the Coenzyme and Formation of the Transketolase Active Center. IUBMB Life 2005, 57, 491–497. [Google Scholar] [CrossRef]
- Sundström, M.; Lindqvist, Y.; Schneider, G. Three-dimensional structure of apotransketolase flexible loops at the active site enable cofactor binding. FEBS Lett. 1992, 313, 229–231. [Google Scholar] [CrossRef] [Green Version]
- Kochetov, G.A.; Solovjeva, O.N. Structure and functioning mechanism of transketolase. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2014, 1844, 1608–1618. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Steffen, H.; Janser, P.; Wiss, O. Studies on the Reconstitution of Apotransketolase with Thiamine Pyrophosphate and Analogs of the Coenzyme. Eur. J. Biochem. 1972, 30, 533–541. [Google Scholar] [CrossRef]
- Schröder-Tittmann, K.; Meyer, D.; Arens, J.; Wechsler, C.; Tietzel, M.; Golbik, R.; Tittmann, K. Alternating Sites Reactivity Is a Common Feature of Thiamin Diphosphate-Dependent Enzymes As Evidenced by Isothermal Titration Calorimetry Studies of Substrate Binding. Biochemistry 2013, 52, 2505–2507. [Google Scholar] [CrossRef] [PubMed]
- Seifert, F.; Golbik, R.; Brauer, J.; Lilie, H.; Schröder-Tittmann, K.; Hinze, E.; Korotchkina, L.G.; Patel, M.S.; Tittmann, K. Direct Kinetic Evidence for Half-Of-The-Sites Reactivity in the E1 Component of the Human Pyruvate Dehydrogenase Multienzyme Complex through Alternating Sites Cofactor Activation. Biochemistry 2006, 45, 12775–12785. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, H.C.; Dalby, P.A. Novel insights into transketolase activation by cofactor binding identifies two native species subpopulations. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovina, M.V.; Selivanov, V.A.; Kochevova, N.V.; Kochetov, G.A. Kinetic investigation of cooperativity in coenzyme binding by transketolase active sites. Biochemistry 1998, 63, 988–995. [Google Scholar] [PubMed]
- Jordan, F.; Nemeria, N.S.; Sergienko, E. Multiple Modes of Active Center Communication in Thiamin Diphosphate-Dependent Enzymes. Accounts Chem. Res. 2005, 38, 755–763. [Google Scholar] [CrossRef]
- Nemeria, N.S.; Arjunan, P.; Chandrasekhar, K.; Mossad, M.; Tittmann, K.; Furey, W.; Jordan, F. Communication between Thiamin Cofactors in theEscherichia coliPyruvate Dehydrogenase Complex E1 Component Active Centers. J. Biol. Chem. 2010, 285, 11197–11209. [Google Scholar] [CrossRef] [Green Version]
- Schapfl, M.; Baier, S.; Fries, A.; Ferlaino, S.; Waltzer, S.; Müller, M.; Sprenger, G.A. Extended substrate range of thiamine diphosphate-dependent MenD enzyme by coupling of two C–C-bonding reactions. Appl. Microbiol. Biotechnol. 2018, 102, 8359–8372. [Google Scholar] [CrossRef]
- Hernández, K.; Parella, T.; Petrillo, G.; Usón, I.; Wandtke, C.M.; Joglar, J.; Bujons, J.; Clapés, P. Intramolecular Benzoin Reaction Catalyzed by Benzaldehyde Lyase from Pseudomonas Fluorescens Biovar I. Angew. Chem. Int. Ed. 2017, 56, 5304–5307. [Google Scholar] [CrossRef] [Green Version]
- Saravanan, T.; Junker, S.; Kickstein, M.; Hein, S.; Link, M.-K.; Ranglack, J.; Witt, S.; Lorillière, M.; Hecquet, L.; Fessner, W.-D. Donor Promiscuity of a Thermostable Transketolase by Directed Evolution: Efficient Complementation of 1-Deoxy-d -xylulose-5-phosphate Synthase Activity. Angew. Chem. Int. Ed. 2017, 56, 5358–5362. [Google Scholar] [CrossRef]
- Hecquet, L.; Casajus, H.; Lagarde, A.; Leremboure, M.; Miguel, T.D.D.; Nauton, L.; Thery, V.; Fessner, W.-D.; Duguet, N.; Charmantray, F. Enzymatic Synthesis of Aliphatic Acyloins Catalyzed by Thermostable Transketolase. ChemCatChem 2020. [Google Scholar] [CrossRef]
- Yu, H.; López, R.I.H.; Steadman, D.; Méndez-Sánchez, D.; Higson, S.; Cázares-Körner, A.; Sheppard, T.D.; Ward, J.M.; Hailes, H.C.; Dalby, P.A. Engineering transketolase to accept both unnatural donor and acceptor substrates and produce α-hydroxyketones. FEBS J. 2019, 287, 1758–1776. [Google Scholar] [CrossRef]
- Kruger, N.J.; Von Schaewen, A. The oxidative pentose phosphate pathway: Structure and organisation. Curr. Opin. Plant Biol. 2003, 6, 236–246. [Google Scholar] [CrossRef]
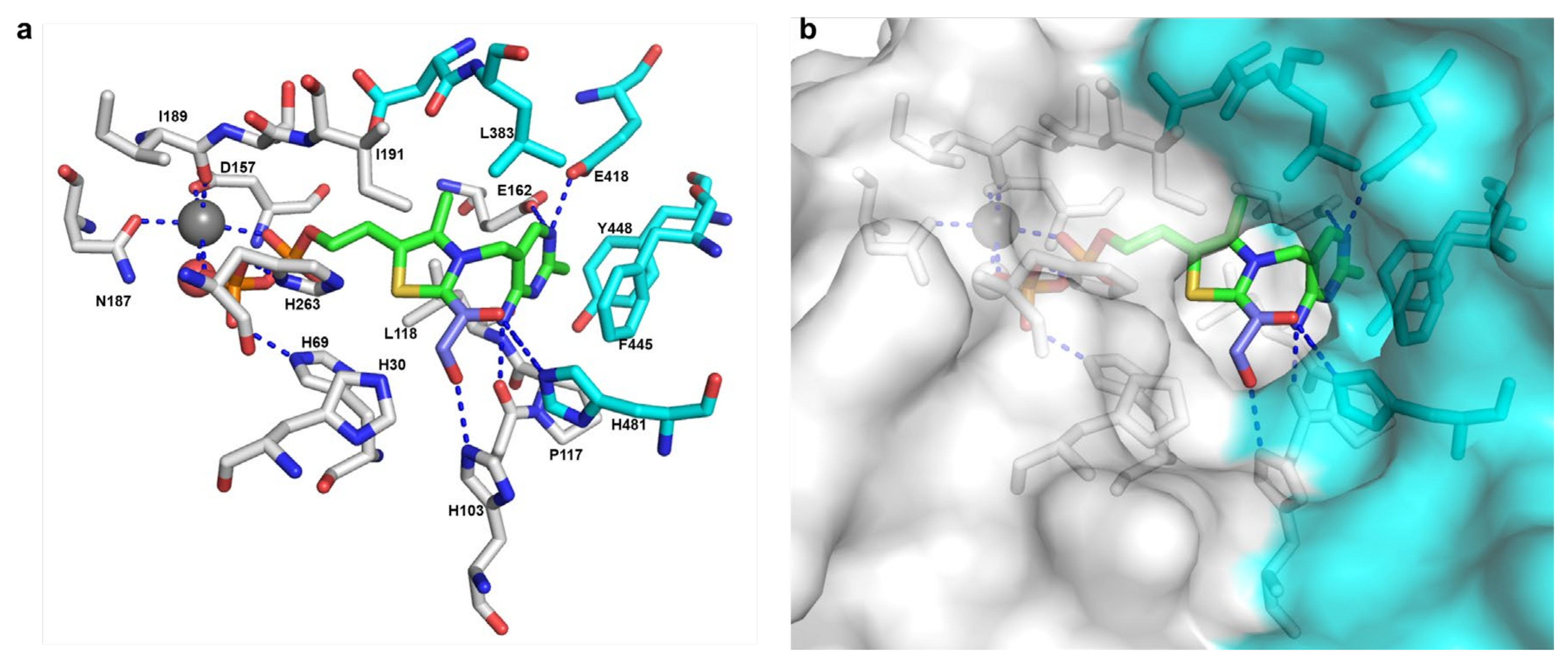
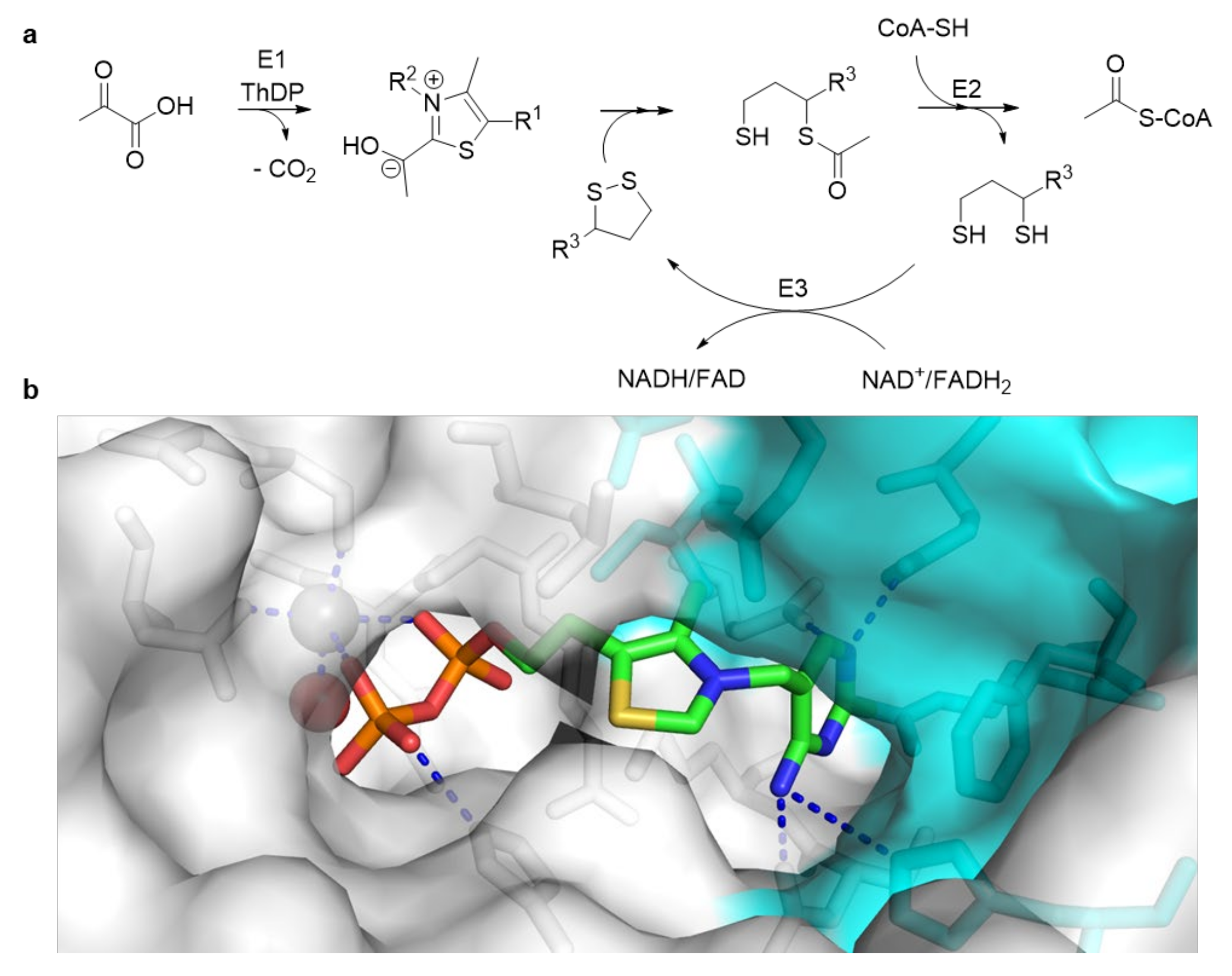
- Vogel, C.; Pleiss, J. The modular structure of ThDP-dependent enzymes. Proteins Struct. Funct. Bioinform. 2014, 82, 2523–2537. [Google Scholar] [CrossRef] [PubMed]
- Widmann, M.; Radloff, R.; Pleiss, J. The Thiamine diphosphate dependent Enzyme Engineering Database: A tool for the systematic analysis of sequence and structure relations. BMC Biochem. 2010, 11, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, C.; Widmann, M.; Pohl, M.; Pleiss, J. A standard numbering scheme for thiamine diphosphate-dependent decarboxylases. BMC Biochem. 2012, 13, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchholz, P.C.F.; Vogel, C.; Reusch, W.; Pohl, M.; Rother, D.; Spieß, I.A.C.; Pleiss, J. BioCatNet: A Database System for the Integration of Enzyme Sequences and Biocatalytic Experiments. ChemBioChem 2016, 17, 2093–2098. [Google Scholar] [CrossRef]
- Yi, J.; Nemeria, N.; McNally, A.; Jordan, F.; Machado, R.S.; Guest, J.R. Effect of Substitutions in the Thiamin Diphosphate-Magnesium Fold on the Activation of the Pyruvate Dehydrogenase Complex fromEscherichia coliby Cofactors and Substrate. J. Biol. Chem. 1996, 271, 33192–33200. [Google Scholar] [CrossRef] [Green Version]
- Behal, R.; Buxton, D.; Robertson, J.; Olson, M. Regulation of the pyruvate dehydrogenase multienzyme complex. Annu. Rev. Nutr. 1993, 13, 497–520. [Google Scholar] [CrossRef]
- Park, Y.-H.; Wei, W.; Zhou, L.; Nemeria, N.; Jordan, F. Amino-terminal residues 1− 45 of the Escherichia coli pyruvate dehydrogenase complex E1 subunit interact with the E2 subunit and are required for activity of the complex but not for reductive acetylation of the E2 subunit. Biochemistry 2004, 43, 14037–14046. [Google Scholar] [CrossRef]
- Bisswanger, H. Substrate specificity of the pyruvate dehydrogenase complex from Escherichia coli. J. Biol. Chem. 1981, 256, 815–822. [Google Scholar]
- Ke, C.; He, Y.; He, H.; Yang, X.; Li, R.; Yuan, J. A new spectrophotometric assay for measuring pyruvate dehydrogenase complex activity: A comparative evaluation. Anal. Methods 2014, 6, 6381–6388. [Google Scholar] [CrossRef]
- Humphrey, A.J.; Parsons, S.F.; Smith, M.E.; Turner, N.J. Synthesis of a novel N-hydroxypyrrolidine using enzyme catalysed asymmetric carbon–carbon bond synthesis. Tetrahedron Lett. 2000, 41, 4481–4485. [Google Scholar] [CrossRef]
- Effenberger, F.; Null, V.; Ziegler, T. Preparation of optically pure L-2-hydroxyaldehydes with yeast transketolase. Tetrahedron Lett. 1992, 33, 5157–5160. [Google Scholar] [CrossRef] [Green Version]
- Bykova, I.A.; Solovjeva, O.N.; Meshalkina, L.E.; Kovina, M.V.; Kochetov, G.A. One-Substrate Transketolase-Catalyzed Reaction. Biochem. Biophys. Res. Commun. 2001, 280, 845–847. [Google Scholar] [CrossRef] [PubMed]
- Hilal, S.H.; Bornander, L.L.; Carreira, L.A. Hydration Equilibrium Constants of Aldehydes, Ketones and Quinazolines. QSAR Comb. Sci. 2005, 24, 631–638. [Google Scholar] [CrossRef]
- Hibbert, E.G.; Senussi, T.; Smith, M.E.B.; Costelloe, S.J.; Ward, J.M.; Hailes, H.C.; Dalby, P.A. Directed evolution of transketolase substrate specificity towards an aliphatic aldehyde. J. Biotechnol. 2008, 134, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Médici, R.; Stammes, H.; Kwakernaak, S.; Otten, L.G.; Hanefeld, U. Assessing the stereoselectivity of Serratia marcescens CECT 977 2,3-butanediol dehydrogenase. Catal. Sci. Technol. 2017, 7, 1831–1837. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product | Conversion (%) | Isolated Yield (%) | ee (%) |
|---|---|---|---|
| 3a | 64 | 5 | 93 |
| 3b | 44 | 43 | 93 |
| 5b | 78 | 68 | 95 |
| 7a | 99 | 57 | 95 |
| 9a | 69 | 22 | 93 |
| Substrate | Enzyme | Variant | U/mg | KM | kcat | kcat/KM | Ref. |
|---|---|---|---|---|---|---|---|
| (mM) | (s−1) | (s−1mM−1) | |||||
| Pyruvate | TKGST | H102L/H474S | n.a. [g] | 16.6 [a] | 0.17 [a] | 0.01 [a] | [40] |
| EcDXS | WT | n.a. [g] | 3.3 [a] | 0.50 [a] | 0.15 [a] | [40] | |
| EcE1 | WT | 2.95 [a] | 4.2 [a] | 10.3 [a] | 2.45 [a] | t.w. [f] | |
| HPA | TKGST | WT | n.a. [g] | 2.3 [a] | 12.7 [a] | 5.50 [a] | [40] |
| ScTK | R528Q/S527T | 0.44 [a] | 53 [a] | n.a. [g] | n.a. [g] | [12] | |
| EcE1 | WT | 0.24 [a] | 100 [a] | 0.83 [a] | 0.008 [a] | t.w. [f] | |
| 2-oxobutyrate | TKGST | H102L/H474S | 0.006 [e] | 3.3 [a] | 0.16 [a] | 0.048 [a] | [40,41] |
| EcE1 | WT | 2.72 [a] | 1.09 [a] | 9.46 [a] | 8.68 [a] | t.w. [f] | |
| EcE1 | WT | 2.33 [e] | t.w. [f] | ||||
| glycolaldehyde | EcTK | H461S | 3.14 [d] | n.a. [g] | n.a. [g] | n.a. [g] | [11] |
| TKGST | L191I | 4.07 [d] | n.a. [g] | n.a. [g] | n.a. [g] | [15] | |
| EcE1 | WT | 3.09 [b] | 25.2 [b] | 10.7 [b] | 0.42 [b] | t.w. [f] | |
| propanal | TKGST | L191I/D470L | 1.48 [d] | n.a. [g] | n.a. [g] | n.a. [g] | [15] |
| TKGST | H102L/H474S/ F435I | 0.007 [c] | n.a. [g] | n.a. [g] | n.a. [g] | [41] | |
| EcE1 | WT | 2.33 [c] | 42.4 [c] | 8.10 [c] | 0.19 [c] | t.w. [f] | |
| isobutanal | ScTK | D477E | n.a. [g] | 66 [d] | 0.6 [d] | 0.009 [d] | [5] |
| EcE1 | WT | 0.78 [b] | 69.4 [b] | 2.70 [b] | 0.039 [b] | t.w. [f] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marsden, S.R.; McMillan, D.G.G.; Hanefeld, U. Assessing the Thiamine Diphosphate Dependent Pyruvate Dehydrogenase E1 Subunit for Carboligation Reactions with Aliphatic Ketoacids. Int. J. Mol. Sci. 2020, 21, 8641. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228641
Marsden SR, McMillan DGG, Hanefeld U. Assessing the Thiamine Diphosphate Dependent Pyruvate Dehydrogenase E1 Subunit for Carboligation Reactions with Aliphatic Ketoacids. International Journal of Molecular Sciences. 2020; 21(22):8641. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228641
Chicago/Turabian StyleMarsden, Stefan R., Duncan G. G. McMillan, and Ulf Hanefeld. 2020. "Assessing the Thiamine Diphosphate Dependent Pyruvate Dehydrogenase E1 Subunit for Carboligation Reactions with Aliphatic Ketoacids" International Journal of Molecular Sciences 21, no. 22: 8641. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228641