A Multi-Strategy Sequencing Workflow in Inherited Retinal Dystrophies: Routine Diagnosis, Addressing Unsolved Cases and Candidate Genes Identification

Abstract
:1. Introduction
2. Results
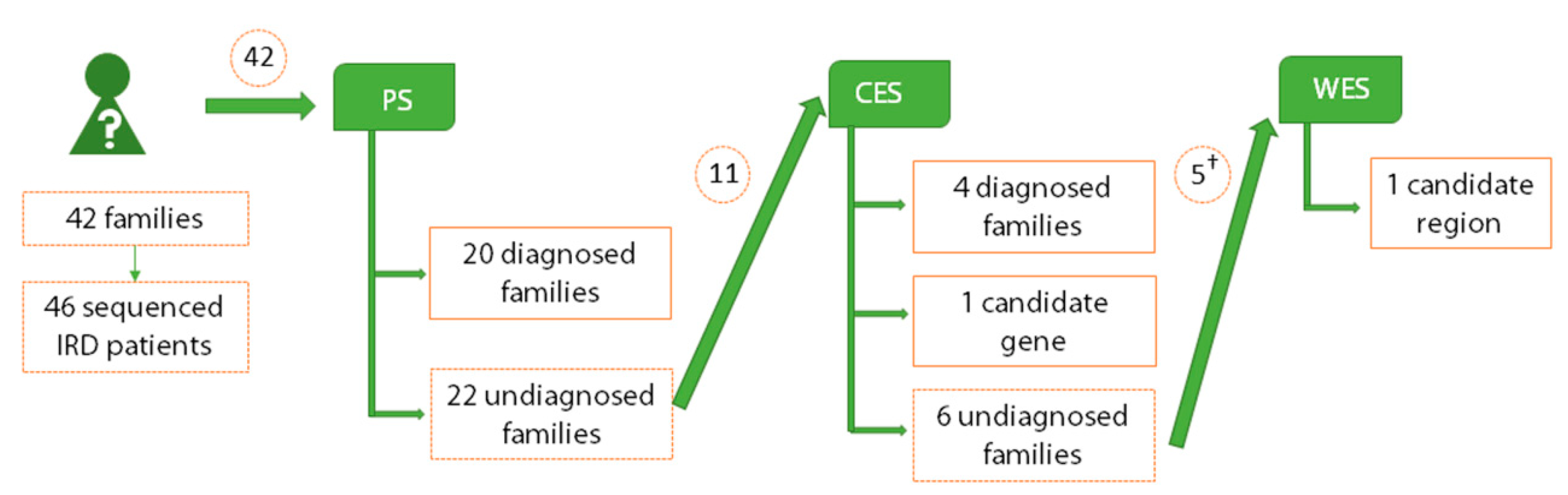
2.1. Genetic Diagnosis and Screening Unsolved Cases by Panel Sequencing
2.2. Detection of Pathogenic Mutations in Retinal Genes by Clinical Exome Sequencing
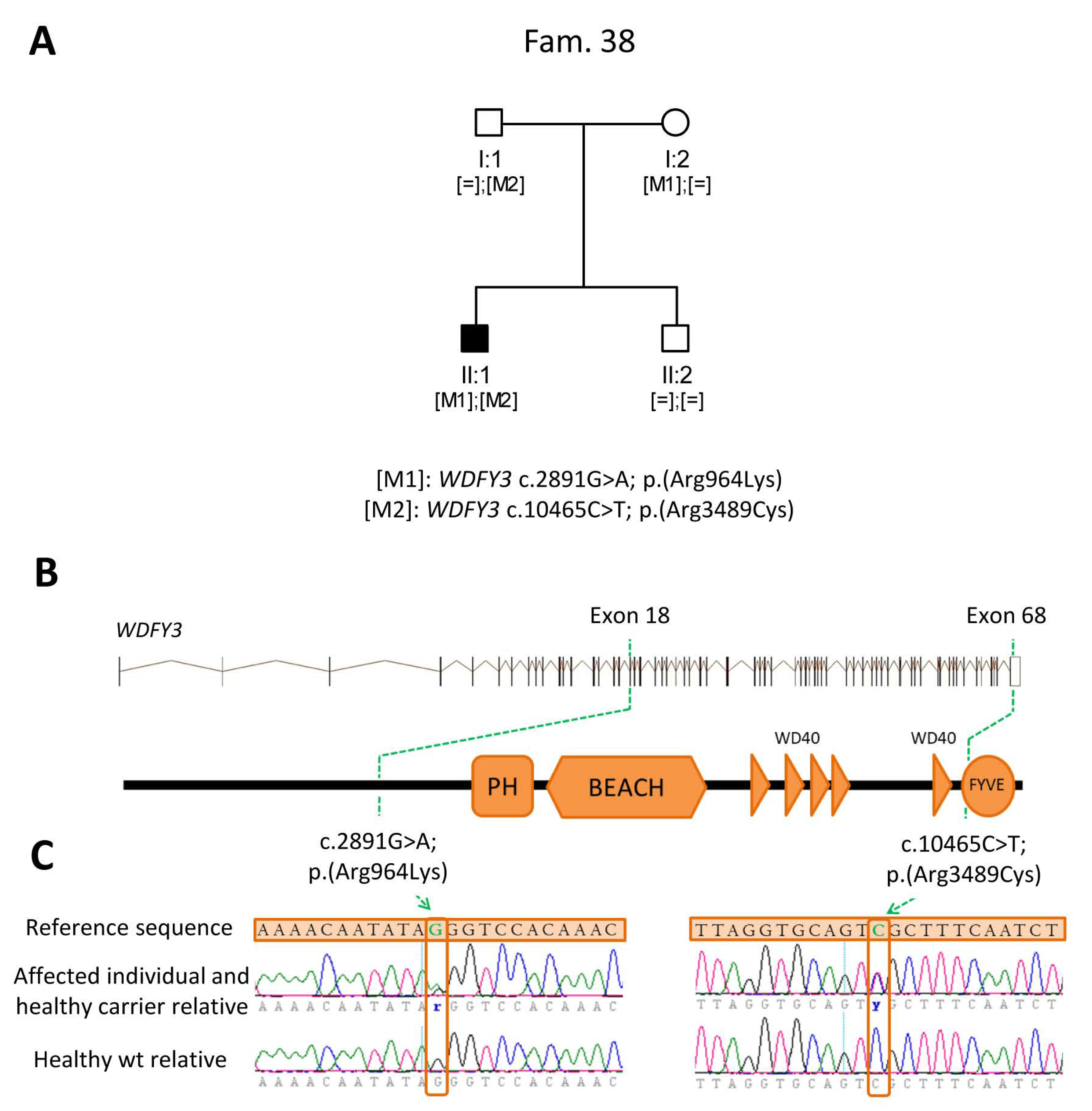
2.3. Identification of Novel Candidate Genes by Clinical Exome Sequencing
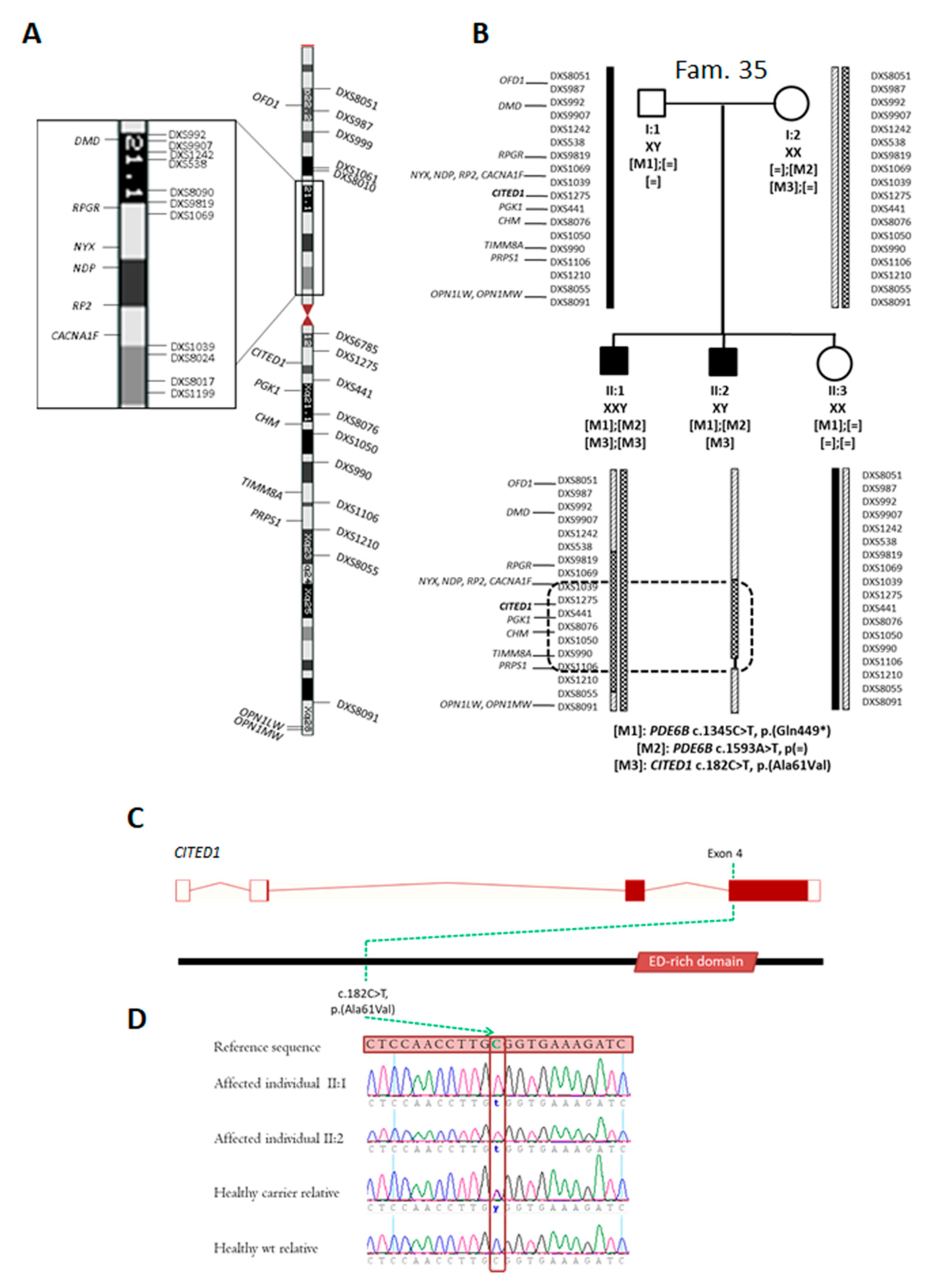
2.4. Identification of Novel Candidate Regions by Whole Exome Sequencing
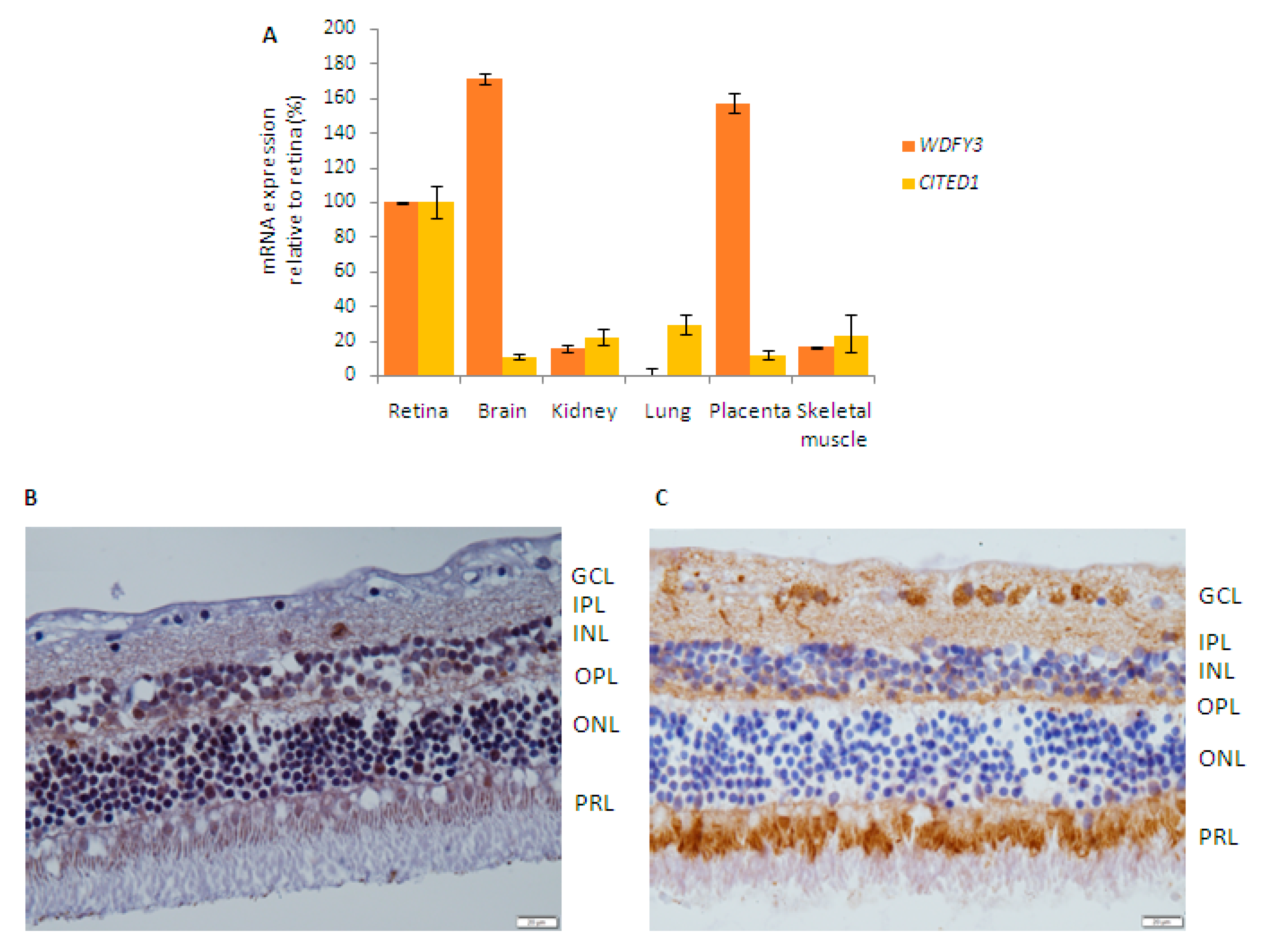
2.5. Retinal Expression of Candidate Genes
2.6. Comparison among Applied Sequencing Methods
3. Discussion
4. Materials and Methods
4.1. Patients Recruitment and Clinical Assessment
4.2. Targeted Panel Sequencing
4.3. Clinical Exome Sequencing
4.4. Whole-Exome Sequencing
4.5. Bioinformatics Analysis
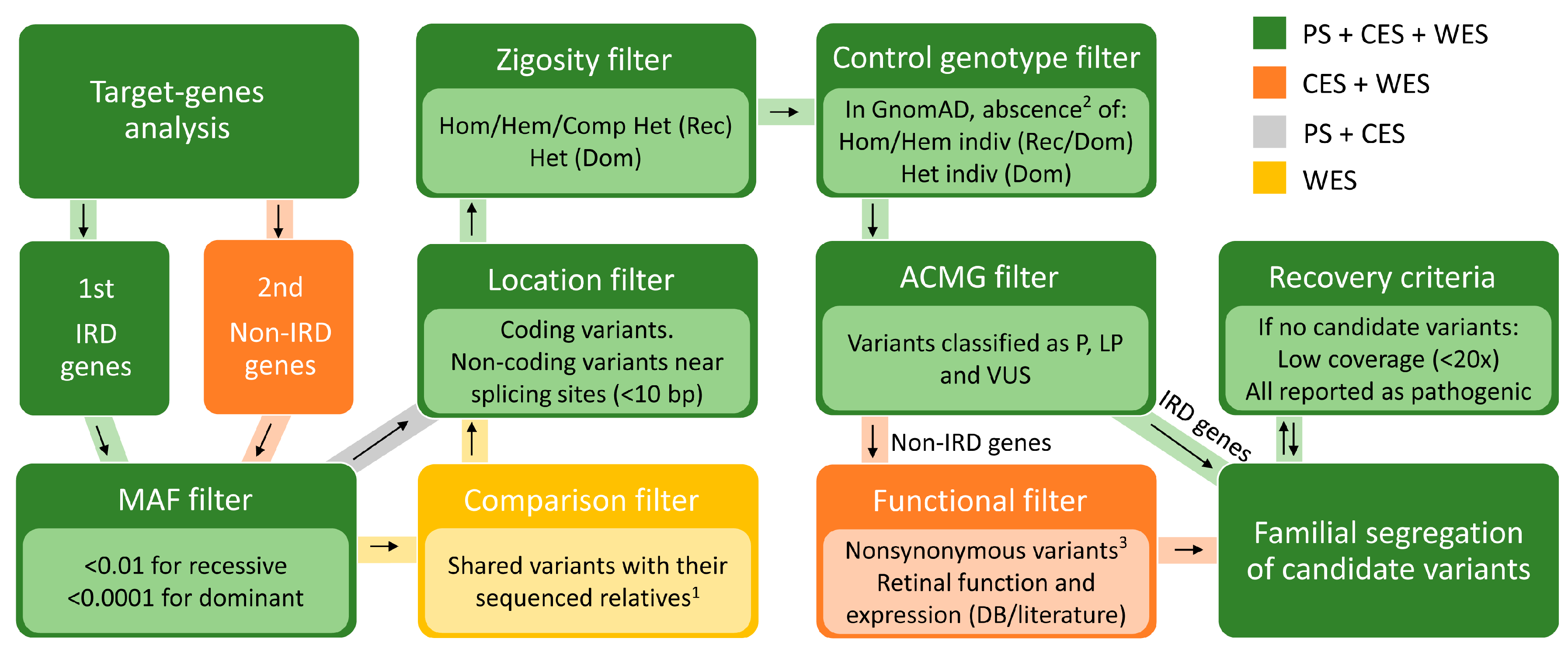
4.6. Prioritization of Identified Variants
4.7. Pathogenicity Evaluation of Variants and Family Segregation Studies
4.8. Expression and Localization Studies in Human Retina
4.9. Comparative Study of the Sequencing Strategies
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CES | Clinical Exome Sequencing |
| CNV | Copy-Number Variations |
| GnomAD | Genome Aggregation Database |
| IRD | Inherited Retinal Dystrophies |
| NGS | Next Generation Sequencing |
| RP | Retinitis Pigmentosa |
| STR | Short Tandem Repeats |
| VUS | Variants of Unknown Significance |
| WES | Whole Exome Sequencing |
| WGS | Whole Genome Sequencing |
References
- Farrar, G.J.; Carrigan, M.; Dockery, A.; Millington-Ward, S.; Palfi, A.; Chadderton, N.; Humphries, M.; Kiang, A.S.; Kenna, P.F.; Humphries, P. Toward an elucidation of the molecular genetics of inherited retinal degenerations. Hum. Mol. Genet. 2017, 26, R2–R11. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Gil, N.; Méndez-Vidal, C.; Romero-Pérez, L.; Pozo, M.G.-D.; La Rúa, E.R.-D.; Dopazo, J.; Borrego, S.; Antiñolo, G. Improving the management of Inherited Retinal Dystrophies by targeted sequencing of a population-specific gene panel. Sci. Rep. 2016, 6, 23910. [Google Scholar] [CrossRef] [PubMed]
- Hafler, B.P. Clinical Progress in Inherited Retinal Degenerations: Gene Therapy Clinical Trials and Advances in Genetic Sequencing. Retina 2017, 37, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Hayward, J.D.; Tailor, V.; Nyanhete, R.; Ahlfors, H.; Gabriel, C.; Jannini, T.B.; Abbou-Rayyah, Y.; Henderson, R.; Nischal, K.K.; et al. The Oculome Panel Test: Next-generation sequencing to diagnose a diverse range of genetic developmental eye disorders. Ophthalmology 2019, 126, 888–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhang, J.; Chen, N.; Wang, L.; Zhang, F.; Ma, Z.; Li, G.; Yang, L. Application of Whole Exome and Targeted Panel Sequencing in the Clinical Molecular Diagnosis of 319 Chinese Families with Inherited Retinal Dystrophy and Comparison Study. Genes 2018, 9, 360. [Google Scholar] [CrossRef] [Green Version]
- Consugar, M.B.; Navarro-Gomez, D.; Place, E.M.; Bujakowska, K.M.; Sousa, M.E.; Fonseca-Kelly, Z.D.; Taub, D.G.; Janessian, M.; Wang, D.Y.; Au, E.D.; et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet. Med. 2015, 17, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Karali, M.; Testa, F.; Brunetti-Pierri, R.; Di Iorio, V.; Pizzo, M.; Melillo, P.; Barillari, M.R.; Torella, A.; Musacchia, F.; D’Angelo, L.; et al. Clinical and Genetic Analysis of a European Cohort with Pericentral Retinitis Pigmentosa. Int. J. Mol. Sci. 2019, 21, 86. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Tao, T.; Zhao, L.; Li, G.; Yang, L. Molecular diagnosis based on comprehensive genetic testing in 800 Chinese families with non-syndromic inherited retinal dystrophies. Clin. Exp. Ophthalmol. 2020. [Google Scholar] [CrossRef]
- Dopazo, J.; Amadoz, A.; Bleda, M.; Garcia-Alonso, L.; Alemán, A.; García-García, F.; Rodriguez, J.A.; Daub, J.T.; Muntané, G.; Rueda, A.; et al. 267 Spanish Exomes Reveal Population-Specific Differences in Disease-Related Genetic Variation. Mol. Biol. Evol. 2016, 33, 1205–1218. [Google Scholar] [CrossRef] [Green Version]
- Schwarze, K.; Buchanan, J.; Taylor, J.C.; Wordsworth, S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet. Med. 2018, 20, 1122–1130. [Google Scholar] [CrossRef] [Green Version]
- Botstein, D.; Risch, N. Discovering genotypes underlying human phenotypes: Past successes for mendelian disease, future approaches for complex disease. Nat. Genet. 2003, 33, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Scholl, U.I.; Ji, W.; Liu, T.; Tikhonova, I.R.; Zumbo, P.; Nayir, A.; Bakkaloglu, A.; Ozen, S.; Sanjad, S.; et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. USA 2009, 106, 19096–19101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollander, A.I.D.; Koenekoop, R.K.; Yzer, S.; Lopez, I.; Arends, M.L.; Voesenek, K.E.J.; Zonneveld, M.N.; Strom, T.M.; Meitinger, T.; Brunner, H.G.; et al. Mutations in the CEP290 (NPHP6) Gene Are a Frequent Cause of Leber Congenital Amaurosis. Am. J. Hum. Genet. 2006, 79, 556–561. [Google Scholar] [CrossRef] [Green Version]
- Bax, N.M.; Sangermano, R.; Roosing, S.; Thiadens, A.A.H.J.; Hoefsloot, L.H.; Born, L.I.V.D.; Phan, M.; Klevering, B.J.; Haaften, C.W.-V.; Braun, T.A.; et al. Heterozygous Deep-Intronic Variants and Deletions inABCA4in Persons with Retinal Dystrophies and One ExonicABCA4Variant. Hum. Mutat. 2015, 36, 43–47. [Google Scholar] [CrossRef]
- Pozo, M.G.-D.; Martín-Sánchez, M.; Bravo-Gil, N.; Méndez-Vidal, C.; Chimenea, Á.; La Rúa, E.R.-D.; Borrego, S.; Antiñolo, G. Searching the second hit in patients with inherited retinal dystrophies and monoallelic variants in ABCA4, USH2A and CEP290 by whole-gene targeted sequencing. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Pozo, M.G.-d.; Mendez-Vidal, C.; Bravo-Gil, N.; Vela-Boza, A.; Dopazo, J.; Borrego, S.; Antinolo, G. Exome sequencing reveals novel and recurrent mutations with clinical significance in inherited retinal dystrophies. PLoS ONE 2014, 9, e116176. [Google Scholar] [CrossRef]
- Jamuar, S.S.; Tan, E.-C. Clinical application of next-generation sequencing for Mendelian diseases. Hum. Genom. 2015, 9, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Celestino-Soper, P.B.S.; Gao, H.; Lynnes, T.C.; Lin, H.; Liu, Y.; Spoonamore, K.G.; Chen, P.-S.; Vatta, M. Validation and Utilization of a Clinical Next-Generation Sequencing Panel for Selected Cardiovascular Disorders. Front. Cardiovasc. Med. 2017, 4, 11. [Google Scholar] [CrossRef] [Green Version]
- Saudi Mendeliome Group Comprehensive gene panels provide advantages over clinical exome sequencing for Mendelian diseases. Genome Biol. 2015, 16, 1–14. [CrossRef] [Green Version]
- Rozet, J.-M.; Gerber, S.; Souied, E.; Perrault, I.; Châtelin, S.; Ghazi, I.; Leowski, C.; Dufier, J.-L.; Munnich, A.; Kaplan, J. Spectrum of ABCR gene mutations in autosomal recessive macular dystrophies. Eur. J. Hum. Genet. 1998, 6, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Lewis, R.A.; Shroyer, N.F.; Singh, N.; Allikmets, R.; Hutchinson, A.; Li, Y.; Lupski, J.R.; Leppert, M.; Dean, M. Genotype/Phenotype Analysis of a Photoreceptor-Specific ATP-Binding Cassette Transporter Gene, ABCR, in Stargardt Disease. Am. J. Hum. Genet. 1999, 64, 422–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riveiro-Alvarez, R.; Lopez-Martinez, M.-A.; Zernant, J.; Aguirre-Lamban, J.; Cantalapiedra, D.; Avila-Fernandez, A.; Gimenez, A.; Lopez-Molina, M.-I.; Garcia-Sandoval, B.; Blanco-Kelly, F.; et al. Outcome of ABCA4 disease-associated alleles in autosomal recessive retinal dystrophies: Retrospective analysis in 420 Spanish families. Ophthalmology 2013, 120, 2332–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaioannou, M.; Ocaka, L.; Bessant, D.; Lois, N.; Bird, A.; Payne, A.; Bhattacharya, S. An analysis of ABCR mutations in British patients with recessive retinal dystrophies. Investig. Ophthalmol. Vis. Sci. 2000, 41, 16–19. [Google Scholar]
- Barragan, I.; Borrego, S.; Pieras, J.I.; Pozo, M.G.-d.; Santoyo, J.; Ayuso, C.; Baiget, M.; Millan, J.M.; Mena, M.; El-Aziz, M.M.A.; et al. Mutation spectrum of EYS in Spanish patients with autosomal recessive retinitis pigmentosa. Hum. Mutat. 2010, 31, E1772–E1800. [Google Scholar] [CrossRef] [Green Version]
- Pozo, M.G.-D.; Borrego, S.; Barragan, I.; Pieras, J.I.; Santoyo-Lopez, J.; Matamala, N.; Naranjo, B.; Dopazo, J.; Antinolo, G. Mutation Screening of Multiple Genes in Spanish Patients with Autosomal Recessive Retinitis Pigmentosa by Targeted Resequencing. PLoS ONE 2011, 6, e27894. [Google Scholar] [CrossRef] [Green Version]
- Eudy, J.D.; Weston, M.D.; Yao, S.; Hoover, D.M.; Rehm, H.L.; Ma-Edmonds, M.; Yan, D.; Ahmad, I.; Cheng, J.J.; Ayuso, C.; et al. Mutation of a Gene Encoding a Protein with Extracellular Matrix Motifs in Usher Syndrome Type IIa. Science 1998, 280, 1753–1757. [Google Scholar] [CrossRef]
- Baux, D.; Larrieu, L.; Blanchet, C.; Hamel, C.; Salah, S.B.; Vielle, A.; Gilbert-Dussardier, B.; Holder, M.; Calvas, P.; Philip, N.; et al. Molecular and in silico analyses of the full-length isoform of usherin identify new pathogenic alleles in Usher type II patients. Hum. Mutat. 2007, 28, 781–789. [Google Scholar] [CrossRef]
- McLaughlin, M.E.; Ehrhart, T.L.; Berson, E.L.; Dryja, T.P. Mutation spectrum of the gene encoding the beta subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 1995, 92, 3249–3253. [Google Scholar] [CrossRef] [Green Version]
- Rivolta, C.; Sweklo, E.A.; Berson, E.L.; Dryja, T.P. Missense Mutation in the USH2A Gene: Association with Recessive Retinitis Pigmentosa without Hearing Loss. Am. J. Hum. Genet. 2000, 66, 1975–1978. [Google Scholar] [CrossRef] [Green Version]
- Martin-Merida, I.; Avila-Fernandez, A.; Del Pozo-Valero, M.; Blanco-Kelly, F.; Zurita, O.; Perez-Carro, R.; Aguilera-Garcia, D.; Riveiro-Alvarez, R.; Arteche, A.; Trujillo-Tiebas, M.J.; et al. Genomic Landscape of Sporadic Retinitis Pigmentosa: Findings from 877 Spanish Cases. Ophthalmology 2019, 126, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Allikmets, R.; Singh, N.; Sun, H.; Shroyer, N.F.; Hutchinson, A.; Chidambaram, A.; Gerrard, B.; Baird, L.; Stauffer, D.; Peiffer, A.; et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 1997, 15, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Kitiratschky, V.B.; Grau, T.; Bernd, A.; Zrenner, E.; Jägle, H.; Renner, A.B.; Kellner, U.; Rudolph, G.; Jacobson, S.G.; Cideciyan, A.V.; et al. ABCA4 gene analysis in patients with autosomal recessive cone and cone rod dystrophies. Eur. J. Hum. Genet. 2008, 16, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Weston, M.D.; Eudy, J.D.; Fujita, S.; Yao, S.-F.; Usami, S.; Cremers, C.; Greenburg, J.; Ramesar, R.; Martini, A.; Moller, C.; et al. Genomic Structure and Identification of Novel Mutations in Usherin, the Gene Responsible for Usher Syndrome Type IIa. Am. J. Hum. Genet. 2000, 66, 1199–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allikmets, R.; Shroyer, N.F.; Singh, N.; Seddon, J.M.; Lewis, R.A.; Bernstein, P.S.; Peiffer, A.; Zabriskie, N.A.; Li, Y.; Hutchinson, A.; et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science 1997, 277, 1805–1807. [Google Scholar] [CrossRef] [Green Version]
- Stoetzel, C.; Laurier, V.; Davis, E.E.; Muller, J.; Rix, S.; Badano, J.L.; Leitch, C.C.; Salem, N.; Chouery, E.; Corbani, S.; et al. BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat. Genet. 2006, 38, 521–524. [Google Scholar] [CrossRef]
- Skorczyk-Werner, A.; Wawrocka, A.; Kochalska, N.; Krawczynski, M.R. Novel CHM mutations in Polish patients with choroideremia—An orphan disease with close perspective of treatment. Orphanet J. Rare Dis. 2018, 13, 221. [Google Scholar] [CrossRef]
- Neveling, K.; Collin, R.W.; Gilissen, C.; van Huet, R.A.; Visser, L.; Kwint, M.P.; Gijsen, S.J.; Zonneveld, M.N.; Wieskamp, N.; de Ligt, J.; et al. Next-generation genetic testing for retinitis pigmentosa. Hum. Mutat. 2012, 33, 963–972. [Google Scholar] [CrossRef]
- Sun, T.; Xu, K.; Ren, Y.; Xie, Y.; Zhang, X.; Tian, L.; Li, Y. Comprehensive Molecular Screening in Chinese Usher Syndrome Patients. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1229–1237. [Google Scholar] [CrossRef] [Green Version]
- Cremers, F.P.; van de Pol, D.J.; van Driel, M.; den Hollander, A.I.; van Haren, F.J.; Knoers, N.V.; Tijmes, N.; Bergen, A.A.; Rohrschneider, K.; Blankenagel, A.; et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum. Mol. Genet. 1998, 7, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Crespi, J.; Buil, J.A.; Bassaganyas, F.; Vela-Segarra, J.I.; Diaz-Cascajosa, J.; Ayala-Ramirez, R.; Zenteno, J.C. A novel mutation confirms MFRP as the gene causing the syndrome of nanophthalmos-renititis pigmentosa-foveoschisis-optic disk drusen. Am. J. Ophthalmol. 2008, 146, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Sandberg, M.A.; Rabe, V.W.; Stillberger, M.; Dryja, T.P.; Berson, E.L. RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am. J. Hum. Genet. 2003, 73, 1131–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmann, T.; Di Gioia, S.A.; Rau, I.; Stöhr, H.; Maksimovic, N.S.; Corbo, J.C.; Renner, A.B.; Zrenner, E.; Kumaramanickavel, G.; Karlstetter, M.; et al. Nonsense Mutations in FAM161A Cause RP28-Associated Recessive Retinitis Pigmentosa. Am. J. Hum. Genet. 2010, 87, 376–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filimonenko, M.; Isakson, P.; Finley, K.D.; Anderson, M.; Jeong, H.; Melia, T.J.; Bartlett, B.J.; Myers, K.M.; Birkeland, H.C.; Lamark, T.; et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell 2010, 38, 265–279. [Google Scholar] [CrossRef] [Green Version]
- Yahata, T.; de Caestecker, M.P.; Lechleider, R.J.; Andriole, S.; Roberts, A.B.; Isselbacher, K.J.; Shioda, T. The MSG1 non-DNA-binding transactivator binds to the p300/CBP coactivators, enhancing their functional link to the Smad transcription factors. J. Boil. Chem. 2000, 275, 8825–8834. [Google Scholar] [CrossRef] [Green Version]
- Howlin, J.; Cirenajwis, H.; Lettiero, B.; Staaf, J.; Lauss, M.; Saal, L.; Borg, A.; Gruvberger-Saal, S.; Jonsson, G. Loss of CITED1, an MITF regulator, drives a phenotype switch In Vitro and can predict clinical outcome in primary melanoma tumours. PeerJ 2015, 3, e788. [Google Scholar] [CrossRef] [Green Version]
- Wen, B.; Li, S.; Li, H.; Chen, Y.; Ma, X.; Wang, J.; Lu, F.; Qu, J.; Hou, L. Microphthalmia-associated transcription factor regulates the visual cycle genes Rlbp1 and Rdh5 in the retinal pigment epithelium. Sci. Rep. 2016, 6, 21208. [Google Scholar] [CrossRef] [Green Version]
- Kadir, R.; Harel, T.; Markus, B.; Perez, Y.; Bakhrat, A.; Cohen, I.; Volodarsky, M.; Feintsein-Linial, M.; Chervinski, E.; Zlotogora, J.; et al. ALFY-Controlled DVL3 Autophagy Regulates Wnt Signaling, Determining Human Brain Size. PLoS Genet. 2016, 12, e1005919. [Google Scholar] [CrossRef] [Green Version]
- Gil, N.I.B.; Pozo, M.G.-D.; Martín-Sánchez, M.; Méndez-Vidal, C.; La Rúa, E.R.-D.; Borrego, S.; Antiñolo, G. Unravelling the genetic basis of simplex Retinitis Pigmentosa cases. Sci. Rep. 2017, 7, srep41937. [Google Scholar] [CrossRef]
- Sánchez, M.J.M.; Valero, P.L.; Pozo, M.G.-D.; Zuñiga, B.P.; Antiñolo, G.; Jiménez, M.R.; Franch, E.R.D.L.R. Posterior microphthalmos, retinitis pigmentosa, and foveoschisis caused by a mutation in the MFRP gene: A familial study. Ophthalmic Genet. 2019, 40, 288–292. [Google Scholar] [CrossRef]
- Kammenga, J.E. The background puzzle: How identical mutations in the same gene lead to different disease symptoms. FEBS J. 2017, 284, 3362–3373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowling, J.E.; Wald, G. Proceedings of the National Academy of Sciences of the United States of America. Nutr. Rev. 2009, 39, 135–138. [Google Scholar] [CrossRef]
- Yao, J.; Jia, L.; Feathers, K.; Lin, C.; Khan, N.W.; Klionsky, D.J.; Ferguson, T.A.; Zacks, D.N. Autophagy-mediated catabolism of visual transduction proteins prevents retinal degeneration. Autophagy 2016, 12, 2439–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Asrag, M.E.; Sergouniotis, P.I.; McKibbin, M.; Plagnol, V.; Sheridan, E.; Waseem, N.; Abdelhamed, Z.; McKeefry, D.; Van Schil, K.; A Poulter, J.; et al. Biallelic mutations in the autophagy regulator DRAM2 cause retinal dystrophy with early macular involvement. Am. J. Hum. Genet. 2015, 96, 948–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, A.J.F.; Miller, J.H.; Suzuki, D.T.; Lewontin, R.C.; Gelbart, W.M. Mutant Types. In An Introduction to Genetic Analysis, 7th ed.; W. H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Mahdieh, N.; Rabbani, B. An Overview of Mutation Detection Methods in Genetic Disorders. Iran. J. Pediatr. 2013, 23, 375–388. [Google Scholar] [PubMed]
- Khan, K.N.; UK Inherited Retinal Disease Consortium; Robson, A.; Mahroo, O.A.R.; Arno, G.; Inglehearn, C.F.; Armengol, M.; Waseem, N.; Holder, G.E.; Carss, K.J.; et al. A clinical and molecular characterisation of CRB1-associated maculopathy. Eur. J. Hum. Genet. 2018, 26, 687–694. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, T.C. A Short History and Description of Drosophila melanogaster Classical Genetics: Chromosome Aberrations, Forward Genetic Screens, and the Nature of Mutations. Genetics 2017, 206, 665–689. [Google Scholar] [CrossRef] [Green Version]
- Macheroux, E.P.; Braunisch, M.C.; Pegler, S.P.; Satanovskij, R.; Riedhammer, K.M.; Günthner, R.; Gross, O.; Nagel, M.; Renders, L.; Hoefele, J. The Hypomorphic Variant p.(Gly624Asp) in COL4A5 as a Possible Cause for an Unexpected Severe Phenotype in a Family With X-Linked Alport Syndrome. Front. Pediatr. 2019, 7, 485. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Pavan, W.J. Transcriptional and signaling regulation in neural crest stem cell-derived melanocyte development: Do all roads lead to Mitf? Cell Res. 2008, 18, 1163–1176. [Google Scholar] [CrossRef]
- Burstedt, M.S.; Sandgren, O.; Holmgren, G.; Forsman-Semb, K. Bothnia dystrophy caused by mutations in the cellular retinaldehyde-binding protein gene (RLBP1) on chromosome 15q26. Investig. Ophthalmol. Vis. Sci. 1999, 40, 995–1000. [Google Scholar]
- Katsanis, N.; Shroyer, N.F.; Lewis, R.; Cavender, J.; Al-Rajhi, A.; Jabak, M.; Lupski, J. Fundus albipunctatus and retinitis punctata albescens in a pedigree with an R150Q mutation in RLBP1. Clin. Genet. 2001, 59, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Simon, A.; Eriksson, U.; Harris, E.; Berson, E.L.; Dryja, T.P. Mutations in the gene encoding 11-cis retinol dehydrogenase cause delayed dark adaptation and fundus albipunctatus. Nat. Genet. 1999, 22, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Kousi, M.; Katsanis, N. Genetic Modifiers and Oligogenic Inheritance. Cold Spring Harb. Perspect. Med. 2015, 5, a017145. [Google Scholar] [CrossRef] [Green Version]
- Pozo, M.G.-D.; Fernández-Suárez, E.; Martín-Sánchez, M.; Bravo-Gil, N.; Méndez-Vidal, C.; La Rúa, E.R.-D.; Borrego, S.; Antiñolo, G. Unmasking Retinitis Pigmentosa complex cases by a whole genome sequencing algorithm based on open-access tools: Hidden recessive inheritance and potential oligogenic variants. J. Transl. Med. 2020, 18, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Niazi, R.; Gonzalez, M.A.; Balciuniene, J.; Evans, P.; Sarmady, M.; Tayoun, A.N.A. The Development and Validation of Clinical Exome-Based Panels Using ExomeSlicer: Considerations and Proof of Concept Using an Epilepsy Panel. J. Mol. Diagn. 2018, 20, 643–652. [Google Scholar] [CrossRef]
- Posey, J.E. Genome sequencing and implications for rare disorders. Orphanet J. Rare Dis. 2019, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- The 1000 Genomes Project Consortium A global reference for human genetic variation. Nat. Cell Biol. 2015, 526, 68–74. [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macdonald, J.R.; Ziman, R.; Yuen, R.K.C.; Feuk, L.; Scherer, S.W. The Database of Genomic Variants: A curated collection of structural variation in the human genome. Nucleic Acids Res. 2014, 42, D986–D992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petryszak, R.; A Fonseca, N.; Füllgrabe, A.; Huerta, L.; Keays, M.; Tang, Y.A.; Brazma, A. The RNASeq-er API—A gateway to systematically updated analysis of public RNA-seq data. Bioinformatics 2017, 33, 2218–2220. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; MacArthur, D.G. The mutational constraint spectrum quantified from variation in 141,456 humans. Yearb. Paediatr. Endocrinol. 2020, 581, 434–443. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.B.; Daly, M.J.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Beroud, G.; Claustres, M.; Beroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.D.M.; Moore, M.W. The International Mouse Phenotyping Consortium: Past and future perspectives on mouse phenotyping. Mamm. Genome 2012, 23, 632–640. [Google Scholar] [CrossRef] [Green Version]
- Fokkema, I.F.A.C.; Taschner, P.E.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; Dunnen, J.T.D. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Boil. J. Comput. Mol. Cell Boil. 2003, 11, 322–331. [Google Scholar] [CrossRef]
- Baldarelli, R.M.; Smith, C.M.; Finger, J.H.; Hayamizu, T.F.; McCright, I.J.; Xu, J.; Shaw, D.R.; Beal, J.S.; Blodgett, O.; Campbell, J.; et al. The mouse Gene Expression Database (GXD): 2021 update. Nucleic Acids Res. 2020, 47, 84. [Google Scholar] [CrossRef] [PubMed]
- Wildeman, M.; Van Ophuizen, E.; Dunnen, J.T.D.; Taschner, P.E. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum. Mutat. 2008, 29, 6–13. [Google Scholar] [CrossRef]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Boil. J. Comput. Mol. Cell Boil. 1997, 4, 232–240. [Google Scholar] [CrossRef]
- Hamosh, A.; Scott, A.F.; Amberger, J.; Bocchini, C.; Valle, D.; McKusick, V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2002, 30, 52–55. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.; Sirotkin, K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999, 9, 677–679. [Google Scholar]
- Cooper, D.N.; Krawczak, M. Human Gene Mutation Database. Hum. Genet. 1996, 98, 629. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; E Chapple, C.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Chang, X.; Wang, K. wANNOVAR: Annotating genetic variants for personal genomes via the web. J. Med. Genet. 2012, 49, 433–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Vidal, C.; Bravo-Gil, N.; Pozo, M.G.-D.; Vela-Boza, A.; Dopazo, J.; Borrego, S.; Antiñolo, G. Novel RP1 mutations and a recurrent BBS1variant explain the co-existence of two distinct retinal phenotypes in the same pedigree. BMC Genet. 2014, 15, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Gene | Variants | Status | ACMG | Segr. (A/H) | Solved | Ref. |
|---|---|---|---|---|---|---|---|
| 2 | ABCA4 | c.3056C>T; p.(Thr1019Met) | Het | 5 | Yes (1/2) | Yes | [21] |
| c.3364G>A; p.(Glu1122Lys) | Het | 5 | [22] | ||||
| 3 | CHM | c.1797C>G; p.(Cys599Trp) | Hem | 3 | No | Yes | This study |
| 4 | ABCA4 | c.3287C>T; p.(Ser1096Leu) | Het | 4 | No | Yes | [23] |
| c.466A>G; p.(Ile156Val) | Het | 3 | [24] | ||||
| 6 | EYS | c.4451G>A; p.(Trp1484*) | Het | 5 | No | Yes | [25] |
| c.5928-2A>G; r.spl | Het | 5 | [26] | ||||
| 10 | USH2A | c.2299delG; p.(Glu767Serfs*21) | Het | 5 | Yes (2/5) | Yes | [27] |
| c.13374delA; p.(Glu4458Aspfs*3) | Het | 5 | [28] | ||||
| 12 | PDE6B | c.2193+1G>A; r.spl | Het | 5 | Yes (2/2) | Yes | [29] |
| c.1572delC; p.(Tyr525Thrfs*50) | Het | 5 | This study | ||||
| 13 | USH2A | c.2276G>T; p.(Cys759Phe) | Het | 5 | No | Yes | [30] |
| c.13531G>A; p.(Ala4511Thr) | Het | 3 | [31] | ||||
| 14 | WHRN | c.1417-8G>A; r.(spl?) | Hom | 3 | Yes (2/4) | No | ClinVar (45653 †) |
| 15 | USH2A | c.12546T>G; p.(Asn4182Lys) | Het | 3 | No | Yes | This study |
| c.13979C>G; p.(Pro4660Arg) | Het | 3 | GnomAD | ||||
| 19 | ABCA4 | c.5882G>A; p.(Gly1961Glu) | Het | 4 | Yes (3/3) | Yes | [32] |
| c.700C>T; p.(Gln234*) | Het | 5 | [33] | ||||
| 21 | USH2A | c.920_923dup; p.(His308Glnfs*16) | Het | 5 | Yes (2/6) | Yes | [34] |
| c.2276G>T; p.(Cys759Phe) | Het | 5 | [30] | ||||
| 23 | ABCA4 | c.3386G>T; p.(Arg1129Leu) | Het | 5 | Yes (1/4) | Yes | [35] |
| c.700C>T; p.(Gln234*) | Het | 5 | [33] | ||||
| 24 | ABCA4 | c.3386G>T; p.(Arg1129Leu) | Het | 5 | No | Yes | [35] |
| c.(2382+1_2383-1)_(2587+1_2588-1)del; p.? (Deletion of exon 16) | Het | 4 | This study | ||||
| 25 | BBS10 | c.273C>G; p.(Cys91Trp) | Hom | 5 | Yes (2/2) | Yes | [36] |
| 26 | CHM | c.83C>G; p.(Ser28*) | Hem | 5 | Yes (1/5) | Yes | [37] |
| 28 | PDE6B | c.1107+3A>G; r.(spl?) | Het | 4 | No | Yes | [38] |
| c.1969A>G; p.(Ile657Val) | Het | 3 | This study | ||||
| 29 | PROM1 | c.(1002+1_1003-1)_(1454+1_1455-1)del; p.? (Deletion of exons 9-12) | Hom | 4 | Yes (3/1) | Yes | This study |
| 30 | USH2A | c.2276G>T; p.(Cys759Phe) | Het | 5 | Yes (1/1) | Yes | [30] |
| c.6967C>T; p.(Arg2323*) | Het | 5 | [39] | ||||
| 31 | PDE6B | c.1923_1969delinsTCTGGG; p.(Asn643Glyfs*29) | Hom | 5 | Yes (1/5) | Yes | [38] |
| 33 | USH2A | c.2276G>T; p.(Cys759Phe) | Het | 5 | Yes (2/2) | Yes | [30] |
| c.14011G>T; p.(Glu4671*) | Het | 5 | [16] | ||||
| 34 | ABCA4 | c.5714+5G>A; r.(spl?) | Het | 5 | Yes (2/3) | Yes | [40] |
| c.223T>G; p.(Cys75Gly) | Het | 4 | [22] | ||||
| 35 | PDE6B | c.1345C>T, p.(Gln449*) | Het | 5 | Yes (2/3) | No | dbSNP ‡ |
| c.1593A>T, p.(=) | Het | 4 | This study |
| ID | Gen | Variants | Status | ACMG | Segr. (A/H) | Solved | Ref. |
|---|---|---|---|---|---|---|---|
| 17 | MFRP | c.498delC; p.(Asn167Thrfs*25) | Hom | 5 | Yes (3/2) | Yes | [41] |
| 36 | RPGR | c.2655_2656del; p.(Glu886Glyfs*192) | Hem | 5 | Yes (2/5) | Yes | [42] |
| 37 | FAM161A | c.1309A>T; p.(Arg437*) | Hom | 5 | Yes (1/4) | Yes | [43] |
| 40 | RP1L1 | c.5821C>T; p.(Gln1941*) | Het | 5 | Yes (1/6) | Yes | ClinVar (361237) |
| c.3412A>G; p.(Lys1138Glu) | Het | 3 | This study |
| Family | 38 | 35 | ||
| Applied strategy | CES | WES | ||
| Sequenced individual | II:1 | II:2 | ||
| Gene | WDFY3 | CITED1 | ||
| Variant | M1 | M2 | M3 | |
| Status | Het | Het | Hem | |
| Segr. (A/H) | Yes (1/3) | Yes (2/3) | ||
| Reference | Novel | Novel | Novel | |
| GnomAD GC (Het/Hem/Hom) | NA | 2/0/0 | 9/2/0 | |
| ACMG | VUS | VUS | VUS | |
| In silico prediction | CIP | PP | PB | |
| Conserv. | ClustalO | V | V | P |
| PhyloP | V (7.49) | V (4.22) | NC | |
| PhastCons | P (0.998) | P (0.995) | P (0.666) | |
| GERP | C (5.5) | C (5.8) | NC (1.3) | |
| Retinal expression | Yes | Yes | ||
| Reported function | Scaffolding protein in autophagy [44] | Transcriptional co-regulator [45] of MITF [46], associated with the pigmentation process and regulator of RLBP1 and RDH5 IRD- genes [47] | ||
| Previously associated with IRD | No | No | ||
| Animal models | DB | Perinatal lethality in mouse. | Placental disorders and neonatal lethality. | |
| Ref. | Eye-related phenotype in Drosophila [48] | NA | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martín-Sánchez, M.; Bravo-Gil, N.; González-del Pozo, M.; Méndez-Vidal, C.; Fernández-Suárez, E.; Rodríguez-de la Rúa, E.; Borrego, S.; Antiñolo, G. A Multi-Strategy Sequencing Workflow in Inherited Retinal Dystrophies: Routine Diagnosis, Addressing Unsolved Cases and Candidate Genes Identification. Int. J. Mol. Sci. 2020, 21, 9355. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249355
Martín-Sánchez M, Bravo-Gil N, González-del Pozo M, Méndez-Vidal C, Fernández-Suárez E, Rodríguez-de la Rúa E, Borrego S, Antiñolo G. A Multi-Strategy Sequencing Workflow in Inherited Retinal Dystrophies: Routine Diagnosis, Addressing Unsolved Cases and Candidate Genes Identification. International Journal of Molecular Sciences. 2020; 21(24):9355. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249355
Chicago/Turabian StyleMartín-Sánchez, Marta, Nereida Bravo-Gil, María González-del Pozo, Cristina Méndez-Vidal, Elena Fernández-Suárez, Enrique Rodríguez-de la Rúa, Salud Borrego, and Guillermo Antiñolo. 2020. "A Multi-Strategy Sequencing Workflow in Inherited Retinal Dystrophies: Routine Diagnosis, Addressing Unsolved Cases and Candidate Genes Identification" International Journal of Molecular Sciences 21, no. 24: 9355. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249355