Structural Basis for Design of New Purine-Based Inhibitors Targeting the Hydrophobic Binding Pocket of Hsp90

 , and
, and
Abstract
:
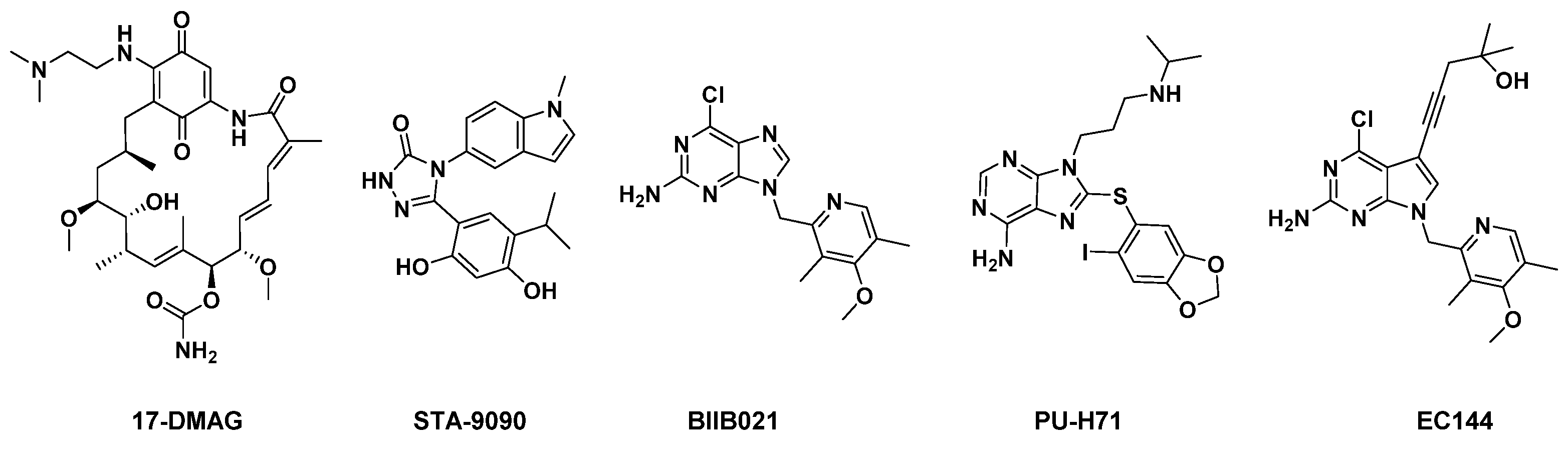
1. Introduction
2. Results and Discussion
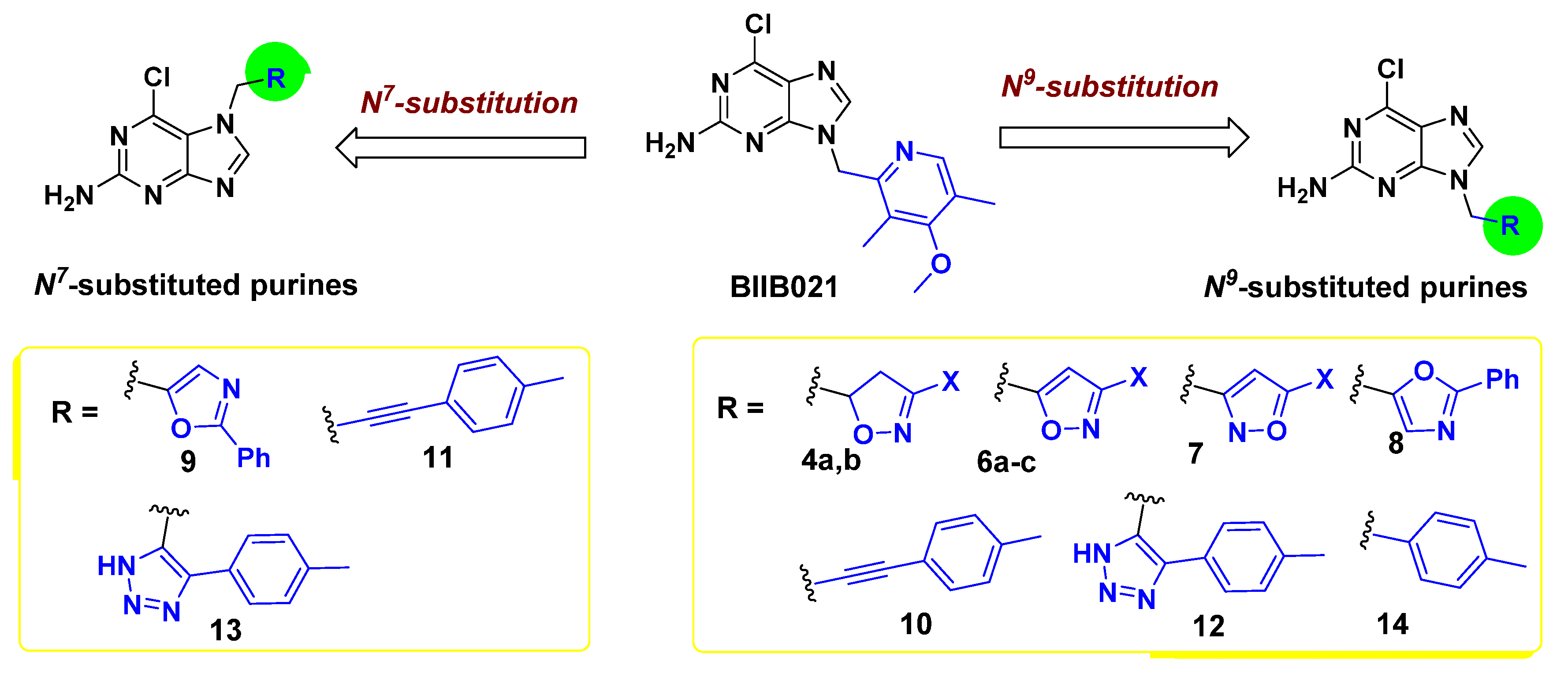
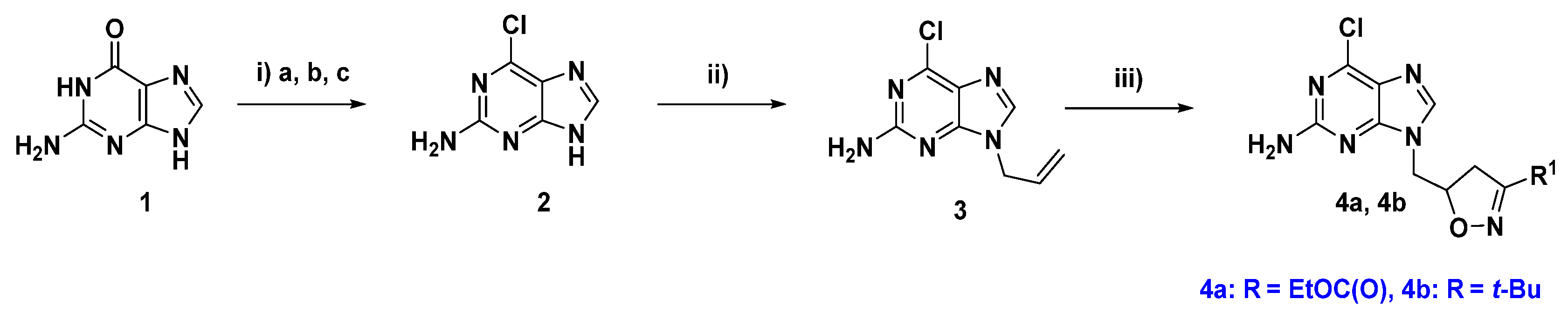
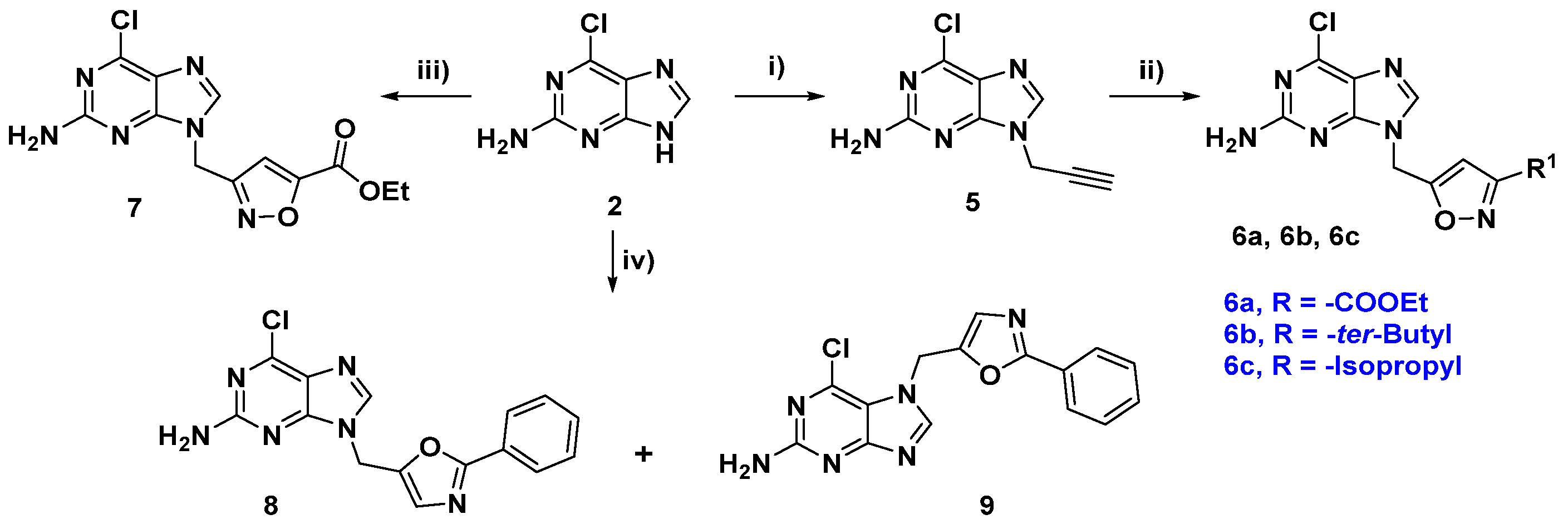
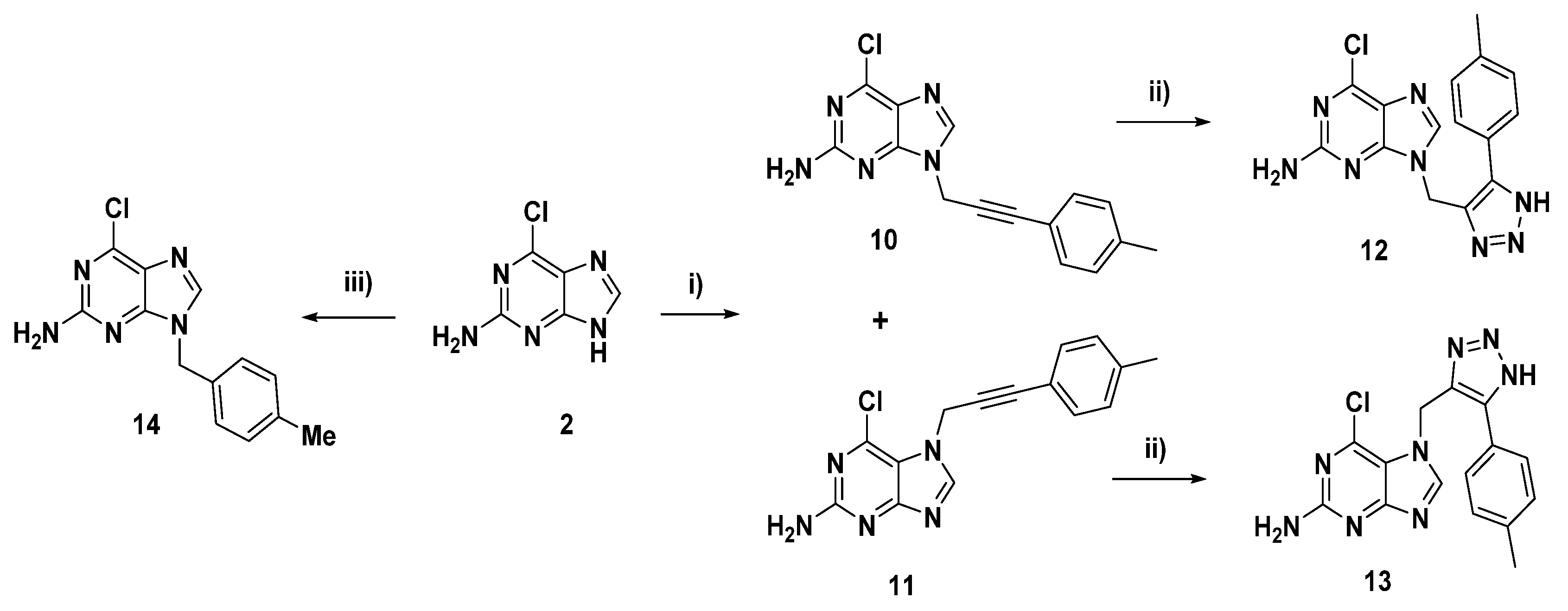
2.1. Chemical Synthesis
2.2. Biological Evaluation
2.2.1. Assessment of Hsp90α Binding Affinity Using FP Assay
2.2.2. In Vitro Antiproliferative Activity
2.3. Biochemical, Cellular, and Structural Evaluation
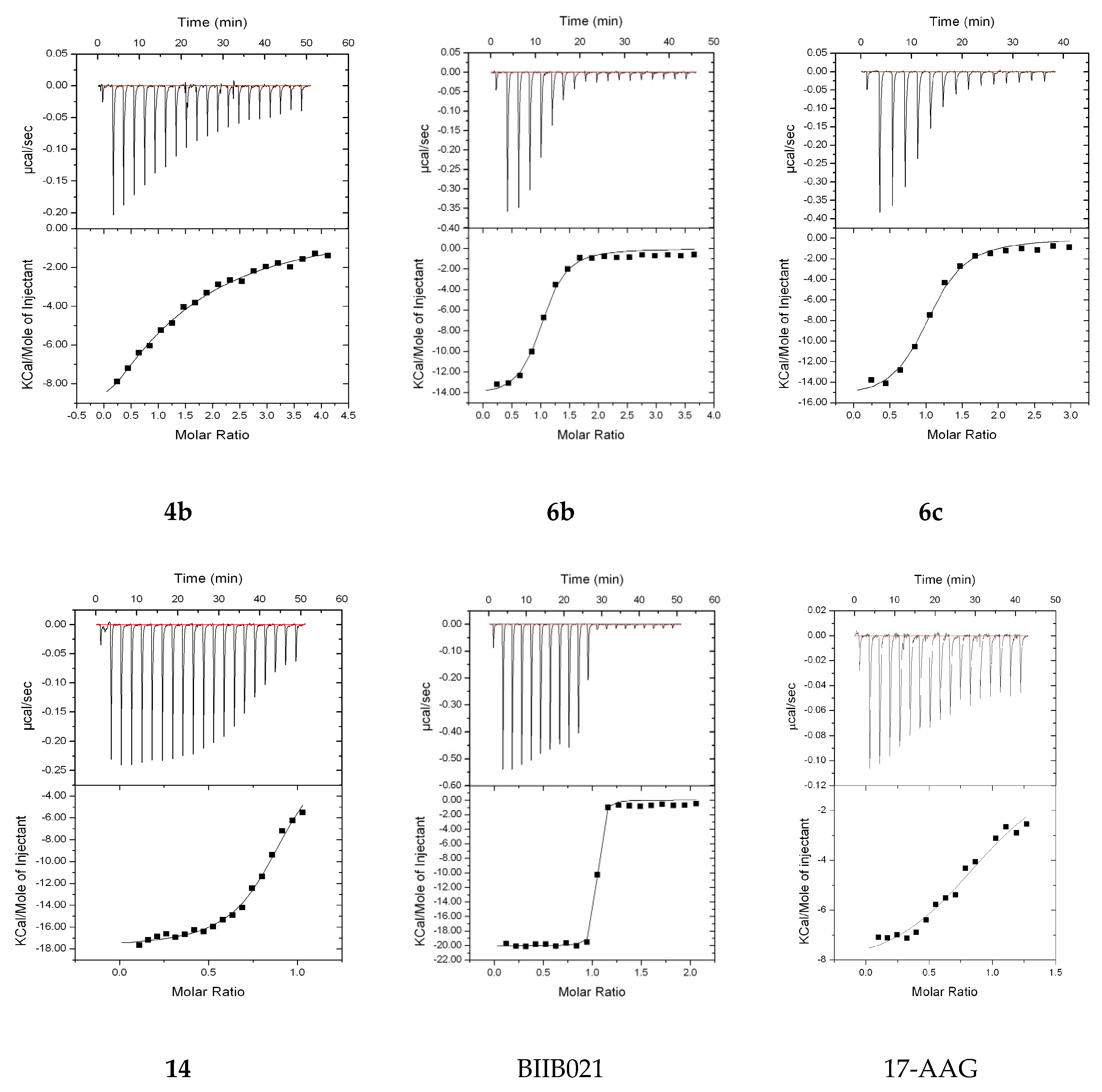
2.3.1. Assessment of mHsp90ND Binding Affinity Using ITC
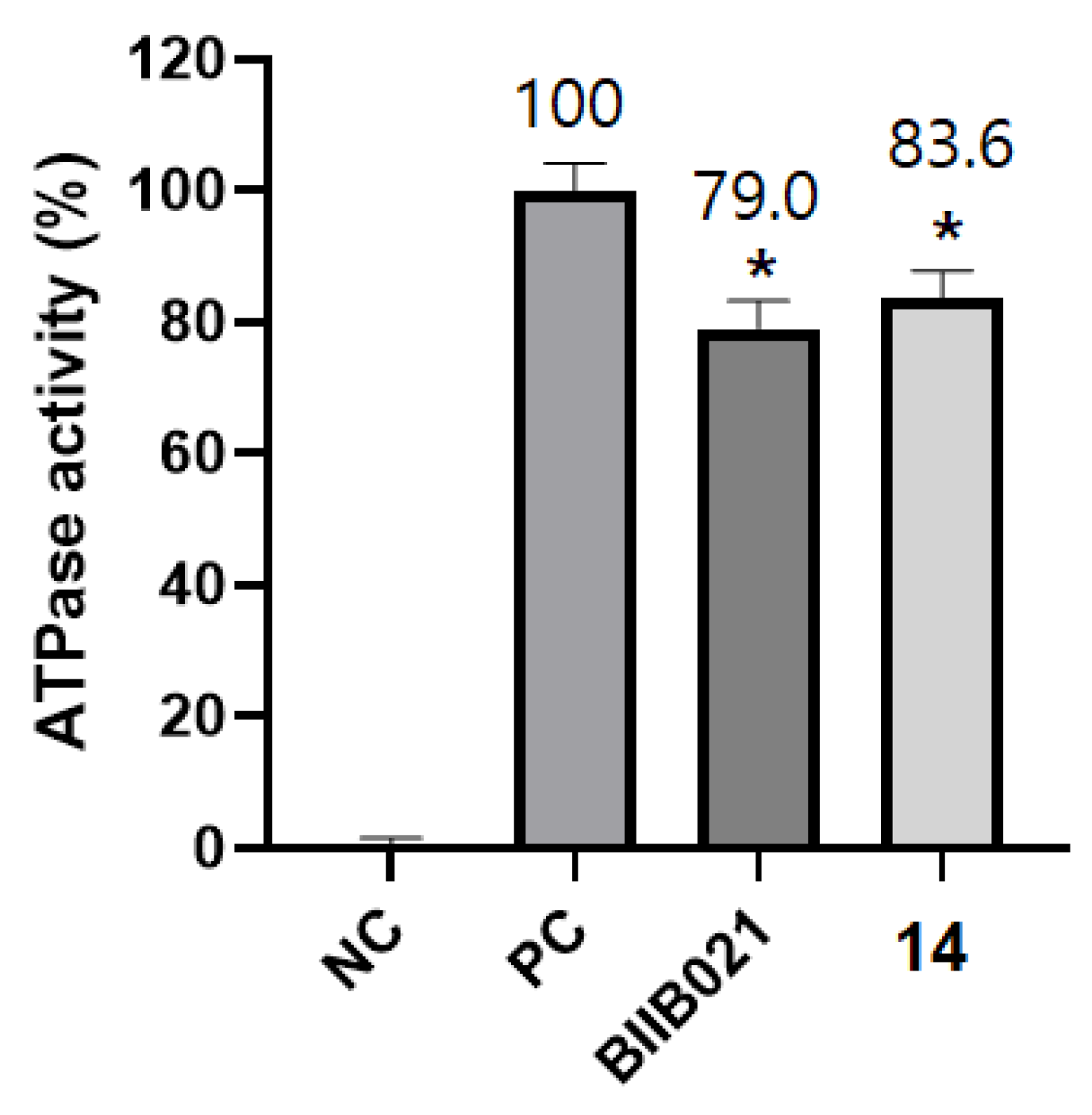
2.3.2. Colorimetric Determination of ATPase Activity of Hsp90
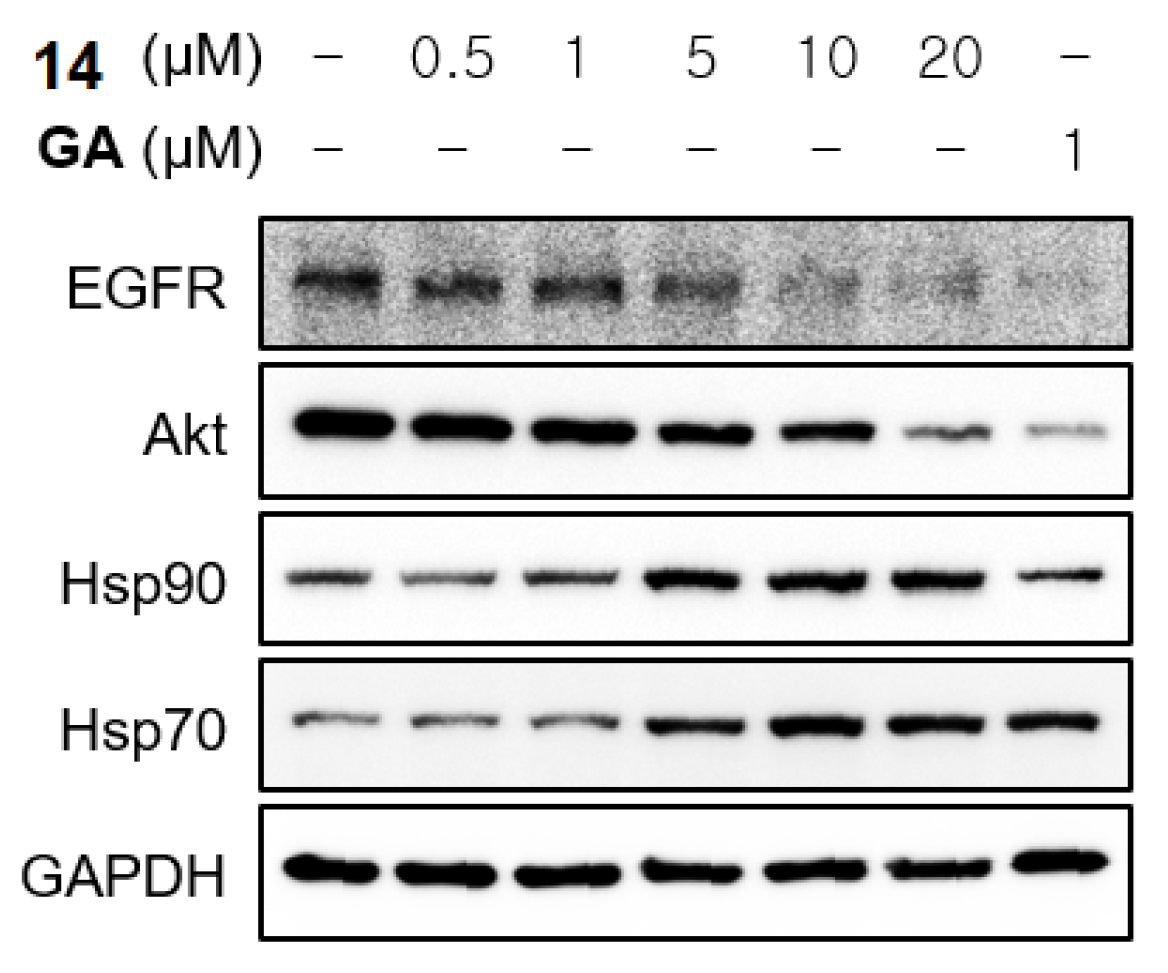
2.3.3. Western Blot Analysis of HSP90 Key Client Protein Expression and Activation
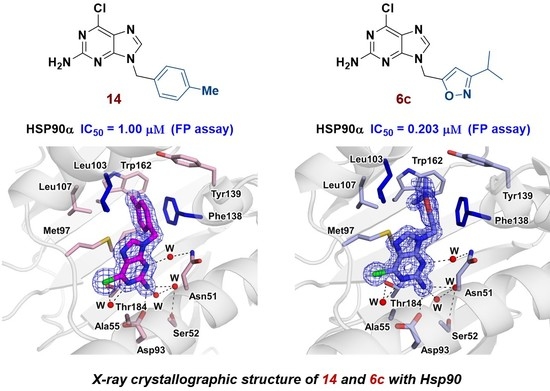
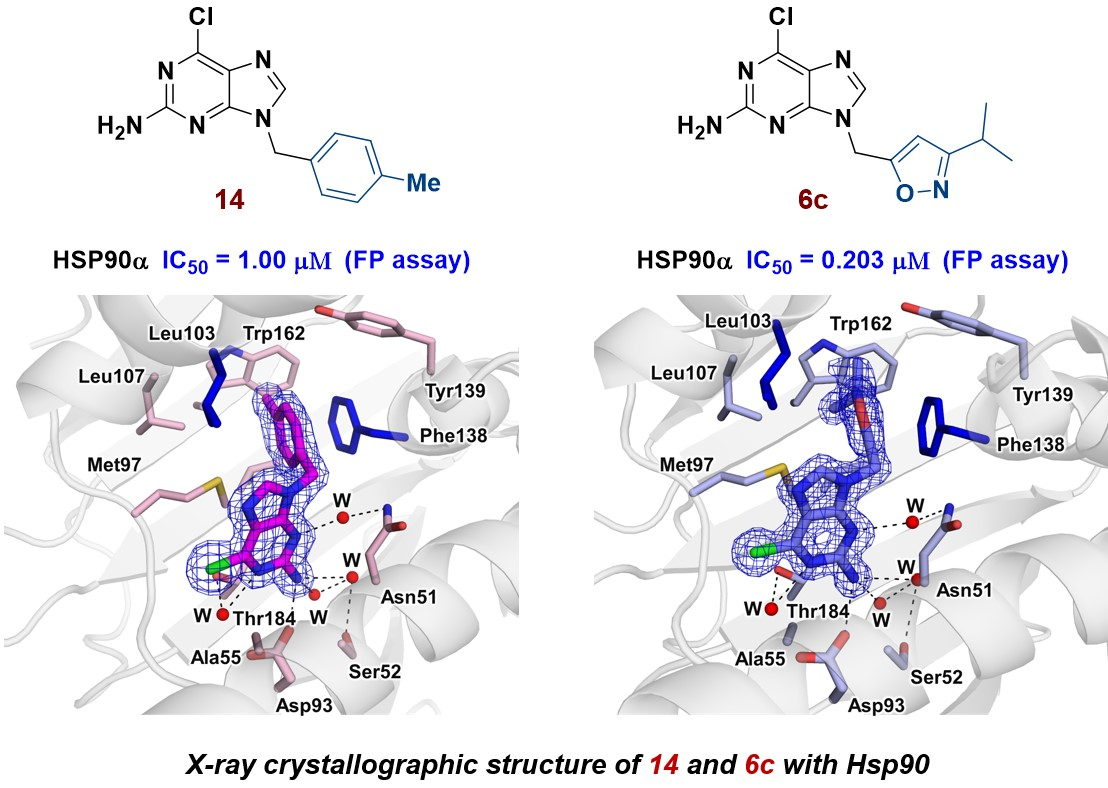
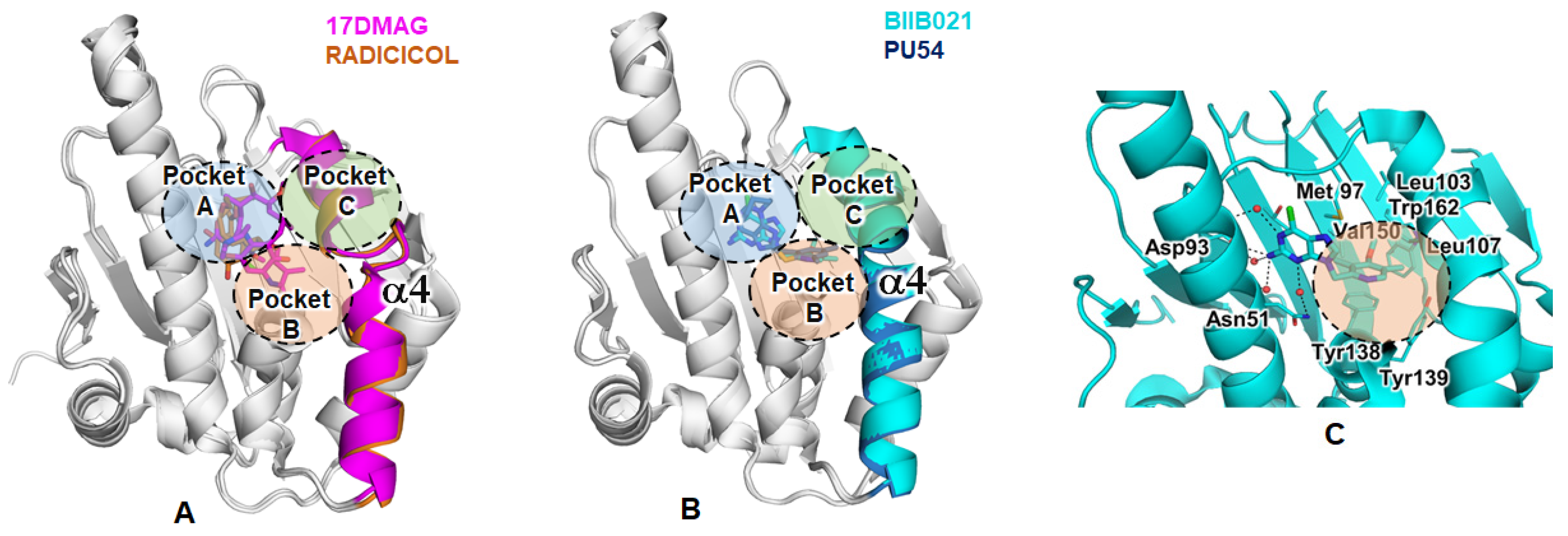
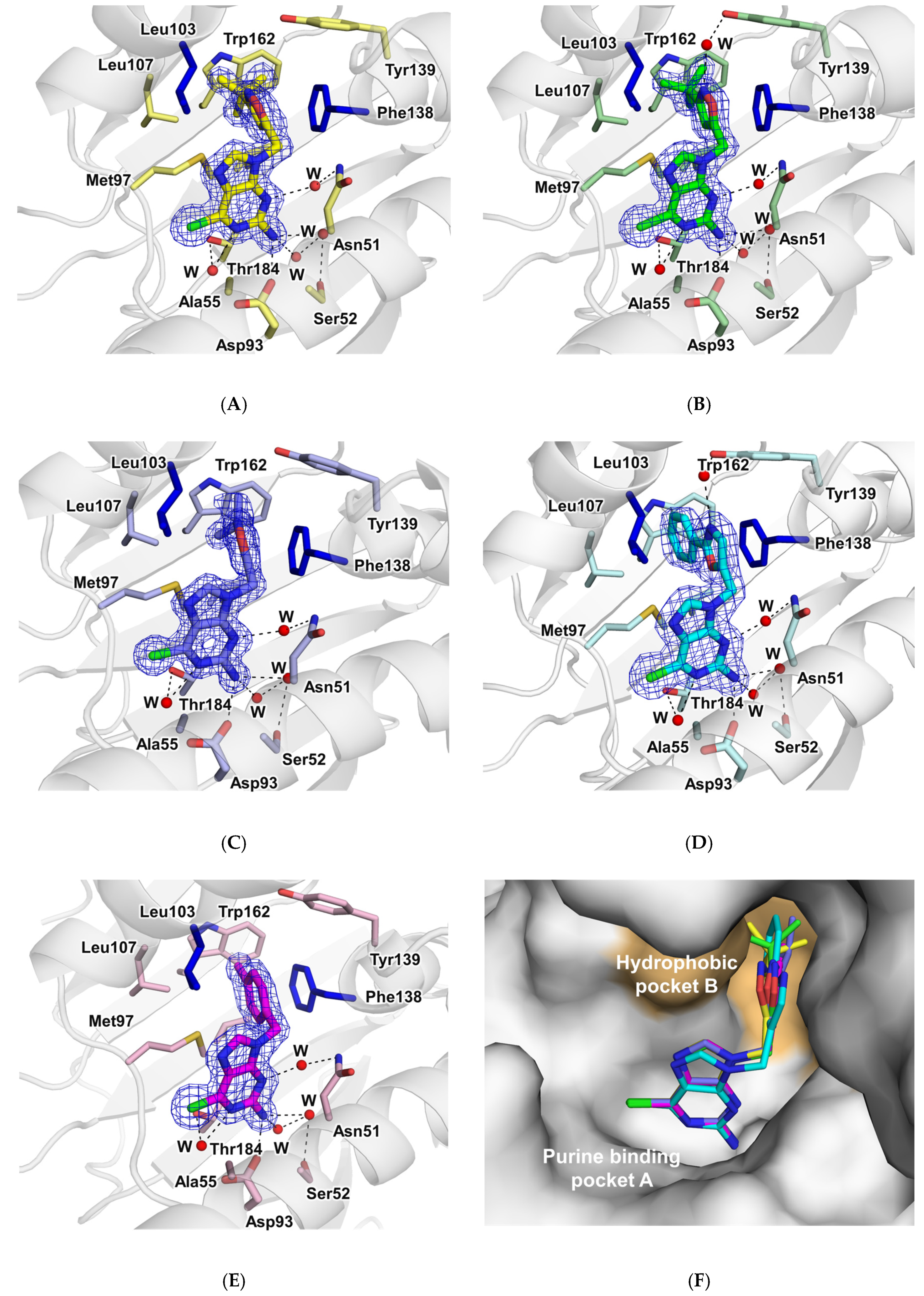
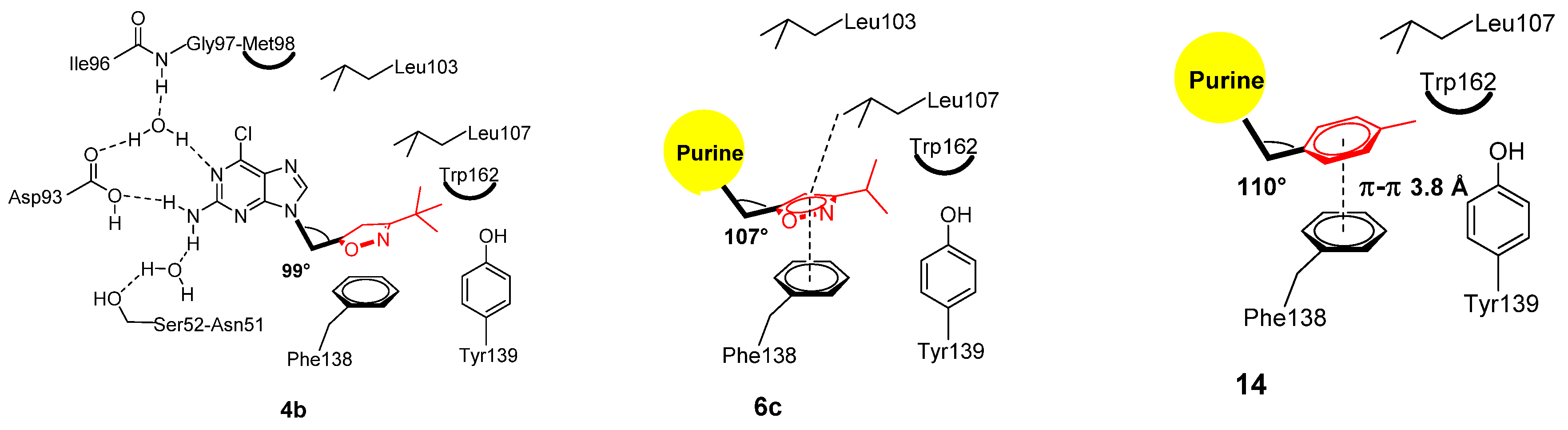
2.3.4. Structural Analysis of the Complexes Using X-ray Crystallography
3. Materials and Methods
3.1. Chemistry
3.1.1. General Methods
3.1.2. Synthesis and Structural Characterization
3.2. Biological Evaluations
3.2.1. Fluorescence Polarization (FP) Assay Using Green BODIPY-GM and FITC-GM
3.2.2. Cell-Based Anticancer Evaluation by MTT Assay
3.3. Biochemical, Cellular, and Structural Analysis
3.3.1. Expression and Protein Purification of mHsp90ND
3.3.2. Binding Affinity Using ITC
3.3.3. Colorimetric Determination of ATPase Activity of Hsp90
3.3.4. Western Blot Assay
3.3.5. Crystallization and Data Collection
3.3.6. Structure Determination and Refinement
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 17-AAG | Allyl-aminogeldanamycin |
| ATP | Adenosine triphosphate |
| 17-DMAG | 17-[[(2-Dimethylamino)ethyl]amino]geldanamycin |
| DMF | Dimethylformamide |
| DMSO | Dimethylsulfoxide |
| DTT | Dithiothreitol |
| FP | Fluorescence polarization |
| GM | Geldanamycin |
| HIF-1α | Hypoxia-inducible factor 1α |
| HNSCC | Head and neck squamous cell carcinoma |
| HRMS | High-resolution mass spectra |
| Hsp90 | Heat shock protein 90 |
| ITC | Isothermal titration calorimetry |
| mHsp90ND | N-terminal domain of mouse Hsp90α |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide |
| NMR | Nuclear magnetic resonance |
| PCR | Polymerase chain reaction |
| PDB | Protein Data Bank |
| PMSF | Phenylmethylsulfonyl fluoride |
| SAR | Structure–activity relationship |
| TCEP | Tris(2-chloroethyl) phosphate |
| TMS | Tetramethylsilane |
References
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef]
- DeZwaan, D.C.; Freeman, B.C. HSP90 manages the ends. Trends Biochem. Sci. 2010, 35, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Swords, R.; Carew, J.S.; Nawrocki, S.; Bhalla, K.; Giles, F. Targeting HSP90 for cancer therapy. Br. J. Cancer 2009, 100, 1523–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neckers, L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol. Med. 2002, 8, S55–S61. [Google Scholar] [CrossRef]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef] [Green Version]
- Ferrarini, M.; Heltai, S.; Zocchi, M.R.; Rugarli, C. Unusual expression and localization of heat-shock proteins in human tumor cells. Int. J. Cancer 1992, 51, 613–619. [Google Scholar] [CrossRef]
- Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M.F.; Fritz, L.C.; Burrows, F.J. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003, 425, 407–410. [Google Scholar] [CrossRef]
- Powers, M.V.; Workman, P. Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr. Relat. Cancer 2006, 13 (Suppl. 1), S125–S135. [Google Scholar] [CrossRef]
- Solit, D.B.; Chiosis, G. Development and application of Hsp90 inhibitors. Drug Discov. Today 2008, 13, 38–43. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhu, H.-P.; Xie, X.; Mao, Q.; Liu, Y.-Q.; He, X.-H.; Peng, C.; Jiang, Q.-L.; Huang, W. Novel HSP90-PI3K Dual Inhibitor Suppresses Melanoma Cell Proliferation by Interfering with HSP90-EGFR Interaction and Downstream Signaling Pathways. Int. J. Mol. Sci. 2020, 21, 1845. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Shin, S.C.; Seo, S.H.; Seo, Y.H.; Jeong, N.; Kim, C.-W.; Kim, E.E.; Keum, G. Synthesis and in vitro antiproliferative activity of C5-benzyl substituted 2-amino-pyrrolo [2,3-d] pyrimidines as potent Hsp90 inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Tummalapalli, S.R.; Rotella, D.P. Progress in the discovery and development of heat shock protein 90 (Hsp90) inhibitors: Miniperspective. J. Med. Chem. 2014, 57, 8718–8728. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Shen, A.; Zhang, C.; Song, Z.; Ai, J.; Liu, H.; Sun, L.; Ding, J.; Geng, M.; Zhang, A. Development of heat shock protein (HSP90) inhibitors to combat resistance to tyrosine kinase inhibitors through HSP90–kinase interactions. J. Med. Chem. 2016, 59, 5563–5586. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.-J.; An, H.; Kim, K.-S.; Kim, H.H.; Jung, J.; Lee, J.M.; Kim, N.-J.; Han, Y.T.; Yun, H.; Lee, S. Design, synthesis, and biological evaluation of novel deguelin-based heat shock protein 90 (HSP90) inhibitors targeting proliferation and angiogenesis. J. Med. Chem. 2012, 55, 10863–10884. [Google Scholar] [CrossRef]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Carbonero, R.; Carnero, A.; Paz-Ares, L. Inhibition of HSP90 molecular chaperones: Moving into the clinic. Lancet Oncol. 2013, 14, e358–e369. [Google Scholar] [CrossRef]
- Jhaveri, K.; Taldone, T.; Modi, S.; Chiosis, G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 742–755. [Google Scholar] [CrossRef] [Green Version]
- Schulte, T.W.; Neckers, L.M. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother. Pharmacol. 1998, 42, 273–279. [Google Scholar] [CrossRef]
- Kaur, G.; Belotti, D.; Burger, A.M.; Fisher-Nielson, K.; Borsotti, P.; Riccardi, E.; Thillainathan, J.; Hollingshead, M.; Sausville, E.A.; Giavazzi, R. Antiangiogenic properties of 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin: An orally bioavailable heat shock protein 90 modulator. Clin. Cancer Res. 2004, 10, 4813–4821. [Google Scholar] [CrossRef] [Green Version]
- Samuni, Y.; Ishii, H.; Hyodo, F.; Samuni, U.; Krishna, M.C.; Goldstein, S.; Mitchell, J.B. Reactive oxygen species mediate hepatotoxicity induced by the Hsp90 inhibitor geldanamycin and its analogs. Free Radic. Biol. Med. 2010, 48, 1559–1563. [Google Scholar] [CrossRef] [Green Version]
- Goldman, J.W.; Raju, R.N.; Gordon, G.A.; El-Hariry, I.; Teofilivici, F.; Vukovic, V.M.; Bradley, R.; Karol, M.D.; Chen, Y.; Guo, W. A first in human, safety, pharmacokinetics, and clinical activity phase I study of once weekly administration of the Hsp90 inhibitor ganetespib (STA-9090) in patients with solid malignancies. BMC Cancer 2013, 13, 152–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renouf, D.J.; Velazquez-Martin, J.P.; Simpson, R.; Siu, L.L.; Bedard, P.L. Ocular toxicity of targeted therapies. J. Clin. Oncol. 2012, 30, 3277–3286. [Google Scholar] [CrossRef] [PubMed]
- Kasibhatla, S.R.; Hong, K.; Biamonte, M.A.; Busch, D.J.; Karjian, P.L.; Sensintaffar, J.L.; Kamal, A.; Lough, R.E.; Brekken, J.; Lundgren, K. Rationally designed high-affinity 2-amino-6-halopurine heat shock protein 90 inhibitors that exhibit potent antitumor activity. J. Med. Chem. 2007, 50, 2767–2778. [Google Scholar] [CrossRef] [PubMed]
- Saif, M.W.; Takimoto, C.; Mita, M.; Banerji, U.; Lamanna, N.; Castro, J.; O’Brien, S.; Stogard, C.; Von Hoff, D. A phase 1, dose-escalation, pharmacokinetic and pharmacodynamic study of BIIB021 administered orally in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Taldone, T.; Patel, P.D.; Patel, M.; Patel, H.J.; Evans, C.E.; Rodina, A.; Ochiana, S.; Shah, S.K.; Uddin, M.; Gewirth, D. Experimental and structural testing module to analyze paralogue-specificity and affinity in the Hsp90 inhibitors series. J. Med. Chem. 2013, 56, 6803–6818. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Van de Water, R.; Hong, K.; Lamer, R.B.; Weichert, K.W.; Sandoval, C.M.; Kasibhatla, S.R.; Boehm, M.F.; Chao, J.; Lundgren, K. EC144 is a potent inhibitor of the heat shock protein 90. J. Med. Chem. 2012, 55, 7786–7795. [Google Scholar] [CrossRef]
- Immormino, R.M.; Kang, Y.; Chiosis, G.; Gewirth, D.T. Structural and quantum chemical studies of 8-aryl-sulfanyl adenine class Hsp90 inhibitors. J. Med. Chem. 2006, 49, 4953–4960. [Google Scholar] [CrossRef]
- Yin, X.; Zhang, H.; Lundgren, K.; Wilson, L.; Burrows, F.; Shores, C.G. BIIB021, a novel Hsp90 inhibitor, sensitizes head and neck squamous cell carcinoma to radiotherapy. Inter. J. Cancer 2010, 126, 1216–1225. [Google Scholar] [CrossRef]
- Zhang, H.; Neely, L.; Lundgren, K.; Yang, Y.C.; Lough, R.; Timple, N.; Burrows, F. BIIB021, a synthetic Hsp90 inhibitor, has broad application against tumors with acquired multidrug resistance. Inter. J. Cancer 2010, 126, 1226–1234. [Google Scholar] [CrossRef]
- El-Damasy, A.K.; Cho, N.C.; Kang, S.B.; Pae, A.N.; Keum, G. ABL kinase inhibitory and antiproliferative activity of novel picolinamide based benzothiazoles. Bioorg. Med. Chem. Lett. 2015, 25, 2162–2168. [Google Scholar] [CrossRef]
- El-Damasy, A.K.; Cho, N.C.; Pae, A.N.; Kim, E.E.; Keum, G. Novel 5-substituted-2-anilinoquinolines with 3-(morpholino or 4-methylpiperazin-1-yl)propoxy moiety as broad spectrum antiproliferative agents: Synthesis, cell based assays and kinase screening. Bioorg. Med. Chem. Lett. 2016, 26, 3307–3312. [Google Scholar] [CrossRef] [PubMed]
- El-Damasy, A.K.; Cho, N.C.; Nam, G.; Pae, A.N.; Keum, G. Discovery of a Nanomolar Multikinase Inhibitor (KST016366): A New Benzothiazole Derivative with Remarkable Broad-Spectrum Antiproliferative Activity. ChemMedChem 2016, 11, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- El-Damasy, A.K.; Seo, S.H.; Cho, N.C.; Pae, A.N.; Kim, E.E.; Keum, G. Design and synthesis of new 2-anilinoquinolines bearing N-methylpicolinamide moiety as potential antiproliferative agents. Chem. Biol. Drug Des. 2017, 89, 98–113. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.H.; Han, B.; Jung, K.H.; Lee, J.H.; El-Damasy, A.K.; Gadhe, C.G.; Kim, S.J.; Yan, H.H.; Park, J.H.; Lee, J.E.; et al. A novel tropomyosin-related kinase A inhibitor, KK5101 to treat pancreatic cancer. Cancer Lett. 2018, 426, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; El-Damasy, A.K.; Seo, S.H.; Gadhe, C.G.; Pae, A.N.; Jeong, N.; Hong, S.S.; Keum, G. Novel 5,6-disubstituted pyrrolo[2,3-d]pyrimidine derivatives as broad spectrum antiproliferative agents: Synthesis, cell based assays, kinase profile and molecular docking study. Bioorg. Med. Chem. 2018, 26, 5596–5611. [Google Scholar] [CrossRef]
- Steklov, M.Y.; Tararov, V.I.; Romanov, G.A.; Mikhailov, S.N. Facile synthesis of 8-azido-6-benzylaminopurine. Nucleosides Nucleotides Nucleic Acids 2011, 30, 503–511. [Google Scholar] [CrossRef]
- McIntosh, M.L.; Naffziger, M.R.; Ashburn, B.O.; Zakharov, L.N.; Carter, R.G. Highly regioselective nitrile oxide dipolar cycloadditions with ortho-nitrophenyl alkynes. Org. Biomol. Chem. 2012, 10, 9204–9213. [Google Scholar] [CrossRef]
- Liu, Y.; Cui, Z.; Liu, B.; Cai, B.; Li, Y.; Wang, Q. Design, synthesis, and herbicidal activities of novel 2-cyanoacrylates containing isoxazole moieties. J. Agric. Food Chem. 2010, 58, 2685–2689. [Google Scholar] [CrossRef]
- Moulick, K.; Clement, C.C.; Aguirre, J.; Kim, J.; Kang, Y.; Felts, S.; Chiosis, G. Synthesis of a red-shifted fluorescence polarization probe for Hsp90. Bioorg. Med. Chem. Lett. 2006, 16, 4515–4518. [Google Scholar] [CrossRef]
- Llauger-Bufi, L.; Felts, S.J.; Huezo, H.; Rosen, N.; Chiosis, G. Synthesis of novel fluorescent probes for the molecular chaperone Hsp90. Bioorg. Med. Chem. Lett. 2003, 13, 3975–3978. [Google Scholar] [CrossRef]
- Howes, R.; Barril, X.; Dymock, B.; Grant, K.; Northfield, C.; Robertson, A.; Surgenor, A.; Wayne, J.; Wright, L.; James, K. A fluorescence polarization assay for inhibitors of Hsp90. Anal. Biochem. 2006, 350, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Felts, S.; Llauger, L.; He, H.; Huezo, H.; Rosen, N.; Chiosis, G. Development of a fluorescence polarization assay for the molecular chaperone Hsp90. J. Biomol. Screen. 2004, 9, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Reaction Biology Corporation. Available online: https://www.reactionbiology.com/services/target-specific-assays/heat-shock-protein-assays (accessed on 13 October 2020).
- Nilapwar, S.; Williams, E.; Fu, C.; Prodromou, C.; Pearl, L.H.; Williams, M.A.; Ladbury, J.E. Structural–thermodynamic relationships of interactions in the N-terminal ATP-binding domain of Hsp90. J. Mol. Biol. 2009, 392, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; He, X.P.; Shen, Q.; Li, J.Y.; Shi, X.X.; Xie, J.; Li, J.; Chen, G.R. Synthesis of (Glycopyranosyl-triazolyl)-purines and Their Inhibitory Activities against Protein Tyrosine Phosphatase 1B (PTP1B). Chem. Biodivers. 2011, 8, 2035–2044. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.; Barril, X.; Dymock, B.; Sheridan, L.; Surgenor, A.; Beswick, M.; Drysdale, M.; Collier, A.; Massey, A.; Davies, N. Structure-activity relationships in purine-based inhibitor binding to HSP90 isoforms. Chem. Biol. 2004, 11, 775–785. [Google Scholar] [CrossRef] [Green Version]
- Brünger, A.T.; Adams, P.D.; Clore, G.M.; DeLano, W.L.; Gros, P.; Grosse-Kunstleve, R.W.; Jiang, J.-S.; Kuszewski, J.; Nilges, M.; Pannu, N.S. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 905–921. [Google Scholar]
- Potterton, E.; Briggs, P.; Turkenburg, M.; Dodson, E. A graphical user interface to the CCP4 program suite. Acta Crystallogr. Sect. D Biol. Crystallogr. 2003, 59, 1131–1137. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound No. | R | Hsp90α | |
|---|---|---|---|
| % Inhibition @10 μM a | IC50, μM b | ||
| 4a | 3-CH3CH2OCO-isoxazoline- | −2.4 | - c |
| 4b | 3-(CH3)3C-isoxazoline- | 35.4 | - c |
| 6a | 3-CH3CH2OCO-isoxazole- | −4.7 | - c |
| 6b | 3-(CH3)3C-isoxazole- | 95.8 | 1.76 |
| 6c | 3-(CH3)2CH-isoxazole- | 77.3 | 0.203 |
| 7 | 5-CH3CH2OCO-isoxazole- | 14.8 | - c |
| 8 | 2-C6H5-oxazole- | 48.7 | - c |
| 9 | 2-C6H5-oxazole- | NI d | - c |
| 10 | 4-CH3-C6H4-C≡C- | 22.2 | - c |
| 11 | 4-CH3-C6H4-C≡C- | NI d | - c |
| 12 | 5-(4-tolyl)-1H-1,2,3-triazole- | NI d | - c |
| 13 | 5-(4-tolyl)-1H-1,2,3-triazole- | NI d | - c |
| 14 | 4-CH3-C6H4- | 95.4 | 1.00 |
| BIIB021 | 96.1 | 0.029 | |
| 17-AAG | 100 | 0.056 | |
| Compound No. | MCF-7 | SK-BR-3 | HCT116 | |||
|---|---|---|---|---|---|---|
| % Inh. at 10 μM | GI50 (μM) | % Inh. at 10 μM | GI50 (μM) | % Inh. at 10 μM | GI50 (μM) | |
| 4b | 4.94 | ND | 13.47 | ND | 18.27 | ND |
| 6b | 15.66 | ND | 45.76 | 26.03 ± 1.36 | 26.03 | ND |
| 6c | 11.92 | ND | 33.17 | ND | −0.60 | ND |
| 8 | 33.27 | ND | 37.46 | 22.54 ± 0.70 | 19.19 | 23.45 ± 2.94 |
| 14 | 55.15 | 16.22 ± 0.71 | 60.93 | 15.24 ± 2.08 | 46.48 | ND |
| BIIB021 | 84.81 | 0.392 ± 0.03 | 71.94 | 0.347 ± 0.023 | 86.20 | 0.227 ± 0.015 |
| 17-AAG | 79.74 | 0.498 ± 0.06 | 74.44 | 0.140 ± 0.012 | 88.89 | 0.205 ± 0.009 |
| Protein (in the Cell) | Ligand/Protein (in the Syringe) | N | KD (µM) | ∆H (Kcal/mol) | ∆S (cal/mol/deg) |
|---|---|---|---|---|---|
| mHsp90ND | 4b | 1.17 | 15.6 | −19.8 ± 3.29 | −44.5 |
| 6b | 0.97 | 0.44 | −14.5 ± 0.4 | −19.4 | |
| 6c | 1.00 | 0.68 | −15.8 ± 0.5 | −24.8 | |
| 14 | 0.89 | 0.41 | −17.8 ± 0.2 | −30.5 | |
| BIIB021 | 0.87 | 0.0083 | −22.7 ± 0.2 | −39.1 | |
| 17-AAG | 0.98 | 2.9 | −8.6 ± 0.4 | −3.66 |
| 4b | 6b | 6c | 8 | 14 | |
|---|---|---|---|---|---|
| Data Collection Statistics | |||||
| X-ray source | PAL5-C | ||||
| Wavelength (Å) | 1.0000 | ||||
| Resolution range (Å) | 50.0–1.55 | 50.0–1.60 | 50.0–1.33 | 50–1.75 | 50–1.65 |
| (1.61–1.55) a | (1.66–1.60) | (1.38–1.33) | (1.81–1.75) | (1.71–1.65) | |
| Space group | I222 | ||||
| Unit cell parameters | |||||
| a (Å) | 67.35 | 68.14 | 67.10 | 67.61 | 67.60 |
| b (Å) | 89.89 | 89.90 | 90.08 | 89.38 | 98.82 |
| c (Å) | 98.59 | 98.78 | 98.71 | 98.21 | 98.23 |
| α β γ (°) | 90 | 90 | 90 | 90 | 90 |
| Total/unique reflections | 854872/43890 | 786882/40417 | 1934188/68575 | 992546/30364 | 917934/36280 |
| Completeness (%) | 95.7 (82.8) a | 97.3 (92.5) | 99.3 (98.0) | 98.6 (96.2) | 98.5 (96.5) |
| I/σ (I) | 37.0 (4.3) a | 23.5 (1.9) | 55.1 (4.3) | 20.3 (2.9) | 31.1 (3.3) |
| Redundancy | 6.4 (4.2) a | 5.4 (3.0) | 10.8 (7.0) | 5.5 (3.4) | 5.8 (3.9) |
| Rmerge b (%) | 4.0 (20.6) a | 4.8 (34.1) | 5.6 (32.8) | 9.3 (27.7) | 5.3 (25.2) |
| Refinement Statistics | |||||
| Resolution range (Å) | 50.0–1.55 | 50.0–1.60 | 50.0–1.33 | 50–1.75 | 50–1.65 |
| (1.61–1.55) a | (1.66–1.60) | (1.38–1.33) | (1.81–1.75) | (1.71–1.65) | |
| R-value/Rfree c (%) | 20.3/22.3 | 19.4/22.2 | 17.8/19.3 | 19.3/22.7 | 18.6/21.4 |
| No. of protein atoms | 1641 | 1633 | 1641 | 1641 | 1641 |
| No. of water molecules | 330 | 327 | 457 | 216 | 311 |
| No. of ligand molecules | 1 | 1 | 2 | 1 | 2 |
| Average B-factor (Å2) | 22.50 | 25.84 | 18.20 | 29.69 | 24.06 |
| bond lengths (Å)/bond angles (°) | 0.007/0.993 | 0.006/0.831 | 0.006/0.922 | 0.007/0.916 | 0.007/1.079 |
| Ramachandran analysis (%) | |||||
| Favored | 98.07 | 97.54 | 98.55 | 96.62 | 97.10 |
| Allowed | 1.93 | 2.46 | 1.45 | 3.38 | 2.90 |
| Outliers | 0 | 0 | 0 | 0 | 0 |
| PDB ID codes | 7D24 | 7D22 | 7D1V | 7D26 | 7D25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, S.C.; El-Damasy, A.K.; Lee, J.H.; Seo, S.H.; Kim, J.H.; Seo, Y.H.; Lee, Y.; Yu, J.H.; Bang, E.K.; Kim, E.E.; et al. Structural Basis for Design of New Purine-Based Inhibitors Targeting the Hydrophobic Binding Pocket of Hsp90. Int. J. Mol. Sci. 2020, 21, 9377. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249377
Shin SC, El-Damasy AK, Lee JH, Seo SH, Kim JH, Seo YH, Lee Y, Yu JH, Bang EK, Kim EE, et al. Structural Basis for Design of New Purine-Based Inhibitors Targeting the Hydrophobic Binding Pocket of Hsp90. International Journal of Molecular Sciences. 2020; 21(24):9377. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249377
Chicago/Turabian StyleShin, Sang Chul, Ashraf K. El-Damasy, Ju Hyeon Lee, Seon Hee Seo, Ji Hyun Kim, Young Ho Seo, Yuri Lee, Ji Hoon Yu, Eun Kyoung Bang, Eunice EunKyeong Kim, and et al. 2020. "Structural Basis for Design of New Purine-Based Inhibitors Targeting the Hydrophobic Binding Pocket of Hsp90" International Journal of Molecular Sciences 21, no. 24: 9377. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249377