Adaptation of Mitochondrial Substrate Flux in a Mouse Model of Nonalcoholic Fatty Liver Disease

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Whole Body, Liver, and Epididymal Fat Weight
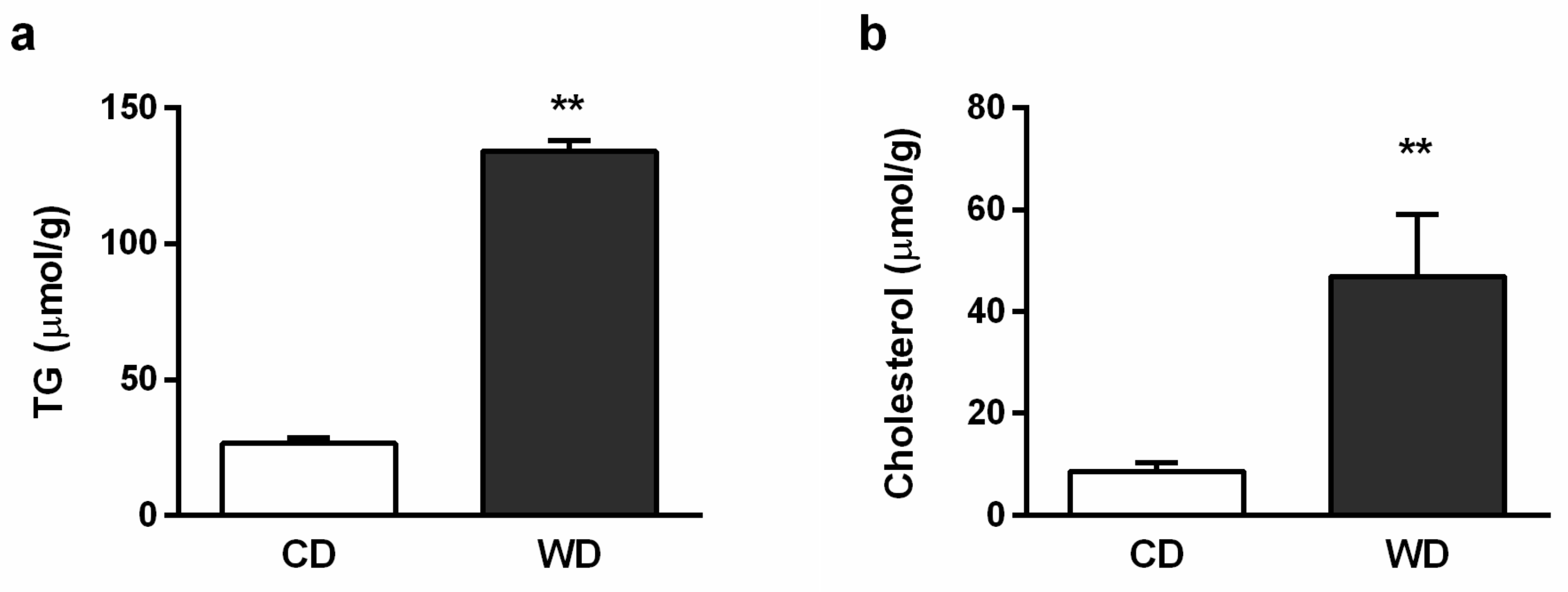
2.2. Blood Analysis
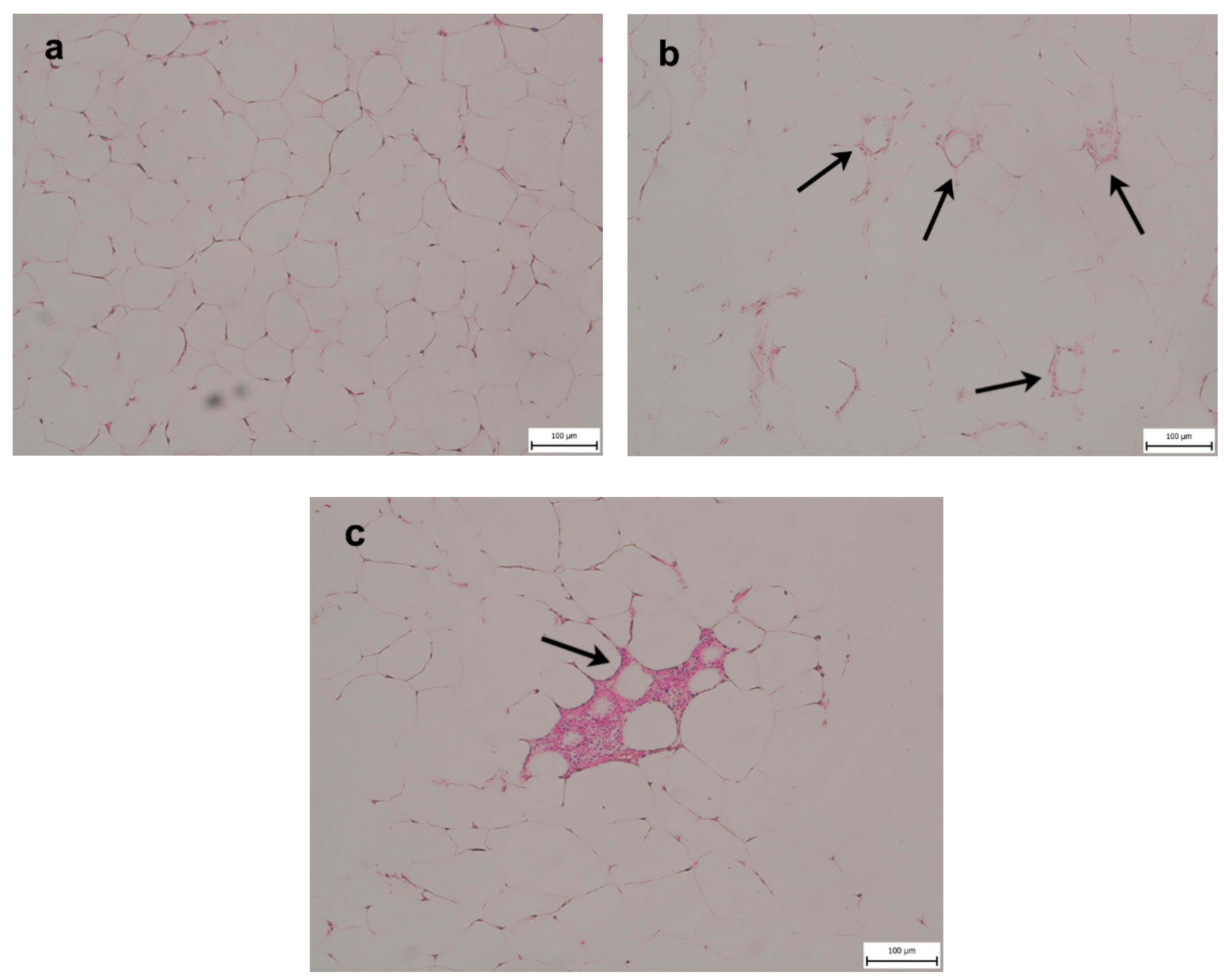
2.3. Epididymal Fat (eWAT)
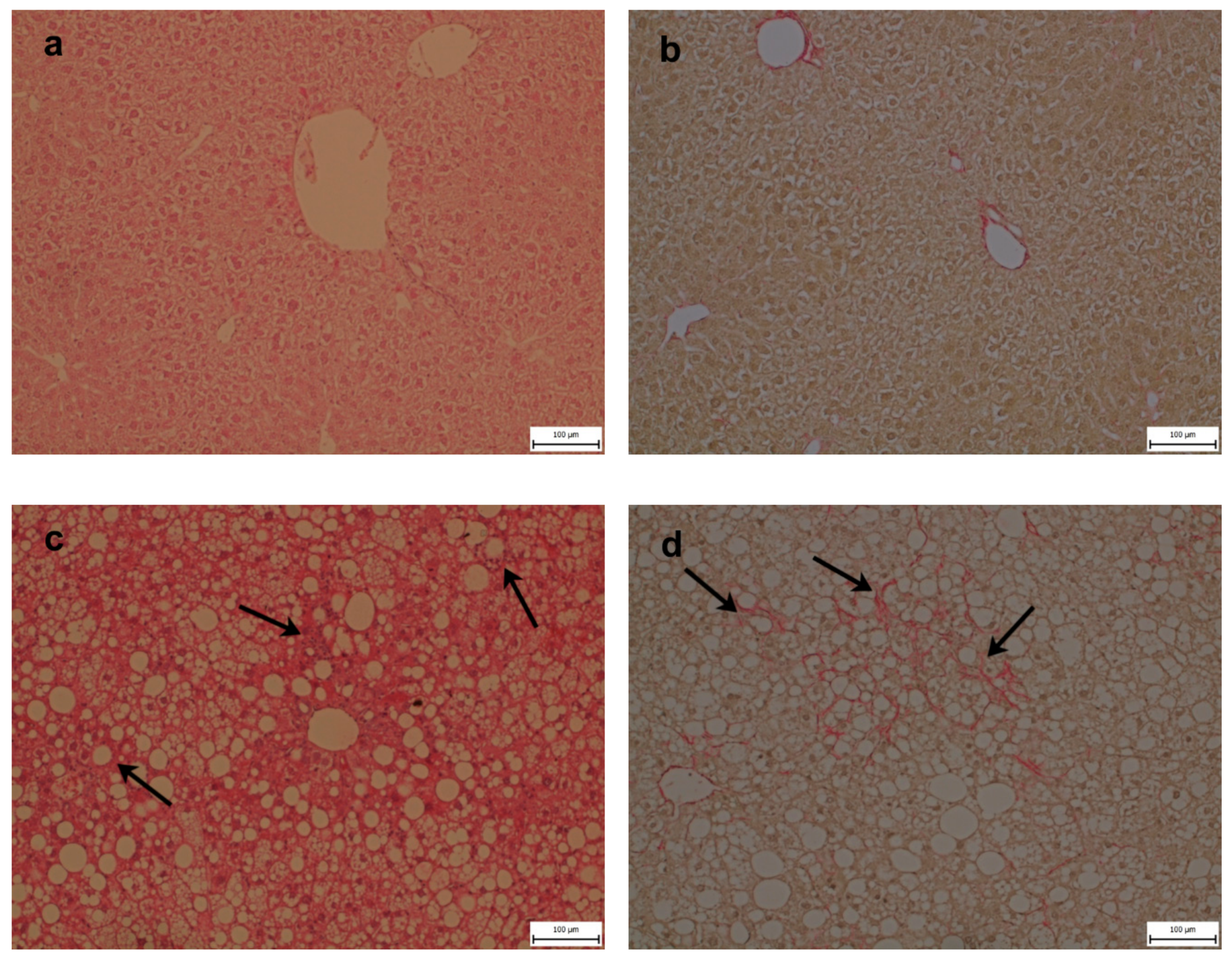
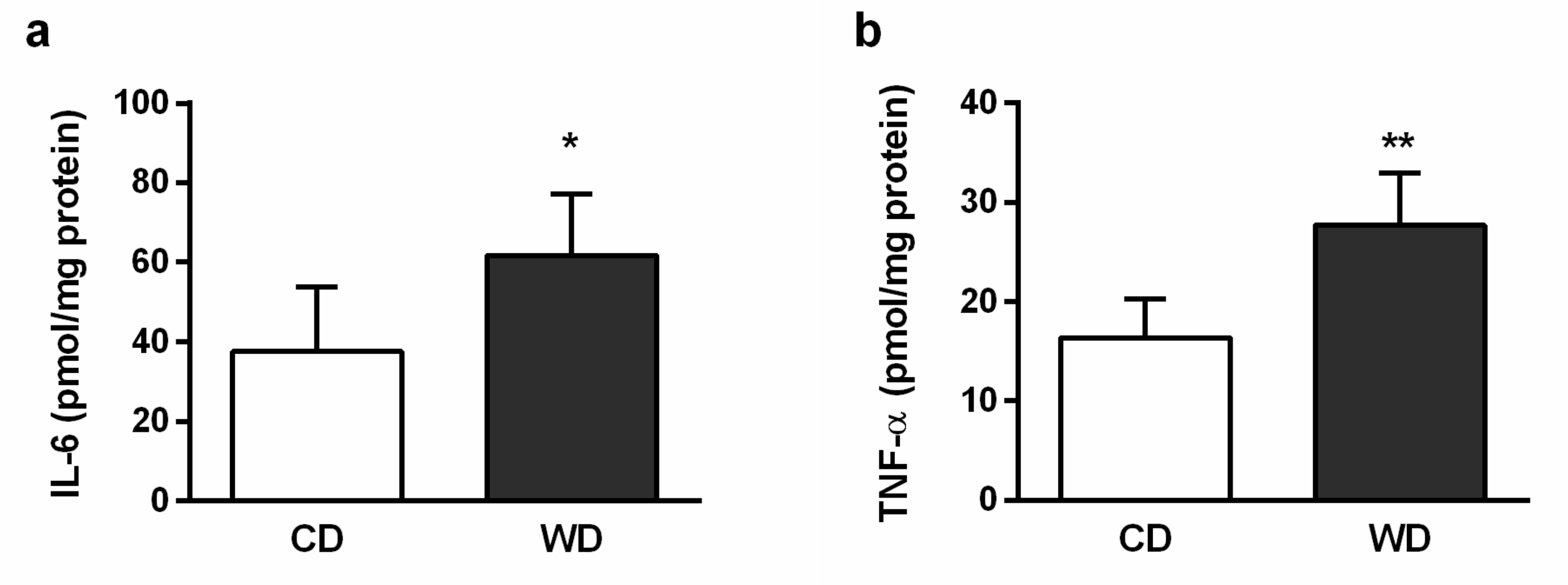
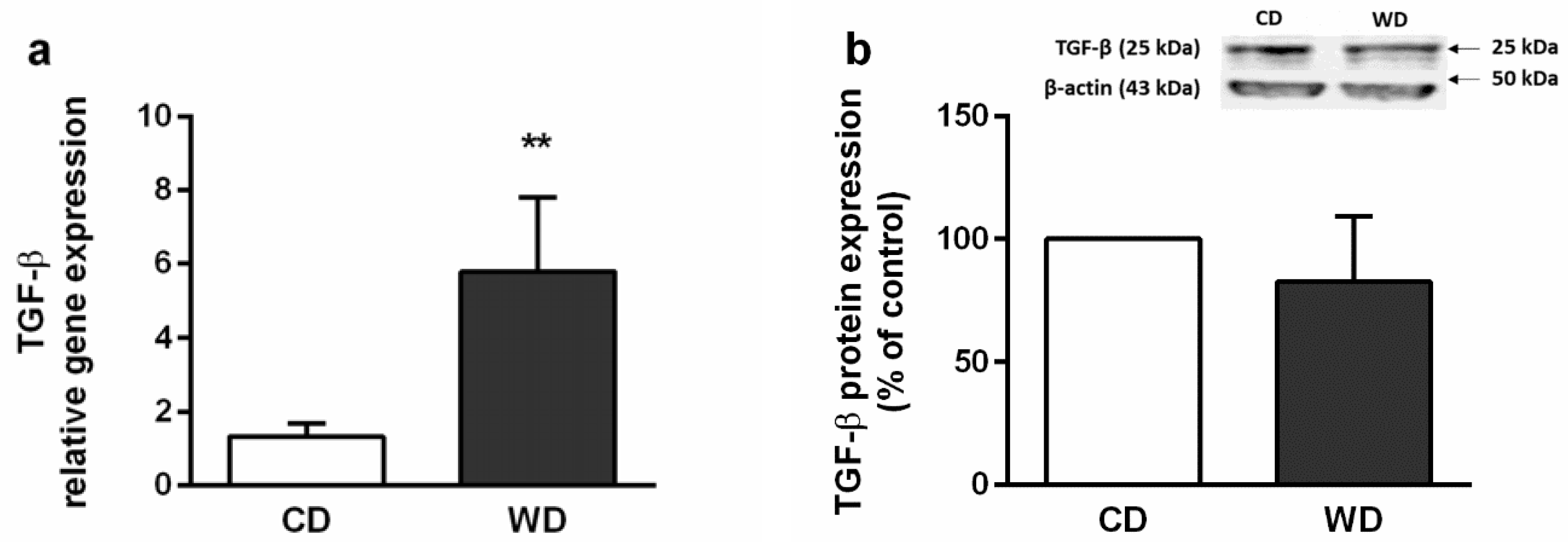
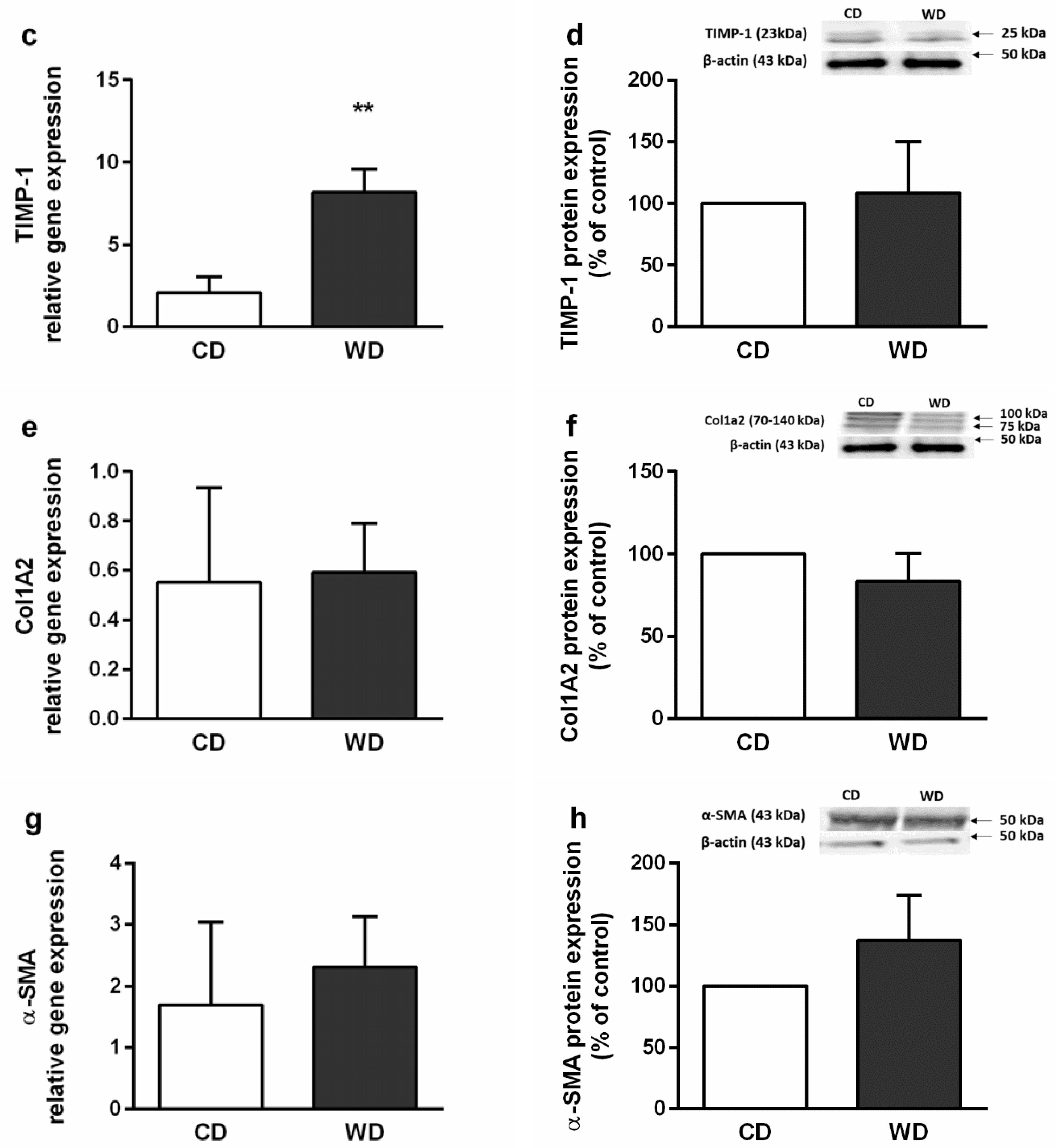
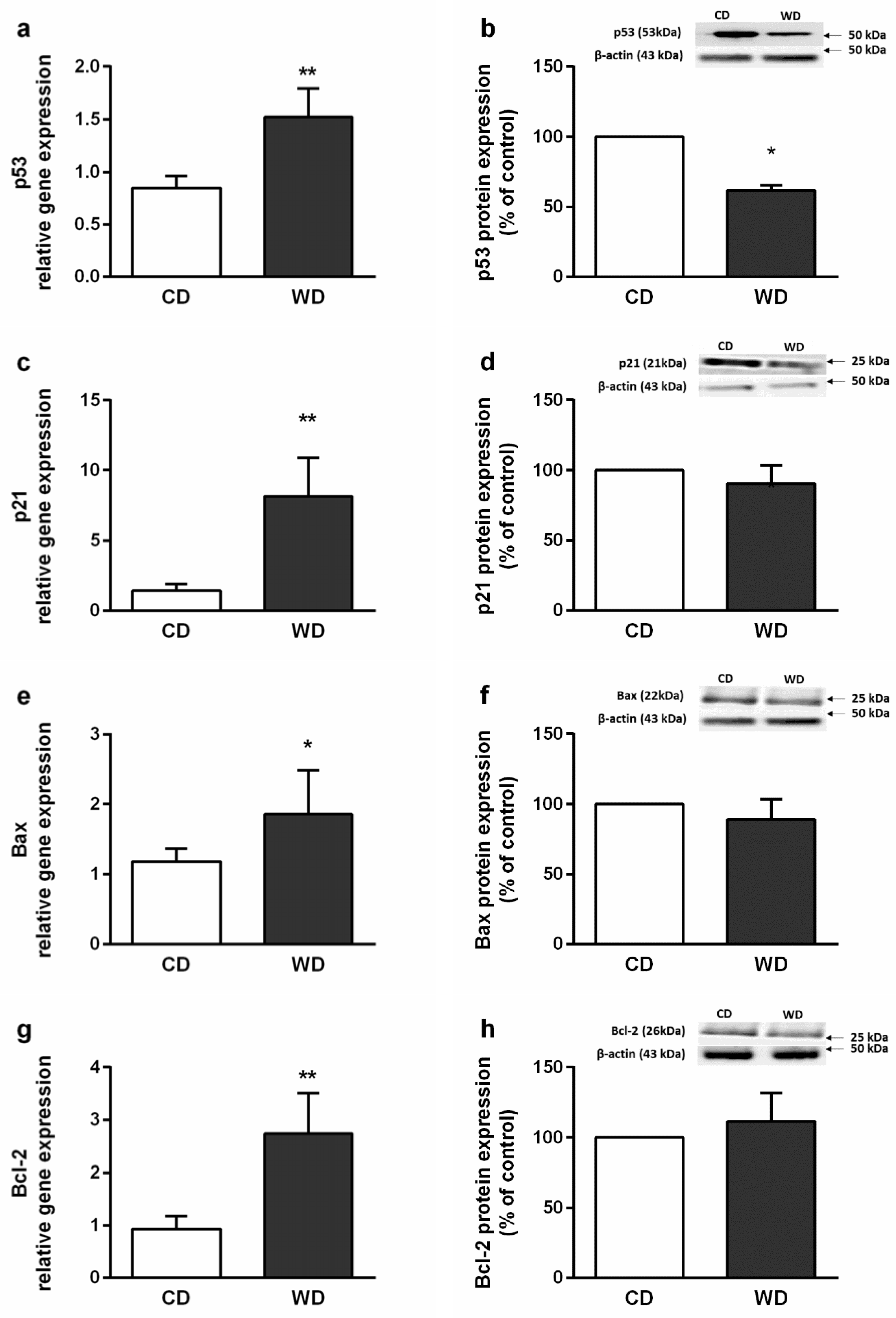
2.4. Steatosis, Inflammation, Fibrosis, and Apoptosis
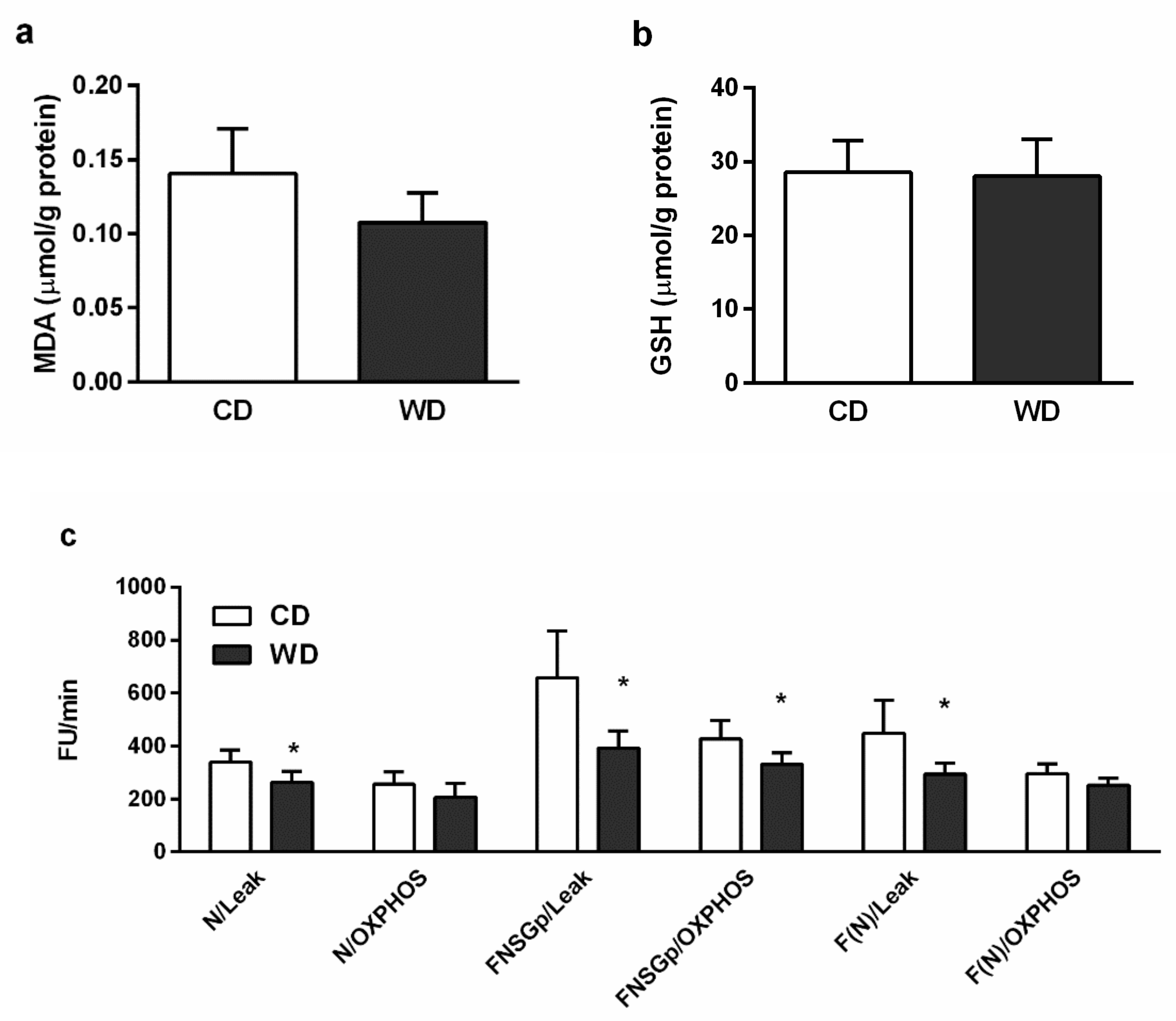
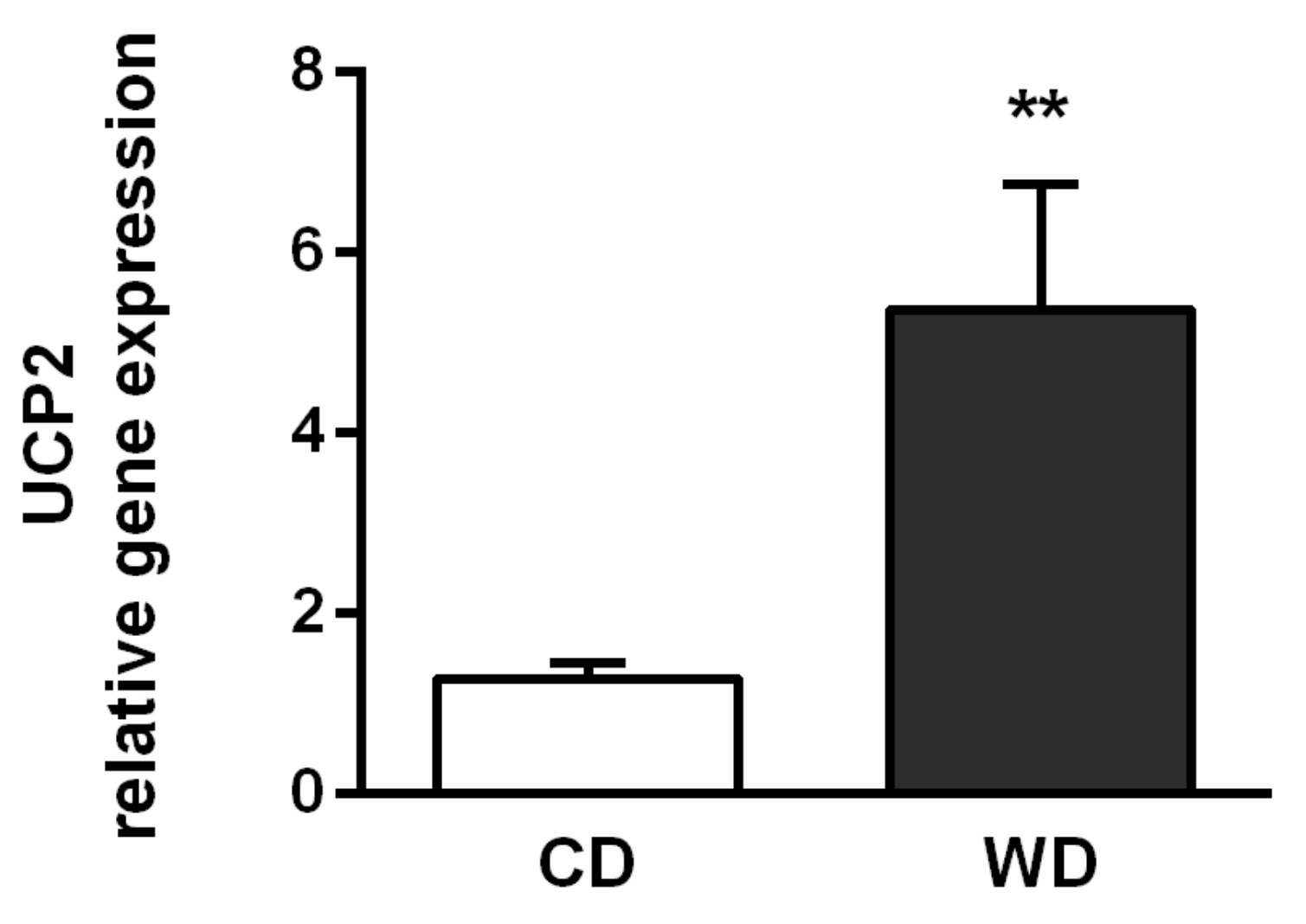
2.5. Oxidative Stress
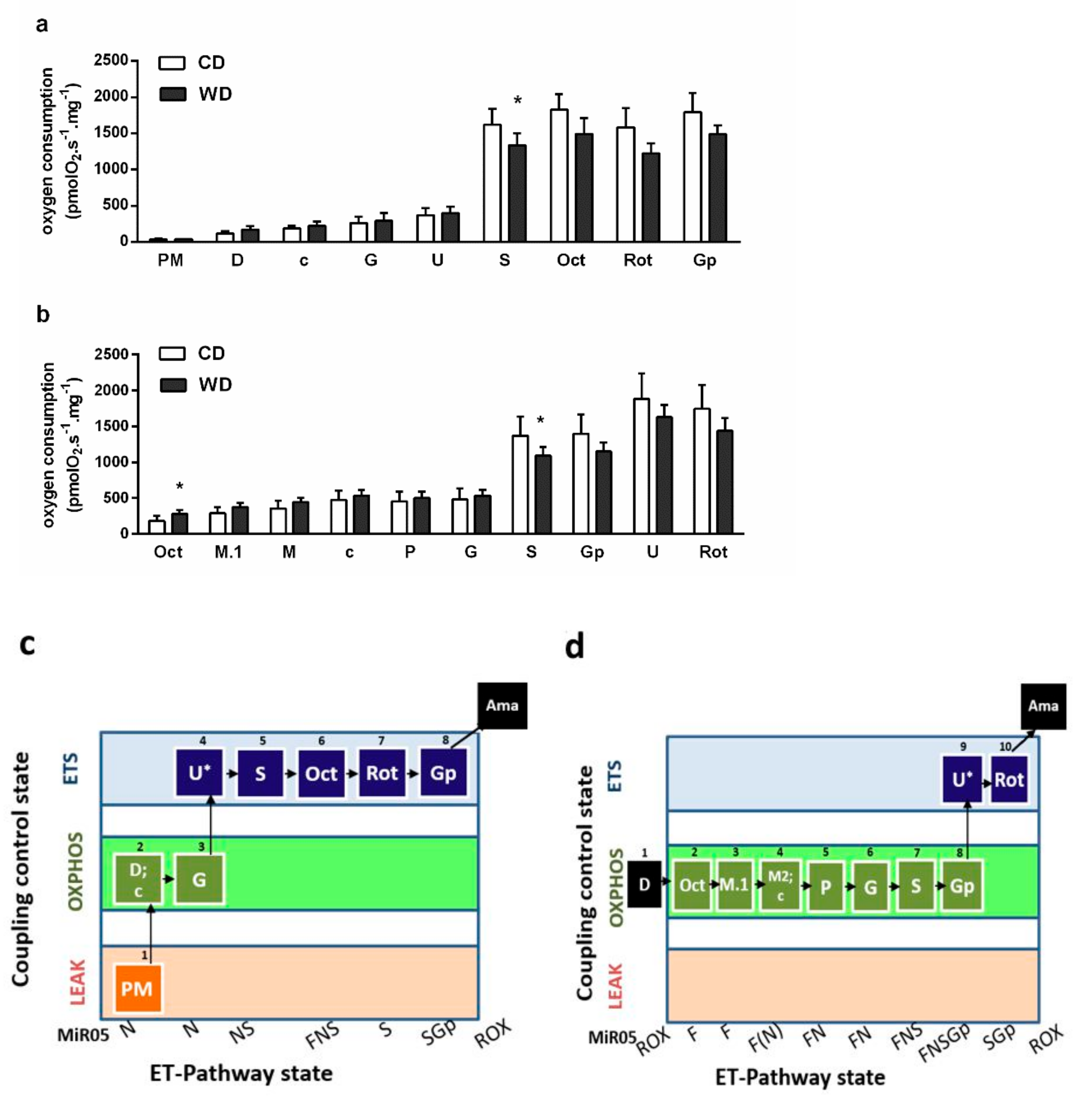
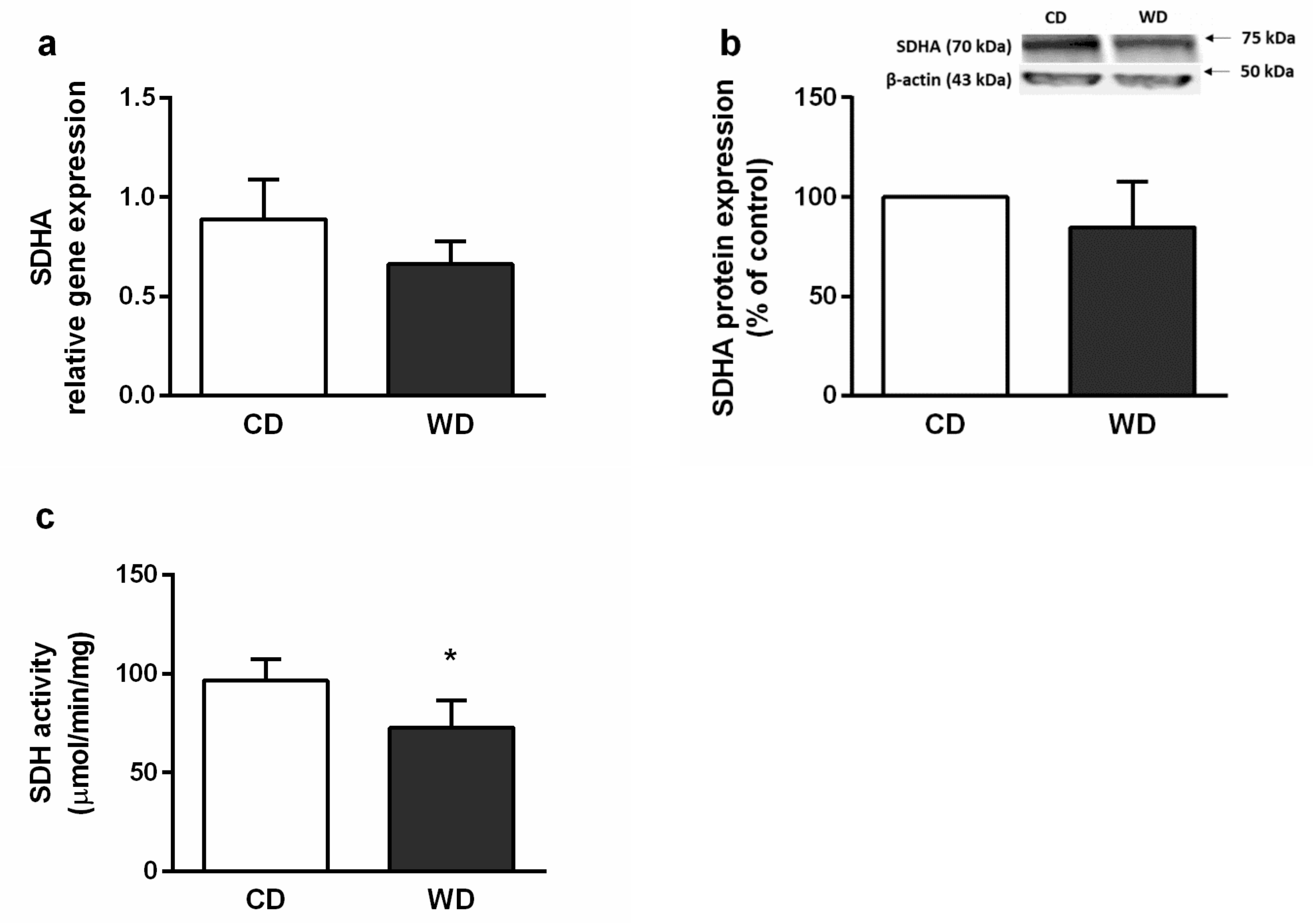
2.6. Mitochondrial Respiration
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α-SMA | Alpha smooth muscle actin |
| ADP | Adenosine diphosphate |
| ALP | Alkaline phosphatase |
| ALT | Alanine transaminase |
| AT | Adipose tissue |
| BUN | Blood urea nitrogen |
| CD | Control diet |
| CoA | Coenzyme A |
| Col1A2 | Collagen type I alpha 2 chain |
| ETS | Electron transfer system |
| eWAT | Epididymal white adipose tissue |
| FADH2 | Reduced flavin adenine dinucleotide |
| FAs | Fatty acids |
| GGT | γ-glutamyl transferase |
| GSH | Reduced glutathione |
| HFD | High-fat diet |
| IL-6 | Interleukin 6 |
| MCD | Methionine-choline deficient diet |
| MDA | Malondialdehyde |
| NADH | Reduced nicotinamide adenine dinucleotide |
| NAFLD | Nonalcoholic fatty liver disease |
| NASH | Nonalcoholic steatohepatitis |
| OXPHOS | Oxidative phosphorylation |
| p21 | Cyclin-dependent kinase inhibitor 1 |
| p53 | Tumor protein p53 |
| ROS | Reactive oxygen species |
| ROX | Residual oxygen consumption |
| SDH | Succinate dehydrogenase |
| SDHA | Succinate dehydrogenase subunit A |
| SUIT | Substrate-uncoupler-inhibitor-titration |
| TBARS | Thiobarbituric acid reactive substances |
| TCA | Tricarboxylic acid cycle |
| TGF-β | Transforming growth factor beta |
| TGs | Triglycerides |
| TIMP-1 | Tissue inhibitor of metalloproteinases-1 |
| TNF-α | Tumor necrosis factor alpha |
| UCP-2 | Uncoupling protein 2 |
| WD | Western-style diet |
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, H. Recent advances in mouse models of obesity- and nonalcoholic steatohepatitis-associated hepatocarcinogenesis. World J. Hepatol. 2015, 7, 2110–2118. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Michelotti, G.A.; Xie, G.; Almeida Pereira, T.; Boursier, J.; Bohnic, B.; Guy, C.D.; Diehl, A.M. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS ONE 2015, 10, e0127991. [Google Scholar] [CrossRef] [Green Version]
- Abenavoli, L.; Falalyeyeva, T.; Boccuto, L.; Tsyryuk, O.; Kobyliak, N. Obeticholic Acid: A New Era in the Treatment of Nonalcoholic Fatty Liver Disease. Pharmaceuticals 2018, 11, 104. [Google Scholar] [CrossRef] [Green Version]
- Melzer, K. Carbohydrate and fat utilization during rest and physical activity. SPEN Eur. J. Clin. Nutr. Metab. 2011, 6, e45–e52. [Google Scholar] [CrossRef] [Green Version]
- Muoio, D.M. Metabolic inflexibility: When mitochondrial indecision leads to metabolic gridlock. Cell 2014, 159, 1253–1262. [Google Scholar] [CrossRef] [Green Version]
- Rutkowski, J.M.; Stern, J.H.; Scherer, P.E. The cell biology of fat expansion. J. Cell Biol. 2015, 208, 501–512. [Google Scholar] [CrossRef] [Green Version]
- Mansouri, A.; Gattolliat, C.-H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [Green Version]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunny, N.E.; Bril, F.; Cusi, K. Mitochondrial Adaptation in Nonalcoholic Fatty Liver Disease: Novel Mechanisms and Treatment Strategies. Trends Endocrinol. Metab. 2017, 28, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.; Orynbayeva, Z. Determination of mitochondrial metabolic phenotype through investigation of the intrinsic inhibition of succinate dehydrogenase. Anal. Biochem. 2018, 552, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Parry, S.A.; Hodson, L. Influence of dietary macronutrients on liver fat accumulation and metabolism. J. Investig. Med. 2017, 65, 1102–1115. [Google Scholar] [CrossRef] [PubMed]
- Hodson, L.; Rosqvist, F.; Parry, S.A. The influence of dietary fatty acids on liver fat content and metabolism. Proc. Nutr. Soc. 2019, 79, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Sadevirta, S.; Zhou, Y.; Kayser, B.; Ali, A.; Ahonen, L.; Lallukka, S.; Pelloux, V.; Gaggini, M.; Jian, C.; et al. Saturated Fat Is More Metabolically Harmful for the Human Liver Than Unsaturated Fat or Simple Sugars. Diabetes Care 2018, 41, 1732–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legeza, B.; Marcolongo, P.; Gamberucci, A.; Varga, V.; Banhegyi, G.; Benedetti, A.; Odermatt, A. Fructose, Glucocorticoids and Adipose Tissue: Implications for the Metabolic Syndrome. Nutrients 2017, 9, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, S.H.; Hirsova, P.; Malhi, H.; Gores, G.J. Animal Models of Nonalcoholic Steatohepatitis: Eat, Delete, and Inflame. Dig. Dis. Sci. 2016, 61, 1325–1336. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Burrington, C.M.; Graff, E.C.; Zhang, J.; Judd, R.L.; Suksaranjit, P.; Kaewpoowat, Q.; Davenport, S.K.; O’Neill, A.M.; Greene, M.W. Metabolic phenotype and adipose and liver features in a high-fat Western diet-induced mouse model of obesity-linked NAFLD. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E418–E439. [Google Scholar] [CrossRef] [Green Version]
- Charlton, M.; Krishnan, A.; Viker, K.; Sanderson, S.; Cazanave, S.; McConico, A.; Masuoko, H.; Gores, G. Fast food diet mouse: Novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G825–G834. [Google Scholar] [CrossRef] [Green Version]
- Stephenson, K.; Kennedy, L.; Hargrove, L.; Demieville, J.; Thomson, J.; Alpini, G.; Francis, H. Updates on Dietary Models of Nonalcoholic Fatty Liver Disease: Current Studies and Insights. J. Liver Res. 2018, 18, 5–17. [Google Scholar] [CrossRef]
- Ronchi, J.A.; Figueira, T.R.; Ravagnani, F.G.; Oliveira, H.C.; Vercesi, A.E.; Castilho, R.F. A spontaneous mutation in the nicotinamide nucleotide transhydrogenase gene of C57BL/6J mice results in mitochondrial redox abnormalities. Free Radic. Biol. Med. 2013, 63, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, A.; Abdullah, T.S.; Mounajjed, T.; Hartono, S.; McConico, A.; White, T.; LeBrasseur, N.; Lanza, I.; Nair, S.; Gores, G.; et al. A longitudinal study of whole body, tissue, and cellular physiology in a mouse model of fibrosing NASH with high fidelity to the human condition. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G666–G680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SUIT Reference Protocol. Available online: https://wiki.oroboros.at/index.php/SUIT_reference_protocol (accessed on 27 October 2019).
- Kleiner, D.E.; Makhlouf, H.R. Histology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis in Adults and Children. Clin. Liver Dis. 2016, 20, 293–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiMarco, J.P.; Hoppel, C. Hepatic mitochondrial function in ketogenic states. Diabetes, starvation, and after growth hormone administration. J. Clin. Investig. 1975, 55, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, L.; van Klinken, J.B.; Pronk, A.C.; van Dam, A.D.; Dirven, E.; Rensen, P.C.; Koning, F.; Willems van Dijk, K.; van Harmelen, V. The limited storage capacity of gonadal adipose tissue directs the development of metabolic disorders in male C57Bl/6J mice. Diabetologia 2015, 58, 1601–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunny, N.E.; Satapati, S.; Fu, X.; He, T.; Mehdibeigi, R.; Spring-Robinson, C.; Duarte, J.; Potthoff, M.J.; Browning, J.D.; Burgess, S.C. Progressive adaptation of hepatic ketogenesis in mice fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1226–E1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prentki, M.; Madiraju, S.R. Glycerolipid metabolism and signaling in health and disease. Endocr. Rev. 2008, 29, 647–676. [Google Scholar] [CrossRef] [Green Version]
- Ponziani, F.R.; Pecere, S.; Gasbarrini, A.; Ojetti, V. Physiology and pathophysiology of liver lipid metabolism. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 1055–1067. [Google Scholar] [CrossRef]
- Kalliokoski, O.; Teilmann, A.C.; Jacobsen, K.R.; Abelson, K.S.; Hau, J. The lonely mouse—Single housing affects serotonergic signaling integrity measured by 8-OH-DPAT-induced hypothermia in male mice. PLoS ONE 2014, 9, e111065. [Google Scholar] [CrossRef] [Green Version]
- Kappel, S.; Hawkins, P.; Mendl, M.T. To Group or Not to Group? Good Practice for Housing Male Laboratory Mice. Animals 2017, 7, 88. [Google Scholar] [CrossRef] [Green Version]
- Bailoo, J.D.; Murphy, E.; Varholick, J.A.; Novak, J.; Palme, R.; Wurbel, H. Evaluation of the effects of space allowance on measures of animal welfare in laboratory mice. Sci. Rep. 2018, 8, 713. [Google Scholar] [CrossRef] [Green Version]
- Nolan, C.J.; Prentki, M. Insulin resistance and insulin hypersecretion in the metabolic syndrome and type 2 diabetes: Time for a conceptual framework shift. Diabetes Vasc. Dis. Res. 2019, 16, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial Oxidative Stress and Antioxidants Balance in Fatty Liver Disease. Hepatol. Commun. 2018, 2, 1425–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumida, Y.; Niki, E.; Naito, Y.; Yoshikawa, T. Involvement of free radicals and oxidative stress in NAFLD/NASH. Free Radic. Res. 2013, 47, 869–880. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmstrom, M.H.; Iglesias-Gutierrez, E.; Zierath, J.R.; Garcia-Roves, P.M. Tissue-specific control of mitochondrial respiration in obesity-related insulin resistance and diabetes. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E731–E739. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.H.; Woo, S.H.; Choi, D.H.; Cho, E.H. Succinate causes alpha-SMA production through GPR91 activation in hepatic stellate cells. Biochem. Biophys. Res. Commun. 2015, 463, 853–858. [Google Scholar] [CrossRef]
- Han, D.; Johnson, H.S.; Rao, M.P.; Martin, G.; Sancheti, H.; Silkwood, K.H.; Decker, C.W.; Nguyen, K.T.; Casian, J.G.; Cadenas, E.; et al. Mitochondrial remodeling in the liver following chronic alcohol feeding to rats. Free Radic. Biol. Med. 2017, 102, 100–110. [Google Scholar] [CrossRef] [Green Version]
- Tretter, L.; Patocs, A.; Chinopoulos, C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1857, 1086–1101. [Google Scholar] [CrossRef]
- Drose, S. Differential effects of complex II on mitochondrial ROS production and their relation to cardioprotective pre- and postconditioning. Biochim. Biophys. Acta (BBA)-Bioenerg. 2013, 1827, 578–587. [Google Scholar] [CrossRef] [Green Version]
- Bezawork-Geleta, A.; Rohlena, J.; Dong, L.; Pacak, K.; Neuzil, J. Mitochondrial Complex II: At the Crossroads. Trends Biochem. Sci. 2017, 42, 312–325. [Google Scholar] [CrossRef]
- Kappler, L.; Hoene, M.; Hu, C.; von Toerne, C.; Li, J.; Bleher, D.; Hoffmann, C.; Bohm, A.; Kollipara, L.; Zischka, H.; et al. Linking bioenergetic function of mitochondria to tissue-specific molecular fingerprints. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E374–E387. [Google Scholar] [CrossRef] [PubMed]
- Ralph, S.J.; Moreno-Sanchez, R.; Neuzil, J.; Rodriguez-Enriquez, S. Inhibitors of succinate: Quinone reductase/Complex II regulate production of mitochondrial reactive oxygen species and protect normal cells from ischemic damage but induce specific cancer cell death. Pharm. Res. 2011, 28, 2695–2730. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robb, E.L.; Hall, A.R.; Prime, T.A.; Eaton, S.; Szibor, M.; Viscomi, C.; James, A.M.; Murphy, M.P. Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 2018, 293, 9869–9879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, J.G.; Softic, S.; Basisty, N.; Rardin, M.J.; Verdin, E.; Gibson, B.W.; Ilkayeva, O.; Newgard, C.B.; Kahn, C.R.; Schilling, B. Temporal dynamics of liver mitochondrial protein acetylation and succinylation and metabolites due to high fat diet and/or excess glucose or fructose. PLoS ONE 2018, 13, e0208973. [Google Scholar] [CrossRef] [Green Version]
- Serena, C.; Ceperuelo-Mallafre, V.; Keiran, N.; Queipo-Ortuno, M.I.; Bernal, R.; Gomez-Huelgas, R.; Urpi-Sarda, M.; Sabater, M.; Perez-Brocal, V.; Andres-Lacueva, C.; et al. Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. ISME J. 2018, 12, 1642–1657. [Google Scholar] [CrossRef] [Green Version]
- Schofield, Z.; Reed, M.A.; Newsome, P.N.; Adams, D.H.; Gunther, U.L.; Lalor, P.F. Changes in human hepatic metabolism in steatosis and cirrhosis. World J. Gastroenterol. 2017, 23, 2685–2695. [Google Scholar] [CrossRef]
- Satapati, S.; Sunny, N.E.; Kucejova, B.; Fu, X.; He, T.T.; Mendez-Lucas, A.; Shelton, J.M.; Perales, J.C.; Browning, J.D.; Burgess, S.C. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012, 53, 1080–1092. [Google Scholar] [CrossRef] [Green Version]
- Cotter, D.G.; Ercal, B.; Huang, X.; Leid, J.M.; D’Avignon, D.A.; Graham, M.J.; Dietzen, D.J.; Brunt, E.M.; Patti, G.J.; Crawford, P.A. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J. Clin. Investig. 2014, 124, 5175–5190. [Google Scholar] [CrossRef] [Green Version]
- Mannisto, V.T.; Simonen, M.; Hyysalo, J.; Soininen, P.; Kangas, A.J.; Kaminska, D.; Matte, A.K.; Venesmaa, S.; Kakela, P.; Karja, V.; et al. Ketone body production is differentially altered in steatosis and non-alcoholic steatohepatitis in obese humans. Liver Int. 2015, 35, 1853–1861. [Google Scholar] [CrossRef]
- Vice, E.; Privette, J.D.; Hickner, R.C.; Barakat, H.A. Ketone body metabolism in lean and obese women. Metabolism 2005, 54, 1542–1545. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.A.; Deja, S.; Satapati, S.; Fu, X.; Burgess, S.C.; Browning, J.D. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight 2019. [Google Scholar] [CrossRef] [PubMed]
- Abdurrachim, D.; Woo, C.C.; Teo, X.Q.; Chan, W.X.; Radda, G.K.; Lee, P.T.H. A new hyperpolarized 13C ketone body probe reveals an increase in acetoacetate utilization in the diabetic rat heart. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Kand’ar, R.; Zakova, P.; Lotkova, H.; Kucera, O.; Cervinkova, Z. Determination of reduced and oxidized glutathione in biological samples using liquid chromatography with fluorimetric detection. J. Pharm. Biomed. Anal. 2007, 43, 1382–1387. [Google Scholar] [CrossRef]
- Kucera, O.; Rousar, T.; Stankova, P.; Hanackova, L.; Lotkova, H.; Podhola, M.; Cervinkova, Z. Susceptibility of rat non-alcoholic fatty liver to the acute toxic effect of acetaminophen. J. Gastroenterol. Hepatol. 2012, 27, 323–330. [Google Scholar] [CrossRef]
- Gnaiger, E.; Aasander Frostner, E.; Abdul Karim, N.; Abdel-Rahman, E.A.; Abumrad, N.A.; Acuna-Castroviejo, D.; Adiele, R.C.; Amati, F. Mitochondrial respiratory states and rates. MitoFit Prepr. Arch. 2019. [Google Scholar] [CrossRef]
- Hartwig, S.; Kotzka, J.; Lehr, S. Isolation and quality control of functional mitochondria. Methods Mol. Biol. 2015, 1264, 9–23. [Google Scholar] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | CD | WD |
|---|---|---|
| Body weight, g | 30.5 ± 3.27 | 45.17 ± 4.02 ** |
| Absolute liver weight, g | 1.42 ± 0.18 | 3.58 ± 0.88 ** |
| Relative liver weight, % | 4.65 ± 0.46 | 7.83 ± 1.38 ** |
| Epididymal fat weight, g | 0.80 ± 0.25 | 2.17 ± 0.27 ** |
| ALT, µkat/L | 0.45 ± 0.08 | 5.23 ± 2.38 ** |
| ALP, µkat/L | 0.62 ± 0.42 | 1.50 ± 0.55 |
| Cholesterol, mmol/L | 2.52 ± 0.45 | 6.82 ± 0.75 ** |
| Bile acids, µmol/L | < 1 | 10.00 ± 3.16 ** |
| Total bilirubin, µmol/L | 3.25 ± 0.96 | 3.75 ± 0.50 |
| BUN, mmol/L | 6.55 ± 1.01 | 4.58 ± 0.83 ** |
| albumin, g/L | 26.67 ± 7.17 | 28.00 ± 2.92 |
| GGT, µkat/L | < 0.1 | < 0.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staňková, P.; Kučera, O.; Peterová, E.; Lotková, H.; Maseko, T.E.; Nožičková, K.; Červinková, Z. Adaptation of Mitochondrial Substrate Flux in a Mouse Model of Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 1101. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031101
Staňková P, Kučera O, Peterová E, Lotková H, Maseko TE, Nožičková K, Červinková Z. Adaptation of Mitochondrial Substrate Flux in a Mouse Model of Nonalcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2020; 21(3):1101. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031101
Chicago/Turabian StyleStaňková, Pavla, Otto Kučera, Eva Peterová, Halka Lotková, Tumisang Edward Maseko, Kateřina Nožičková, and Zuzana Červinková. 2020. "Adaptation of Mitochondrial Substrate Flux in a Mouse Model of Nonalcoholic Fatty Liver Disease" International Journal of Molecular Sciences 21, no. 3: 1101. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031101