JAK/STAT Activation: A General Mechanism for Bone Development, Homeostasis, and Regeneration


Abstract
:1. Introduction
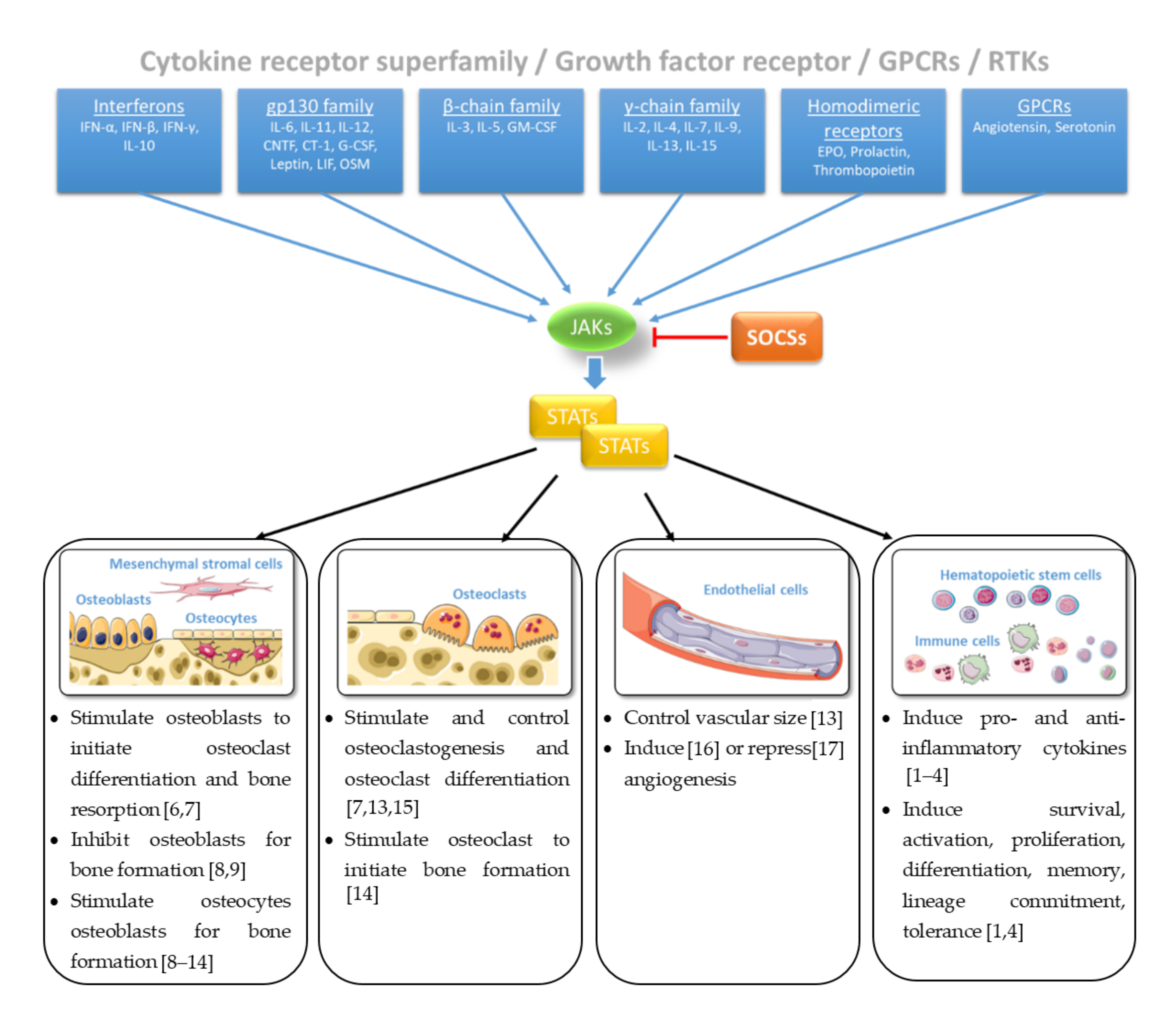
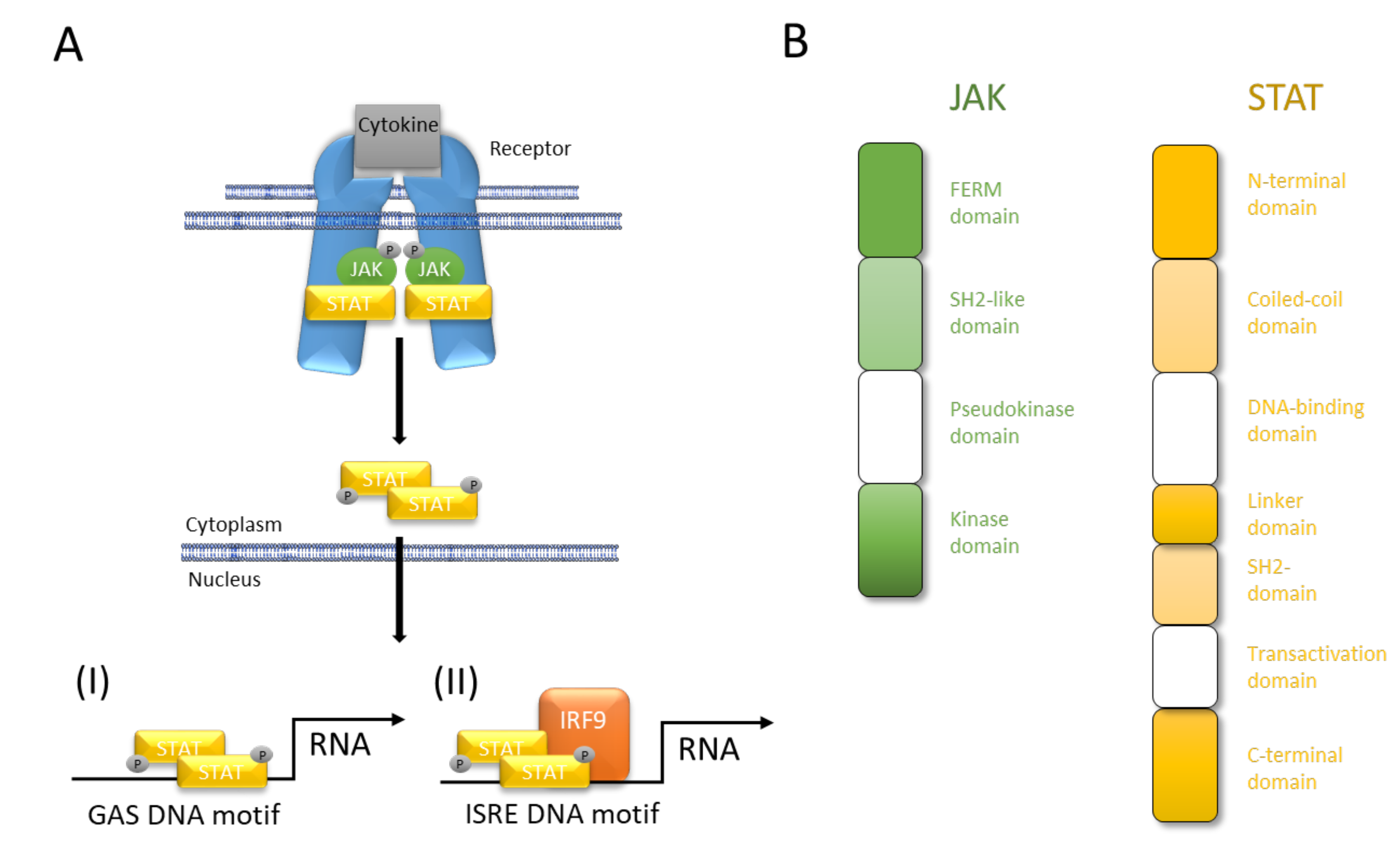
2. JAK/STAT Pathway at a Glance
3. Guiding Bone Development by Combining JAKs and STATs
4. JAK/STAT Signaling in Bone Turnover: From Homeostasis to Osteoporosis
5. Inhibiting the Two-Faced JAK/STAT Pathway Regenerates Bones
6. Concluding Remarks and Future Prospects
Funding
Acknowledgments
Conflicts of Interest
References
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and Consequences of Jak-Stat Signaling in the Immune System. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Leonard, W.J.; O’Shea, J.J. Jaks and Stats: Biological Implications. Annu. Rev. Immunol. 1998, 16, 293–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The Molecular Details of Cytokine Signaling Via the Jak/Stat Pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casanova, J.L.; Holland, S.M.; Notarangelo, L.D. Inborn Errors of Human Jaks and Stats. Immunity 2012, 36, 515–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sims, N.A. The Jak1/Stat3/Socs3 Axis in Bone Development, Physiology, and Pathology. Exp. Mol. Med. 2020, 52, 1185–1197. [Google Scholar] [CrossRef]
- Onan, D.; Allan, E.H.; Quinn, J.M.; Gooi, J.H.; Pompolo, S.; Sims, N.A.; Gillespie, M.T.; Martin, T.J. The Chemokine Cxcl1 Is a Novel Target Gene of Parathyroid Hormone (Pth)/Pth-Related Protein in Committed Osteoblasts. Endocrinology 2009, 150, 2244–2253. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, C.A.; Gubrij, I.; Lin, S.C.; Saylors, R.L.; Manolagas, S.C. Stat3 Activation in Stromal/Osteoblastic Cells Is Required for Induction of the Receptor Activator of Nf-Kappab Ligand and Stimulation of Osteoclastogenesis by Gp130-Utilizing Cytokines or Interleukin-1 but Not 1,25-Dihydroxyvitamin D3 or Parathyroid Hormone. J. Biol. Chem. 1999, 274, 19301–19308. [Google Scholar]
- Sims, N.A. Cell-Specific Paracrine Actions of Il-6 Family Cytokines from Bone, Marrow and Muscle That Control Bone Formation and Resorption. Int. J. Biochem. Cell Biol. 2016, 79, 14–23. [Google Scholar] [CrossRef]
- Walker, E.C.; McGregor, N.E.; Poulton, I.J.; Solano, M.; Pompolo, S.; Fernandes, T.J.; Constable, M.J.; Nicholson, G.C.; Zhang, J.G.; Nicola, N.A.; et al. Oncostatin M Promotes Bone Formation Independently of Resorption When Signaling through Leukemia Inhibitory Factor Receptor in Mice. J. Clin. Investig. 2010, 120, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Ni, L.; Yu, J.; Gui, X.; Lu, Z.; Wang, X.; Guo, H.; Zhou, Y. Overexpression of Rpn2 Promotes Osteogenic Differentiation of Hbmscs through the Jak/Stat3 Pathway. FEBS Open Bio 2020, 10, 158–167. [Google Scholar] [CrossRef] [Green Version]
- McGregor, N.E.; Murat, M.; Elango, J.; Poulton, I.J.; Walker, E.C.; Crimeen-Irwin, B.; Ho, P.W.M.; Gooi, J.H.; Martin, T.J.; Sims, N.A. Il-6 Exhibits Both Cis- and Trans-Signaling in Osteocytes and Osteoblasts, but Only Trans-Signaling Promotes Bone Formation and Osteoclastogenesis. J. Biol. Chem. 2019, 294, 7850–7863. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.W.; Brennan, H.J.; Vrahnas, C.; Poulton, I.J.; McGregor, N.E.; Standal, T.; Walker, E.C.; Koh, T.T.; Nguyen, H.; Walsh, N.C.; et al. The Primary Function of Gp130 Signaling in Osteoblasts Is to Maintain Bone Formation and Strength, Rather Than Promote Osteoclast Formation. J. Bone Miner. Res. 2014, 29, 1492–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulton, I.J.; McGregor, N.E.; Pompolo, S.; Walker, E.C.; Sims, N.A. Contrasting Roles of Leukemia Inhibitory Factor in Murine Bone Development and Remodeling Involve Region-Specific Changes in Vascularization. J. Bone Miner. Res. 2012, 27, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.C.; McGregor, N.E.; Poulton, I.J.; Pompolo, S.; Allan, E.H.; Quinn, J.M.; Gillespie, M.T.; Martin, T.J.; Sims, N.A. Cardiotrophin-1 Is an Osteoclast-Derived Stimulus of Bone Formation Required for Normal Bone Remodeling. J. Bone Miner. Res. 2008, 23, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Kobayashi, Y.; Uehara, S.; Suzuki, T.; Koide, M.; Yamashita, T.; Nakamura, M.; Takahashi, N.; Kato, H.; Udagawa, N.; et al. A Jak1/2 Inhibitor, Baricitinib, Inhibits Osteoclastogenesis by Suppressing Rankl Expression in Osteoblasts in Vitro. PLoS ONE 2017, 12, e0181126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujio, Y.; Maeda, M.; Mohri, T.; Obana, M.; Iwakura, T.; Hayama, A.; Yamashita, T.; Nakayama, H.; Azuma, J. Glycoprotein 130 Cytokine Signal as a Therapeutic Target against Cardiovascular Diseases. J. Pharmacol. Sci. 2011, 117, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Nur, H.; Rao, L.; Frassanito, M.A.; De Raeve, H.; Ribatti, D.; Mfopou, J.K.; Van Valckenborgh, E.; De Bruyne, E.; Vacca, A.; Vanderkerken, K.; et al. Stimulation of Invariant Natural Killer T Cells by Alpha-Galactosylceramide Activates the Jak-Stat Pathway in Endothelial Cells and Reduces Angiogenesis in the 5t33 Multiple Myeloma Model. Br. J. Haematol. 2014, 167, 651–663. [Google Scholar] [CrossRef]
- Russell, S.M.; Tayebi, N.; Nakajima, H.; Riedy, M.C.; Roberts, J.L.; Aman, M.J.; Migone, T.S.; Noguchi, M.; Markert, M.L.; Buckley, R.H.; et al. Mutation of Jak3 in a Patient with Scid: Essential Role of Jak3 in Lymphoid Development. Science 1995, 270, 797–800. [Google Scholar] [CrossRef] [Green Version]
- Macchi, P.; Villa, A.; Giliani, S.; Sacco, M.G.; Frattini, A.; Porta, F.; Ugazio, A.G.; Johnston, J.A.; Candotti, F.; O’Shea, J.J.; et al. Mutations of Jak-3 Gene in Patients with Autosomal Severe Combined Immune Deficiency (Scid). Nature 1995, 377, 65–68. [Google Scholar] [CrossRef]
- Zhang, D.; Wlodawer, A.; Lubkowski, J. Crystal Structure of a Complex of the Intracellular Domain of Interferon Lambda Receptor 1 (Ifnlr1) and the Ferm/Sh2 Domains of Human Jak1. J. Mol. Biol. 2016, 428, 4651–4668. [Google Scholar] [CrossRef]
- Floss, D.M.; Klocker, T.; Schroder, J.; Lamertz, L.; Mrotzek, S.; Strobl, B.; Hermanns, H.; Scheller, J. Defining the Functional Binding Sites of Interleukin 12 Receptor Beta1 and Interleukin 23 Receptor to Janus Kinases. Mol. Biol. Cell 2016, 27, 2301–2316. [Google Scholar] [CrossRef] [PubMed]
- Wallweber, H.J.; Tam, C.; Franke, Y.; Starovasnik, M.A.; Lupardus, P.J. Structural Basis of Recognition of Interferon-Alpha Receptor by Tyrosine Kinase 2. Nat. Struct. Mol. Biol. 2014, 21, 443–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupardus, P.J.; Skiniotis, G.; Rice, A.J.; Thomas, C.; Fischer, S.; Walz, T.; Garcia, K.C. Structural Snapshots of Full-Length Jak1, a Transmembrane Gp130/Il-6/Il-6ralpha Cytokine Receptor Complex, and the Receptor-Jak1 Holocomplex. Structure 2011, 19, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Kido, S.; Kuriwaka-Kido, R.; Imamura, T.; Ito, Y.; Inoue, D.; Matsumoto, T. Mechanical Stress Induces Interleukin-11 Expression to Stimulate Osteoblast Differentiation. Bone 2009, 45, 1125–1132. [Google Scholar] [CrossRef]
- Ahlen, J.; Andersson, S.; Mukohyama, H.; Roth, C.; Backman, A.; Conaway, H.H.; Lerner, U.H. Characterization of the Bone-Resorptive Effect of Interleukin-11 in Cultured Mouse Calvarial Bones. Bone 2002, 31, 242–251. [Google Scholar] [CrossRef]
- Zhu, J.; Shimizu, E.; Zhang, X.; Partridge, N.C.; Qin, L. Egfr Signaling Suppresses Osteoblast Differentiation and Inhibits Expression of Master Osteoblastic Transcription Factors Runx2 and Osterix. J. Cell Biochem. 2011, 112, 1749–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodig, S.J.; Meraz, M.A.; White, J.M.; Lampe, P.A.; Riley, J.K.; Arthur, C.D.; King, K.L.; Sheehan, K.C.; Yin, L.; Pennica, D.; et al. Disruption of the Jak1 Gene Demonstrates Obligatory and Nonredundant Roles of the Jaks in Cytokine-Induced Biologic Responses. Cell 1998, 93, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Wehde, B.L.; Radler, P.D.; Triplett, A.A.; Wagner, K.U. Generation of Janus Kinase 1 (Jak1) Conditional Knockout Mice. Genesis 2016, 54, 582–588. [Google Scholar] [CrossRef]
- Sabrautzki, S.; Janas, E.; Lorenz-Depiereux, B.; Calzada-Wack, J.; Aguilar-Pimentel, J.A.; Rathkolb, B.; Adler, T.; Cohrs, C.; Hans, W.; Diener, S.; et al. An Enu Mutagenesis-Derived Mouse Model with a Dominant Jak1 Mutation Resembling Phenotypes of Systemic Autoimmune Disease. Am. J. Pathol. 2013, 183, 352–368. [Google Scholar] [CrossRef]
- Mori, T.; Miyamoto, T.; Yoshida, H.; Asakawa, M.; Kawasumi, M.; Kobayashi, T.; Morioka, H.; Chiba, K.; Toyama, Y.; Yoshimura, A. Il-1beta and Tnfalpha-Initiated Il-6-Stat3 Pathway Is Critical in Mediating Inflammatory Cytokines and Rankl Expression in Inflammatory Arthritis. Int. Immunol. 2011, 23, 701–712. [Google Scholar] [CrossRef] [Green Version]
- LaBranche, T.P.; Jesson, M.I.; Radi, Z.A.; Storer, C.E.; Guzova, J.A.; Bonar, S.L.; Thompson, J.M.; Happa, F.A.; Stewart, Z.S.; Zhan, Y.; et al. Jak Inhibition with Tofacitinib Suppresses Arthritic Joint Structural Damage through Decreased Rankl Production. Arthritis Rheum. 2012, 64, 3531–3542. [Google Scholar] [CrossRef] [PubMed]
- Vidal, B.; Cascao, R.; Finnila, M.A.J.; Lopes, I.P.; da Gloria, V.G.; Saarakkala, S.; Zioupos, P.; Canhao, H.; Fonseca, J.E. Effects of Tofacitinib in Early Arthritis-Induced Bone Loss in an Adjuvant-Induced Arthritis Rat Model. Rheumatology 2019, 58, 371. [Google Scholar] [CrossRef] [Green Version]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting Cellular Senescence Prevents Age-Related Bone Loss in Mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Parganas, E.; Wang, D.; Stravopodis, D.; Topham, D.J.; Marine, J.C.; Teglund, S.; Vanin, E.F.; Bodner, S.; Colamonici, O.R.; van Deursen, J.M.; et al. Jak2 Is Essential for Signaling through a Variety of Cytokine Receptors. Cell 1998, 93, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Gotthardt, D.; Trifinopoulos, J.; Sexl, V.; Putz, E.M. Jak/Stat Cytokine Signaling at the Crossroad of Nk Cell Development and Maturation. Front. Immunol. 2019, 10, 2590. [Google Scholar] [CrossRef] [Green Version]
- Krempler, A.; Qi, Y.; Triplett, A.A.; Zhu, J.; Rui, H.; Wagner, K.U. Generation of a Conditional Knockout Allele for the Janus Kinase 2 (Jak2) Gene in Mice. Genesis 2004, 40, 52–57. [Google Scholar] [CrossRef]
- Park, S.Y.; Saijo, K.; Takahashi, T.; Osawa, M.; Arase, H.; Hirayama, N.; Miyake, K.; Nakauchi, H.; Shirasawa, T.; Saito, T. Developmental Defects of Lymphoid Cells in Jak3 Kinase-Deficient Mice. Immunity 1995, 3, 771–782. [Google Scholar] [CrossRef] [Green Version]
- Nosaka, T.; van Deursen, J.M.; Tripp, R.A.; Thierfelder, W.E.; Witthuhn, B.A.; McMickle, A.P.; Doherty, P.C.; Grosveld, G.C.; Ihle, J.N. Defective Lymphoid Development in Mice Lacking Jak3. Science 1995, 270, 800–802. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.D.; Kuo, F.I.; Smith, P.A. Targeting the Janus Kinase Family in Autoimmune Skin Diseases. Front. Immunol. 2019, 10, 2342. [Google Scholar] [CrossRef]
- Lokau, J.; Schoeder, V.; Haybaeck, J.; Garbers, C. Jak-Stat Signaling Induced by Interleukin-6 Family Cytokines in Hepatocellular Carcinoma. Cancers 2019, 11, 1704. [Google Scholar] [CrossRef] [Green Version]
- Meraz, M.A.; White, J.M.; Sheehan, K.C.; Bach, E.A.; Rodig, S.J.; Dighe, A.S.; Kaplan, D.H.; Riley, J.K.; Greenlund, A.C.; Campbell, D.; et al. Targeted Disruption of the Stat1 Gene in Mice Reveals Unexpected Physiologic Specificity in the Jak-Stat Signaling Pathway. Cell 1996, 84, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Koga, T.; Isobe, M.; Kern, B.E.; Yokochi, T.; Chin, Y.E.; Karsenty, G.; Taniguchi, T.; Takayanagi, H. Stat1 Functions as a Cytoplasmic Attenuator of Runx2 in the Transcriptional Program of Osteoblast Differentiation. Genes Dev. 2003, 17, 1979–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajima, K.; Takaishi, H.; Takito, J.; Tohmonda, T.; Yoda, M.; Ota, N.; Kosaki, N.; Matsumoto, M.; Ikegami, H.; Nakamura, T.; et al. Inhibition of Stat1 Accelerates Bone Fracture Healing. J. Orthop. Res. 2010, 28, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Li, S.; Cha, E.; Schindler, C. Immune Response in Stat2 Knockout Mice. Immunity 2000, 13, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Noguchi, K.; Shi, W.; Tanaka, T.; Matsumoto, M.; Yoshida, N.; Kishimoto, T.; Akira, S. Targeted Disruption of the Mouse Stat3 Gene Leads to Early Embryonic Lethality. Proc. Natl. Acad. Sci. USA 1997, 94, 3801–3804. [Google Scholar] [CrossRef] [Green Version]
- Itoh, S.; Udagawa, N.; Takahashi, N.; Yoshitake, F.; Narita, H.; Ebisu, S.; Ishihara, K. A Critical Role for Interleukin-6 Family-Mediated Stat3 Activation in Osteoblast Differentiation and Bone Formation. Bone 2006, 39, 505–512. [Google Scholar] [CrossRef]
- Zhou, H.; Newnum, A.B.; Martin, J.R.; Li, P.; Nelson, M.T.; Moh, A.; Fu, X.Y.; Yokota, H.; Li, J. Osteoblast/Osteocyte-Specific Inactivation of Stat3 Decreases Load-Driven Bone Formation and Accumulates Reactive Oxygen Species. Bone 2011, 49, 404–411. [Google Scholar] [CrossRef]
- Atsumi, T.; Ishihara, K.; Kamimura, D.; Ikushima, H.; Ohtani, T.; Hirota, S.; Kobayashi, H.; Park, S.J.; Saeki, Y.; Kitamura, Y.; et al. A Point Mutation of Tyr-759 in Interleukin 6 Family Cytokine Receptor Subunit Gp130 Causes Autoimmune Arthritis. J. Exp. Med. 2002, 196, 979–990. [Google Scholar] [CrossRef] [Green Version]
- Yong, P.F.; Freeman, A.F.; Engelhardt, K.R.; Holland, S.; Puck, J.M.; Grimbacher, B. An Update on the Hyper-Ige Syndromes. Arthritis Res. Ther. 2012, 14, 228. [Google Scholar] [CrossRef] [Green Version]
- Holland, S.M.; DeLeo, F.R.; Elloumi, H.Z.; Hsu, A.P.; Uzel, G.; Brodsky, N.; Freeman, A.F.; Demidowich, A.; Davis, J.; Turner, M.L.; et al. Stat3 Mutations in the Hyper-Ige Syndrome. N. Engl. J. Med. 2007, 357, 1608–1619. [Google Scholar] [CrossRef]
- Minegishi, Y.; Saito, M.; Tsuchiya, S.; Tsuge, I.; Takada, H.; Hara, T.; Kawamura, N.; Ariga, T.; Pasic, S.; Stojkovic, O.; et al. Dominant-Negative Mutations in the DNA-Binding Domain of Stat3 Cause Hyper-Ige Syndrome. Nature 2007, 448, 1058–1062. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Schlessinger, K.; Zhu, X.; Meffre, E.; Quimby, F.; Levy, D.E.; Darnell, J.E., Jr. Essential Role of Stat3 in Postnatal Survival and Growth Revealed by Mice Lacking Stat3 Serine 727 Phosphorylation. Mol. Cell Biol. 2004, 24, 407–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corry, K.A.; Zhou, H.; Brustovetsky, T.; Himes, E.R.; Bivi, N.; Horn, M.R.; Kitase, Y.; Wallace, J.M.; Bellido, T.; Brustovetsky, N.; et al. Stat3 in Osteocytes Mediates Osteogenic Response to Loading. Bone Rep. 2019, 11, 100218. [Google Scholar] [CrossRef]
- Kaplan, M.H.; Sun, Y.L.; Hoey, T.; Grusby, M.J. Impaired Il-12 Responses and Enhanced Development of Th2 Cells in Stat4-Deficient Mice. Nature 1996, 382, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Woitge, H.W.; Braut, A.; Kronenberg, M.S.; Lichtler, A.C.; Mina, M.; Kream, B.E. Expression and Activity of Osteoblast-Targeted Cre Recombinase Transgenes in Murine Skeletal Tissues. Int. J. Dev. Biol. 2004, 48, 645–653. [Google Scholar] [CrossRef] [Green Version]
- Hall, M.D.; Murray, C.A.; Valdez, M.J.; Perantoni, A.O. Mesoderm-Specific Stat3 Deletion Affects Expression of Sox9 Yielding Sox9-Dependent Phenotypes. PLoS Genet. 2017, 13, e1006610. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Welte, T.; Troiano, N.; Maher, S.E.; Fu, X.Y.; Bothwell, A.L. Osteoporosis with Increased Osteoclastogenesis in Hematopoietic Cell-Specific Stat3-Deficient Mice. Biochem. Biophys. Res. Commun. 2005, 328, 800–807. [Google Scholar] [CrossRef]
- Marine, J.C.; McKay, C.; Wang, D.; Topham, D.J.; Parganas, E.; Nakajima, H.; Pendeville, H.; Yasukawa, H.; Sasaki, A.; Yoshimura, A.; et al. Socs3 Is Essential in the Regulation of Fetal Liver Erythropoiesis. Cell 1999, 98, 617–627. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.W.; Robb, L.; Rakar, S.; Hartley, L.; Cluse, L.; Nicola, N.A.; Metcalf, D.; Hilton, D.J.; Alexander, W.S. Placental Defects and Embryonic Lethality in Mice Lacking Suppressor of Cytokine Signaling 3. Proc. Natl. Acad. Sci. USA 2001, 98, 9324–9329. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.K.; Egan, P.J.; Croker, B.A.; O’Donnell, K.; Sims, N.A.; Drake, S.; Kiu, H.; McManus, E.J.; Alexander, W.S.; Roberts, A.W.; et al. Socs-3 Negatively Regulates Innate and Adaptive Immune Mechanisms in Acute Il-1-Dependent Inflammatory Arthritis. J. Clin. Investig. 2006, 116, 1571–1581. [Google Scholar] [CrossRef]
- Cho, D.C.; Brennan, H.J.; Johnson, R.W.; Poulton, I.J.; Gooi, J.H.; Tonkin, B.A.; McGregor, N.E.; Walker, E.C.; Handelsman, D.J.; Martin, T.J.; et al. Bone Corticalization Requires Local Socs3 Activity and Is Promoted by Androgen Action Via Interleukin-6. Nat. Commun. 2017, 8, 806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; D’Cruz, A.A.; Hansen, J.; Croker, B.A.; Lawlor, K.E.; Sims, N.A.; Wicks, I.P. Deleting Suppressor of Cytokine Signaling-3 in Chondrocytes Reduces Bone Growth by Disrupting Mitogen-Activated Protein Kinase Signaling. Osteoarthr. Cartil. 2019, 27, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Thierfelder, W.E.; van Deursen, J.M.; Yamamoto, K.; Tripp, R.A.; Sarawar, S.R.; Carson, R.T.; Sangster, M.Y.; Vignali, D.A.; Doherty, P.C.; Grosveld, G.C.; et al. Requirement for Stat4 in Interleukin-12-Mediated Responses of Natural Killer and T Cells. Nature 1996, 382, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Robinson, G.W.; Wagner, K.U.; Garrett, L.; Wynshaw-Boris, A.; Hennighausen, L. Stat5a Is Mandatory for Adult Mammary Gland Development and Lactogenesis. Genes Dev. 1997, 11, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Udy, G.B.; Towers, R.P.; Snell, R.G.; Wilkins, R.J.; Park, S.H.; Ram, P.A.; Waxman, D.J.; Davey, H.W. Requirement of Stat5b for Sexual Dimorphism of Body Growth Rates and Liver Gene Expression. Proc. Natl. Acad. Sci. USA 1997, 94, 7239–7244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.M.; Park, K.H.; Hwang, J.S.; Lee, M.; Yoon, D.S.; Ryu, H.A.; Jung, H.S.; Park, K.W.; Kim, J.; Park, S.W.; et al. Inhibition of Stat5a Promotes Osteogenesis by Dlx5 Regulation. Cell Death Dis 2018, 9, 1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirose, J.; Masuda, H.; Tokuyama, N.; Omata, Y.; Matsumoto, T.; Yasui, T.; Kadono, Y.; Hennighausen, L.; Tanaka, S. Bone Resorption Is Regulated by Cell-Autonomous Negative Feedback Loop of Stat5-Dusp Axis in the Osteoclast. J. Exp. Med. 2014, 211, 153–163. [Google Scholar] [CrossRef]
- Kaplan, M.H.; Schindler, U.; Smiley, S.T.; Grusby, M.J. Stat6 Is Required for Mediating Responses to Il-4 and for Development of Th2 Cells. Immunity 1996, 4, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Tanaka, T.; Shi, W.; Matsumoto, M.; Minami, M.; Kashiwamura, S.; Nakanishi, K.; Yoshida, N.; Kishimoto, T.; Akira, S. Essential Role of Stat6 in Il-4 Signalling. Nature 1996, 380, 627–630. [Google Scholar] [CrossRef]
- Shimoda, K.; van Deursen, J.; Sangster, M.Y.; Sarawar, S.R.; Carson, R.T.; Tripp, R.A.; Chu, C.; Quelle, F.W.; Nosaka, T.; Vignali, D.A.; et al. Lack of Il-4-Induced Th2 Response and Ige Class Switching in Mice with Disrupted Stat6 Gene. Nature 1996, 380, 630–633. [Google Scholar] [CrossRef]
- Risner, K.; Ahmed, A.; Bakovic, A.; Kortchak, S.; Bhalla, N.; Narayanan, A. Efficacy of Fda-Approved Anti-Inflammatory Drugs against Venezuelan Equine Encephalitis Virus Infection. Viruses 2019, 11, 1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neubauer, H.; Cumano, A.; Muller, M.; Wu, H.; Huffstadt, U.; Pfeffer, K. Jak2 Deficiency Defines an Essential Developmental Checkpoint in Definitive Hematopoiesis. Cell 1998, 93, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Shuai, K.; Stark, G.R.; Kerr, I.M.; Darnell, J.E., Jr. A Single Phosphotyrosine Residue of Stat91 Required for Gene Activation by Interferon-Gamma. Science 1993, 261, 1744–1746. [Google Scholar] [CrossRef] [PubMed]
- Raz, R.; Durbin, J.E.; Levy, D.E. Acute Phase Response Factor and Additional Members of the Interferon-Stimulated Gene Factor 3 Family Integrate Diverse Signals from Cytokines, Interferons, and Growth Factors. J. Biol. Chem. 1994, 269, 24391–24395. [Google Scholar]
- Liao, Z.; Gu, L.; Vergalli, J.; Mariani, S.A.; De Dominici, M.; Lokareddy, R.K.; Dagvadorj, A.; Purushottamachar, P.; McCue, P.A.; Trabulsi, E.; et al. Structure-Based Screen Identifies a Potent Small Molecule Inhibitor of Stat5a/B with Therapeutic Potential for Prostate Cancer and Chronic Myeloid Leukemia. Mol. Cancer Ther. 2015, 14, 1777–1793. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Seong, S.; Kim, J.H.; Kim, K.; Kim, I.; Jeong, B.C.; Nam, K.I.; Kim, K.K.; Hennighausen, L.; Kim, N. Stat5 Is a Key Transcription Factor for Il-3-Mediated Inhibition of Rankl-Induced Osteoclastogenesis. Sci. Rep. 2016, 6, 30977. [Google Scholar] [CrossRef]
- Darvin, P.; Joung, Y.H.; Yang, Y.M. Jak2-Stat5b Pathway and Osteoblast Differentiation. JAKSTAT 2013, 2, e24931. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Levy, D.E. Comparative Evolutionary Genomics of the Stat Family of Transcription Factors. JAKSTAT 2012, 1, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, P. Physiology of Bone. Endocr. Dev. 2009, 16, 32–48. [Google Scholar]
- Rucci, N. Molecular Biology of Bone Remodelling. Clin. Cases Miner. Bone Metab. 2008, 5, 49–56. [Google Scholar] [PubMed]
- Einhorn, T.A. The Cell and Molecular Biology of Fracture Healing. Clin. Orthop. Relat. Res. 1998, 355, S7–S21. [Google Scholar] [CrossRef] [PubMed]
- Cullinane, D.M. The Role of Osteocytes in Bone Regulation: Mineral Homeostasis Versus Mechanoreception. J. Musculoskelet. Neuronal. Interact. 2002, 2, 242–244. [Google Scholar]
- Chang, B.; Quan, Q.; Li, Y.; Qiu, H.; Peng, J.; Gu, Y. Treatment of Osteoporosis, with a Focus on 2 Monoclonal Antibodies. Med. Sci. Monit. 2018, 24, 8758–8766. [Google Scholar] [CrossRef] [PubMed]
- Weivoda, M.M.; Youssef, S.J.; Oursler, M.J. Sclerostin Expression and Functions Beyond the Osteocyte. Bone 2017, 96, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crotti, T.N.; Sharma, S.M.; Fleming, J.D.; Flannery, M.R.; Ostrowski, M.C.; Goldring, S.R.; McHugh, K.P. Pu.1 and Nfatc1 Mediate Osteoclastic Induction of the Mouse Beta3 Integrin Promoter. J. Cell Physiol. 2008, 215, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Hayashi, M.; Sasaki, F.; Nakashima, T. Rankl Biology: Bone Metabolism, the Immune System, and Beyond. Inflamm. Regen. 2020, 40, 2. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K.; Takayanagi, H. Osteoimmunology. Cold Spring Harb. Perspect. Med. 2019, 9. [Google Scholar] [CrossRef]
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. Rank Is Essential for Osteoclast and Lymph Node Development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef]
- Black, D.M.; Rosen, C.J. Clinical Practice. Postmenopausal Osteoporosis. N. Engl. J. Med. 2016, 374, 254–262. [Google Scholar] [CrossRef]
- Adam, S.; Simon, N.; Steffen, U.; Andes, F.T.; Scholtysek, C.; Muller, D.I.H.; Weidner, D.; Andreev, D.; Kleyer, A.; Culemann, S.; et al. Jak Inhibition Increases Bone Mass in Steady-State Conditions and Ameliorates Pathological Bone Loss by Stimulating Osteoblast Function. Sci. Transl. Med. 2020, 12, eaay4447. [Google Scholar] [CrossRef]
- Li, J. Jak-Stat and Bone Metabolism. JAKSTAT 2013, 2, e23930. [Google Scholar] [CrossRef] [PubMed]
- Gaber, T.; Brinkman, A.C.K.; Pienczikowski, J.; Diesing, K.; Damerau, A.; Pfeiffenberger, M.; Lang, A.; Ohrndorf, S.; Burmester, G.R.; Buttgereit, F.; et al. Impact of Janus Kinase Inhibition with Tofacitinib on Fundamental Processes of Bone Healing. Int. J. Mol. Sci. 2020, 21, 865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellido, T.; Borba, V.Z.; Roberson, P.; Manolagas, S.C. Activation of the Janus Kinase/Stat (Signal Transducer and Activator of Transcription) Signal Transduction Pathway by Interleukin-6-Type Cytokines Promotes Osteoblast Differentiation. Endocrinology 1997, 138, 3666–3676. [Google Scholar] [CrossRef] [PubMed]
- Sowerwine, K.J.; Shaw, P.A.; Gu, W.; Ling, J.C.; Collins, M.T.; Darnell, D.N.; Anderson, V.L.; Davis, J.; Hsu, A.; Welch, P.; et al. Bone Density and Fractures in Autosomal Dominant Hyper Ige Syndrome. J. Clin. Immunol. 2014, 34, 260–264. [Google Scholar] [CrossRef] [Green Version]
- Schett, G. Review: Immune Cells and Mediators of Inflammatory Arthritis. Autoimmunity 2008, 41, 224–229. [Google Scholar] [CrossRef]
- Schett, G. Autoimmunity as a Trigger for Structural Bone Damage in Rheumatoid Arthritis. Mod. Rheumatol. 2017, 27, 193–197. [Google Scholar] [CrossRef]
- Terashima, A.; Takayanagi, H. Overview of Osteoimmunology. Calcif. Tissue Int. 2018, 102, 503–511. [Google Scholar] [CrossRef]
- Gravallese, E.M.; Manning, C.; Tsay, A.; Naito, A.; Pan, C.; Amento, E.; Goldring, S.R. Synovial Tissue in Rheumatoid Arthritis Is a Source of Osteoclast Differentiation Factor. Arthritis Rheum. 2000, 43, 250–258. [Google Scholar] [CrossRef]
- Takayanagi, H.; Iizuka, H.; Juji, T.; Nakagawa, T.; Yamamoto, A.; Miyazaki, T.; Koshihara, Y.; Oda, H.; Nakamura, K.; Tanaka, S. Involvement of Receptor Activator of Nuclear Factor Kappab Ligand/Osteoclast Differentiation Factor in Osteoclastogenesis from Synoviocytes in Rheumatoid Arthritis. Arthritis Rheum. 2000, 43, 259–269. [Google Scholar] [CrossRef]
- Danks, L.; Komatsu, N.; Guerrini, M.M.; Sawa, S.; Armaka, M.; Kollias, G.; Nakashima, T.; Takayanagi, H. Rankl Expressed on Synovial Fibroblasts Is Primarily Responsible for Bone Erosions During Joint Inflammation. Ann. Rheum. Dis. 2016, 75, 1187–1195. [Google Scholar] [CrossRef]
- Fiehn, C.; Kruger, K. Treatment Algorithm for Rheumatoid Arthritis: According to the S2e Guidelines 2018. Z. Rheumatol. 2019, 78, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Landewe, R.; Bijlsma, J.; Burmester, G.; Chatzidionysiou, K.; Dougados, M.; Nam, J.; Ramiro, S.; Voshaar, M.; van Vollenhoven, R.; et al. Eular Recommendations for the Management of Rheumatoid Arthritis with Synthetic and Biological Disease-Modifying Antirheumatic Drugs: 2016 Update. Ann. Rheum. Dis. 2017, 76, 960–977. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.; Hall, S.; FitzGerald, O.; van der Heijde, D.; Merola, J.F.; Avila-Zapata, F.; Cieslak, D.; Graham, D.; Wang, C.; Menon, S.; et al. Tofacitinib or Adalimumab Versus Placebo for Psoriatic Arthritis. N. Engl. J. Med. 2017, 377, 1537–1550. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C.; Keystone, E.C.; van der Heijde, D.; Weinblatt, M.E.; Del Carmen Morales, L.; Reyes Gonzaga, J.; Yakushin, S.; Ishii, T.; Emoto, K.; Beattie, S.; et al. Baricitinib Versus Placebo or Adalimumab in Rheumatoid Arthritis. N. Engl. J. Med. 2017, 376, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.B. Safety Profile of Jak Inhibitors: A Focus on Baricitinib. Lancet Rheumatol. 2020, 2, e313–e314. [Google Scholar] [CrossRef]
- Genovese, M.C.; Smolen, J.S.; Takeuchi, T.; Burmester, G.; Brinker, D.; Rooney, T.P.; Zhong, J.; Daojun, M.; Saifan, C.; Cardoso, A.; et al. Safety Profile of Baricitinib for the Treatment of Rheumatoid Arthritis over a Median of 3 Years of Treatment: An Updated Integrated Safety Analysis. Lancet Rheumatol. 2020, 2, e347–e357. [Google Scholar] [CrossRef]
- Wollenhaupt, J.; Lee, E.B.; Curtis, J.R.; Silverfield, J.; Terry, K.; Soma, K.; Mojcik, C.; DeMasi, R.; Strengholt, S.; Kwok, K.; et al. Safety and Efficacy of Tofacitinib for up to 9.5 Years in the Treatment of Rheumatoid Arthritis: Final Results of a Global, Open-Label, Long-Term Extension Study. Arthritis Res. Ther. 2019, 21, 89. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.B.; van Vollenhoven, R.F.; Winthrop, K.L.; Zerbini, C.A.F.; Tanaka, Y.; Bessette, L.; Zhang, Y.; Khan, N.; Hendrickson, B.; Enejosa, J.V.; et al. Safety Profile of Upadacitinib in Rheumatoid Arthritis: Integrated Analysis from the Select Phase Iii Clinical Programme. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef]
- Fragoulis, G.E.; McInnes, I.B.; Siebert, S. Jak-Inhibitors. New Players in the Field of Immune-Mediated Diseases, Beyond Rheumatoid Arthritis. Rheumatology 2019, 58, i43–i54. [Google Scholar] [CrossRef] [Green Version]
- Ly, K.; Beck, K.M.; Smith, M.P.; Orbai, A.M.; Liao, W. Tofacitinib in the Management of Active Psoriatic Arthritis: Patient Selection and Perspectives. Psoriasis 2019, 9, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Biggioggero, M.; Becciolini, A.; Crotti, C.; Agape, E.; Favalli, E.G. Upadacitinib and Filgotinib: The Role of Jak1 Selective Inhibition in the Treatment of Rheumatoid Arthritis. Drugs Context. 2019, 8, 212595. [Google Scholar] [CrossRef] [PubMed]
- Kucharz, E.J.; Stajszczyk, M.; Kotulska-Kucharz, A.; Batko, B.; Brzosko, M.; Jeka, S.; Leszczynski, P.; Majdan, M.; Olesinska, M.; Samborski, W.; et al. Tofacitinib in the Treatment of Patients with Rheumatoid Arthritis: Position Statement of Experts of the Polish Society for Rheumatology. Reumatologia 2018, 56, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Raedler, L.A. Jakafi (Ruxolitinib): First Fda-Approved Medication for the Treatment of Patients with Polycythemia Vera. Am. Health Drug Benefits 2015, 8, 75–79. [Google Scholar] [PubMed]
- Plosker, G.L. Ruxolitinib: A Review of Its Use in Patients with Myelofibrosis. Drugs 2015, 75, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Becker, H.; Engelhardt, M.; von Bubnoff, N.; Wasch, R. Ruxolitinib. Recent Results Cancer Res. 2014, 201, 249–257. [Google Scholar]
- Markham, A.; Keam, S.J. Peficitinib: First Global Approval. Drugs 2019, 79, 887–891. [Google Scholar] [CrossRef]
- Tarrant, J.M.; Galien, R.; Li, W.; Goyal, L.; Pan, Y.; Hawtin, R.; Zhang, W.; Van der Aa, A.; Taylor, P.C. Filgotinib, a Jak1 Inhibitor, Modulates Disease-Related Biomarkers in Rheumatoid Arthritis: Results from Two Randomized, Controlled Phase 2b Trials. Rheumatol. Ther. 2020, 7, 173–190. [Google Scholar] [CrossRef] [Green Version]
- Duggan, S.; Keam, S.J. Upadacitinib: First Approval. Drugs 2019, 79, 1819–1828. [Google Scholar] [CrossRef]
- Fleischmann, R.; Pangan, A.L.; Song, I.H.; Mysler, E.; Bessette, L.; Peterfy, C.; Durez, P.; Ostor, A.J.; Li, Y.; Zhou, Y.; et al. Upadacitinib Versus Placebo or Adalimumab in Patients with Rheumatoid Arthritis and an Inadequate Response to Methotrexate: Results of a Phase Iii, Double-Blind, Randomized Controlled Trial. Arthritis Rheumatol. 2019, 71, 1788–1800. [Google Scholar] [CrossRef]
- Fleischmann, R.M.; Genovese, M.C.; Enejosa, J.V.; Mysler, E.; Bessette, L.; Peterfy, C.; Durez, P.; Ostor, A.; Li, Y.; Song, I.H. Safety and Effectiveness of Upadacitinib or Adalimumab Plus Methotrexate in Patients with Rheumatoid Arthritis over 48 Weeks with Switch to Alternate Therapy in Patients with Insufficient Response. Ann. Rheum. Dis. 2019, 78, 1454–1462. [Google Scholar] [CrossRef] [Green Version]
- Van der Heijde, D.; Dougados, M.; Chen, Y.C.; Greenwald, M.; Drescher, E.; Klar, R.; Xie, L.; de la Torre, I.; Rooney, T.P.; Witt, S.L.; et al. Effects of Baricitinib on Radiographic Progression of Structural Joint Damage at 1 Year in Patients with Rheumatoid Arthritis and an Inadequate Response to Conventional Synthetic Disease-Modifying Antirheumatic Drugs. RMD Open 2018, 4, e000662. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijde, D.; Strand, V.; Tanaka, Y.; Keystone, E.; Kremer, J.; Zerbini, C.A.F.; Cardiel, M.H.; Cohen, S.; Nash, P.; Song, Y.W.; et al. Tofacitinib in Combination with Methotrexate in Patients with Rheumatoid Arthritis: Clinical Efficacy, Radiographic, and Safety Outcomes from a Twenty-Four-Month, Phase III Study. Arthritis Rheumatol. 2019, 71, 878–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokota, K.; Sato, K.; Miyazaki, T.; Kitaura, H.; Kayama, H.; Miyoshi, F.; Araki, Y.; Akiyama, Y.; Takeda, K.; Mimura, T. Combination of Tumor Necrosis Factor Alpha and Interleukin-6 Induces Mouse Osteoclast-Like Cells with Bone Resorption Activity Both In Vitro and In Vivo. Arthritis Rheumatol. 2014, 66, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Ohira, T. Mechanisms and Therapeutic Targets for Bone Damage in Rheumatoid Arthritis, in Particular the Rank-Rankl System. Curr. Opin. Pharmacol. 2018, 40, 110–119. [Google Scholar] [CrossRef]
- Poiana, C.; Capatina, C. Osteoporosis and Fracture Risk in Patients with Type 2 Diabetes Mellitus. Acta Endocrinol. 2019, 15, 231–236. [Google Scholar] [CrossRef]
- Lin, Y.C.; Wu, J.; Kuo, S.F.; Cheung, Y.C.; Sung, C.M.; Fan, C.M.; Chen, F.P.; Mhuircheartaigh, J.N. Vertebral Fractures in Type 2 Diabetes Patients: Utility of Trabecular Bone Score and Relationship with Serum Bone Turnover Biomarkers. J. Clin. Densitom. 2019, 23, 37–43. [Google Scholar] [CrossRef]
- Van Onna, M.; Ozturk, B.; Starmans, M.; Peeters, R.; Boonen, A. Disease and Management Beliefs of Elderly Patients with Rheumatoid Arthritis and Comorbidity: A Qualitative Study. Clin. Rheumatol. 2018, 37, 2367–2372. [Google Scholar] [CrossRef] [Green Version]
- Nicolau, J.; Lequerre, T.; Bacquet, H.; Vittecoq, O. Rheumatoid Arthritis, Insulin Resistance, and Diabetes. Joint Bone Spine 2017, 84, 411–416. [Google Scholar] [CrossRef]
- Dougados, M. Comorbidities in Rheumatoid Arthritis. Curr. Opin. Rheumatol. 2016, 28, 282–288. [Google Scholar] [CrossRef]
- Jiang, P.; Li, H.; Li, X. Diabetes Mellitus Risk Factors in Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Clin. Exp. Rheumatol. 2015, 33, 115–121. [Google Scholar]
- Adami, G.; Fassio, A.; Rossini, M.; Caimmi, C.; Giollo, A.; Orsolini, G.; Viapiana, O.; Gatti, D. Osteoporosis in Rheumatic Diseases. Int. J. Mol. Sci. 2019, 20, 5867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, C.J. The Epidemiology and Pathogenesis of Osteoporosis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dungan, K., Grossman, A., Hershman, J.M., Hofland, H.J., Kaltsas, G., et al., Eds.; Wilson: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Chidrawar, S.M.; Khan, N.; Chan, Y.L.; Nayak, L.; Moss, P.A. Ageing Is Associated with a Decline in Peripheral Blood Cd56bright Nk Cells. Immun. Ageing 2006, 3, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, M.; Nishioka, M.; Hanaki, N.; Miyauchi, T.; Kashiwagi, Y.; Kawasaki, Y.; Miki, H.; Kagawa, H.; Ioki, H.; Nakamura, Y. Postoperative Host Responses in Elderly Patients after Gastrointestinal Surgery. Hepatogastroenterology 2006, 53, 730–735. [Google Scholar] [PubMed]
- Kang, S.C.; Matsutani, T.; Choudhry, M.A.; Schwacha, M.G.; Rue, L.W.; Bland, K.I.; Chaudry, I.H. Are the Immune Responses Different in Middle-Aged and Young Mice Following Bone Fracture, Tissue Trauma and Hemorrhage? Cytokine 2004, 26, 223–230. [Google Scholar] [CrossRef]
- Smith, R.M. Immunity, Trauma and the Elderly. Injury 2007, 38, 1401–1404. [Google Scholar] [CrossRef]
- Woodland, D.L.; Blackman, M.A. Immunity and Age: Living in the Past? Trends Immunol. 2006, 27, 303–307. [Google Scholar] [CrossRef]
- Hadjiargyrou, M.; O’Keefe, R.J. The Convergence of Fracture Repair and Stem Cells: Interplay of Genes, Aging, Environmental Factors and Disease. J. Bone Miner. Res. 2014, 29, 2307–2322. [Google Scholar] [CrossRef]
- Bogoch, E.R.; Moran, E.L. Bone Abnormalities in the Surgical Treatment of Patients with Rheumatoid Arthritis. Clin. Orthop. Relat. Res. 1999, 366, 8–21. [Google Scholar] [CrossRef]
- Busti, A.J.; Hooper, J.S.; Amaya, C.J.; Kazi, S. Effects of Perioperative Antiinflammatory and Immunomodulating Therapy on Surgical Wound Healing. Pharmacotherapy 2005, 25, 1566–1591. [Google Scholar] [CrossRef]
- Dominiak, B.; Oxberry, W.; Chen, P. Study on a Nonhealing Fracture from a Patient with Systemic Lupus Erythematosus and Its Pathogenetic Mechanisms. Ultrastruct. Pathol. 2005, 29, 107–120. [Google Scholar] [CrossRef]
- Ramachandran, M. Basic Orthopaedic Sciences: The Stanmore Guide, 2nd ed.; Chapman and Hall/CRC: Boca Raton, FL, USA, 2018. [Google Scholar]
- Cho, T.J.; Gerstenfeld, L.C.; Einhorn, T.A. Differential Temporal Expression of Members of the Transforming Growth Factor Beta Superfamily during Murine Fracture Healing. J. Bone Miner. Res. 2002, 17, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, C.; Alpern, E.; Miclau, T.; Helms, J.A. Does Adult Fracture Repair Recapitulate Embryonic Skeletal Formation? Mech. Dev. 1999, 87, 57–66. [Google Scholar] [CrossRef]
- Kolar, P.; Schmidt-Bleek, K.; Schell, H.; Gaber, T.; Toben, D.; Schmidmaier, G.; Perka, C.; Buttgereit, F.; Duda, G.N. The Early Fracture Hematoma and Its Potential Role in Fracture Healing. Tissue Eng. Part B Rev. 2010, 16, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Silva, M.; Bahk, W.J.; McKellop, H.; Lieberman, J.R. Effect of Repeated Irrigation and Debridement on Fracture Healing in an Animal Model. J. Orthop. Res. 2002, 20, 1197–1204. [Google Scholar] [CrossRef]
- Hoff, P.; Gaber, T.; Strehl, C.; Schmidt-Bleek, K.; Lang, A.; Huscher, D.; Burmester, G.R.; Schmidmaier, G.; Perka, C.; Duda, G.N.; et al. Immunological Characterization of the Early Human Fracture Hematoma. Immunol. Res. 2016, 64, 1195–1206. [Google Scholar] [CrossRef]
- Kolar, P.; Gaber, T.; Perka, C.; Duda, G.N.; Buttgereit, F. Human Early Fracture Hematoma Is Characterized by Inflammation and Hypoxia. Clin. Orthop. Relat. Res. 2011, 469, 3118–3126. [Google Scholar] [CrossRef] [Green Version]
- Hoff, P.; Gaber, T.; Strehl, C.; Jakstadt, M.; Hoff, H.; Schmidt-Bleek, K.; Lang, A.; Rohner, E.; Huscher, D.; Matziolis, G.; et al. A Pronounced Inflammatory Activity Characterizes the Early Fracture Healing Phase in Immunologically Restricted Patients. Int. J. Mol. Sci. 2017, 18, 583. [Google Scholar] [CrossRef]
- Wagegg, M.; Gaber, T.; Lohanatha, F.L.; Hahne, M.; Strehl, C.; Fangradt, M.; Tran, C.L.; Schonbeck, K.; Hoff, P.; Ode, A.; et al. Hypoxia Promotes Osteogenesis but Suppresses Adipogenesis of Human Mesenchymal Stromal Cells in a Hypoxia-Inducible Factor-1 Dependent Manner. PLoS ONE 2012, 7, e46483. [Google Scholar] [CrossRef] [Green Version]
- Levy, O.; Ruvinov, E.; Reem, T.; Granot, Y.; Cohen, S. Highly Efficient Osteogenic Differentiation of Human Mesenchymal Stem Cells by Eradication of Stat3 Signaling. Int. J. Biochem. Cell Biol. 2010, 42, 1823–1830. [Google Scholar] [CrossRef]
- Claes, L.; Recknagel, S.; Ignatius, A. Fracture Healing under Healthy and Inflammatory Conditions. Nat. Rev. Rheumatol. 2012, 8, 133–143. [Google Scholar] [CrossRef]
- Bechman, K.; Yates, M.; Galloway, J.B. The New Entries in the Therapeutic Armamentarium: The Small Molecule Jak Inhibitors. Pharmacol. Res. 2019, 147, 104392. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Model System | Genes Modified | Species | Bone Phenotype | References |
|---|---|---|---|---|
| Janus kinases (JAKs) | ||||
| Jak1−/− | Jak1 deletion | Mouse | Small bone mass in contrast to wild-type mice; Perinatal lethal; Stunted embryos; Involved in bone formation | [27,28] |
| MMTV-Cre.Jak1fl/fl | ||||
| Jak1S645P+/− | Jak1 activation | Mouse | Low bone mass levels in trabecular and cortical bone; Bone formation and resorption is increased | [29] |
| Tofacitinib treatment | Jak1/3 inhibition | Mouse, rat | Protected against bone resorption by inflammation | [30,31,32] |
| Ruxolitinib treatment | Jak1/2 inhibition | Mouse | Protected against age-related bone resorption | [33] |
| Jak2−/− | Jak2 deletion | Mouse | Jak2-null mice die before bone formation starts; Lethality of anemia at E12.5 (erythropoiesis is absent); Involved in bone formation | [34,35,36] |
| Jak3−/− | Jak3 deletion | Mouse | Born normally; No gross abnormality | [37,38] |
| Tyk2−/− | Tyk2 deletion | Mouse | Viable and fertile mice; No obvious phenotype; Involved in bone formation | [39,40] |
| Signal transducers and activators of transcription (STATs) | ||||
| Stat1−/− | Stat1 deletion | Mouse | KO mice are indistinguishable compared to wild-type mice; Higher bone mass → osteopetrotic bone phenotype; Bone exhibits excessive osteoclastogenesis; Normal epiphyseal growth plate and longitudinal bone length; Characteristics: Pro-inflammatory, antagonize proliferation | [41,42,43] |
| Stat2−/− | Stat2 deletion | Mouse | Viable and fertile mice; No gross abnormality | [44] |
| Stat3−/− | Stat3 deletion in all cells | Mouse | Involved in early embryonic development; Lethality at E6.5–7.5; Selective inactivation causes osteoporosis; Surface mineralization reduced; Characteristics: Pro-proliferative, anti-inflammatory | [45,46,47,48] |
| Hyper-IgE syndrome | Stat3-DNA binding reduced in all cells | Mouse | Low bone mineral density; Recurrent fractures; Craniofacial and skeletal abnormalities | [49,50,51] |
| SA/SA and SA/− | Reduced Stat3 phosphorylation in all cells | Mouse | Perinatal lethality: 75%; SA/SA phenotype is normal; Stat3 phosphorylation in SA/− is reduced; Reduced skeletal size | [52] |
| Dmp1Cre.Stat3fl/fl | Stat3 deletion in osteocytes | Mouse | Low bone mass and reduced bone formation rate; Bone formation response to mechanical forced reduced | [53] |
| Col1α1(2.3 kb) Cre; Stat3flox/flox | Stat3 deletion in osteoblasts and osteocytes | Mouse | Low trabecular bone mass and bone formation rate reduced; Normal bone length; Bone formation response to mechanical forced reduced | [46,47,54,55] |
| Col1α1(3.6 kb) Cre; Stat3flox/flox | Stat3 deletion in chondrocytes, osteoblasts, and osteocytes | Mouse | Skeletal size is very small with low trabecular bone mass; Bone formation rate reduced and osteoclast formation increased | [47,55] |
| Prrx1Cre; Stat3flox/flox | Stat3 deletion in chondrocytes, osteoblasts, and osteocytes | Mouse | Skeletal size reduced; Postnatal limb curvature | [56] |
| TCre.Stat3f/f | Stat3 deletion in mesoderm-derived cells | Mouse | Shortened limbs at birth; Postnatal limb curvature | [56] |
| Tie2(Tek)Cre.Stat3f/f | Stat3 deletion in hematopoietic and endothelial cells | Mouse | Skeletal size and bone mass are reduced; Bone formation rate reduced with increased resorption | [57] |
| Socs3−/− | Socs3 deletion; elevated Stat3 signaling in all cells | Mouse | Embryonic lethality | [58,59] |
| VavCre.Socs3f/f | Elevated Stat3 signaling in endothelial and hematopoietic cells | Mouse | Joint inflammation; Low bone mass; Increased osteoblast and osteoclast formation | [60] |
| Dmp1Cre.Socs3f/f | Elevated Stat3 signaling in osteocytes | Mouse | Cortical porosity increased →delayed development of cortical bone; Increased bone formation and resorption | [61] |
| Dmp1Cre.Socs3f/f. IL6−/− | Elevated Stat3 signaling in osteocytes; no downstream of IL-6 | Mouse | Cortical porosity increased →delayed development of cortical bone | [61] |
| Col2Cre.Socs3f/f | Elevated Stat3 signaling in chondrocytes, osteoblasts and osteocytes | Mouse | Cortical porosity increased; Bone size reduced | [62] |
| Stat4−/− | Stat4−/− deletion | Mouse | Viable and fertile mice; No gross abnormality | [63] |
| Stat5a/b−/− | Double mutation | Mouse | KO mice show obviously defective bone development; Smaller Stat5a/5b (male and female) KO mice and Stat5b (male) KO mice compared to wild-type mice | [64,65] |
| Stat5a−/− | Stat5a deletion | Mouse | Increased bone mass; Increased trabecular bone density and cortical bone formation; Prevented age-related bone loss | [66] |
| Cathepsin K–Cre−/−Stat5fl/fl | Osteoclast-specific deletion | Mouse | Reduced bone mass | [67] |
| Stat6−/− | Stat6 deletion | Mouse | Viable and fertile mice; No gross abnormality compared to their wild-type controls | [68,69,70] |
| JAK Inhibitor | Specificity | FDA and/or EMA Approved | Indication (Trial) | Refs. |
|---|---|---|---|---|
| Tofacitinib | JAK1/JAK3 > JAK2, TYK2 | RA, PsA, JIA, UC | SpA, Ps, AA, AD, SLE, DLE, CS, CD, DM, dSc | [109,110,111,112] |
| Ruxolitinib | JAK1/JAK2 > TYK2 | PCV, MF, GVHD | RA, Ps, AA, BLL, TLL, CD, AD, Vit, HPS | [109,113,114,115] |
| Baricitinib | JAK1/JAK2 | RA | JIA, SLE, AA, GCA, AD, Ps, | [109,111,116] |
| Peficitinib | Pan-JAK | RA | Ps, UC | [109,116] |
| Filgotinib | JAK1 | RA | CD, Small bowel CD, Fistulizing CD, UC, CLE, NIU, PsA, AS, SS, Uveitis | [109,111,117] |
| Itacitinib | JAK1 | - | RA, GVHD, UC, Ps, ALL | [109] |
| SHR0302 | JAK1 > JAK2, JAK3 | - | RA, AS, GVHD, AD, UC, CD, AA | [109] |
| PF-04965842 | JAK1 | AD | Ps | [109] |
| Upadacitinib | JAK1 | RA | PsA, AS, UC, AD, CD, GCA, JIA, SpA, SLE | [109,111,118] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damerau, A.; Gaber, T.; Ohrndorf, S.; Hoff, P. JAK/STAT Activation: A General Mechanism for Bone Development, Homeostasis, and Regeneration. Int. J. Mol. Sci. 2020, 21, 9004. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239004
Damerau A, Gaber T, Ohrndorf S, Hoff P. JAK/STAT Activation: A General Mechanism for Bone Development, Homeostasis, and Regeneration. International Journal of Molecular Sciences. 2020; 21(23):9004. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239004
Chicago/Turabian StyleDamerau, Alexandra, Timo Gaber, Sarah Ohrndorf, and Paula Hoff. 2020. "JAK/STAT Activation: A General Mechanism for Bone Development, Homeostasis, and Regeneration" International Journal of Molecular Sciences 21, no. 23: 9004. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239004