A Case Report of a Japanese Boy with Morquio A Syndrome: Effects of Enzyme Replacement Therapy Initiated at the Age of 24 Months

 ,
,
Abstract
:
1. Introduction
2. Patient
3. Method
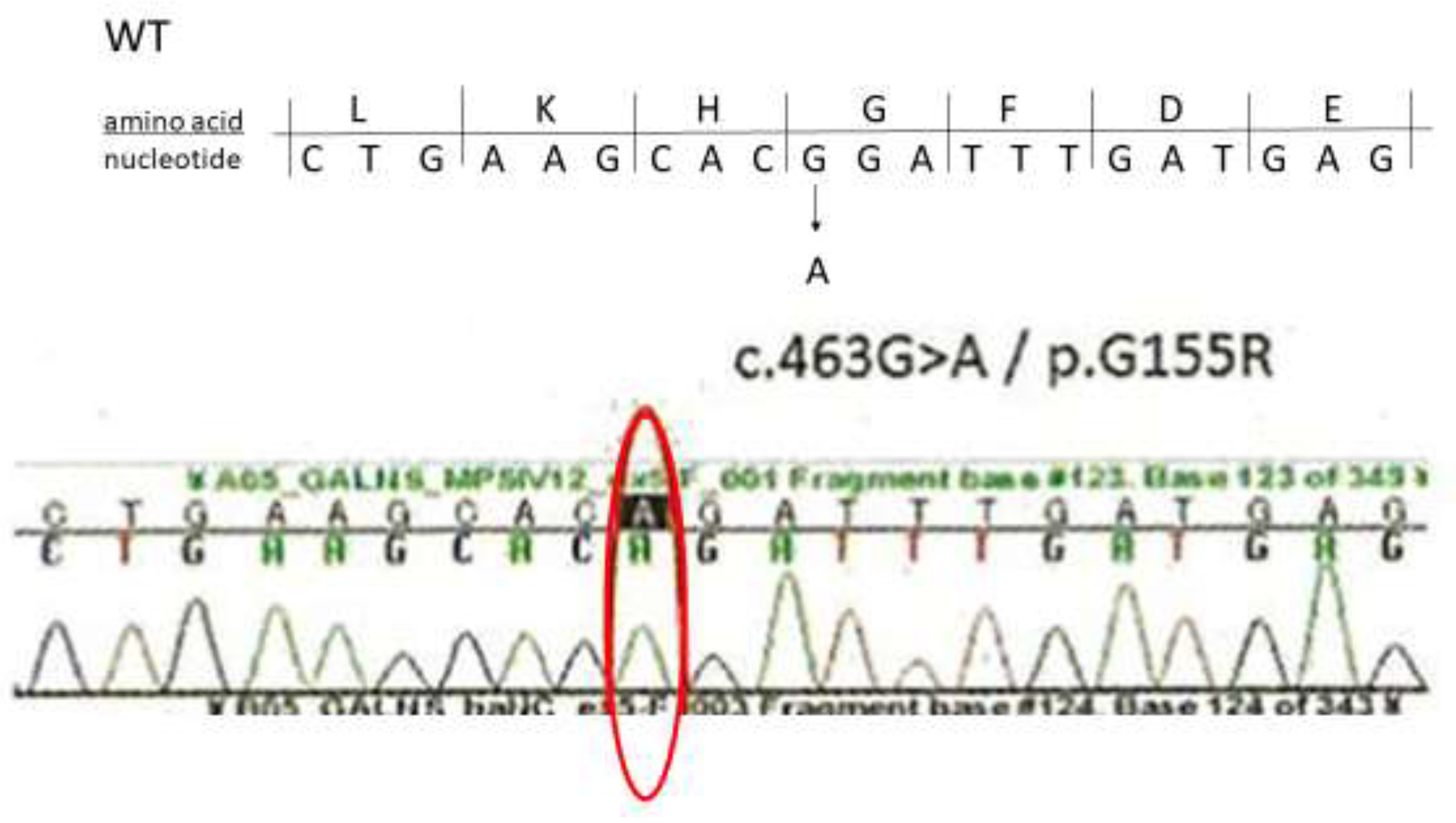
4. Diagnosis
5. Enzyme Replacement Therapy
6. Clinical Findings Before and After Initiation of the ERT
6.1. Urinary GAG Analysis and Other Laboratory Tests
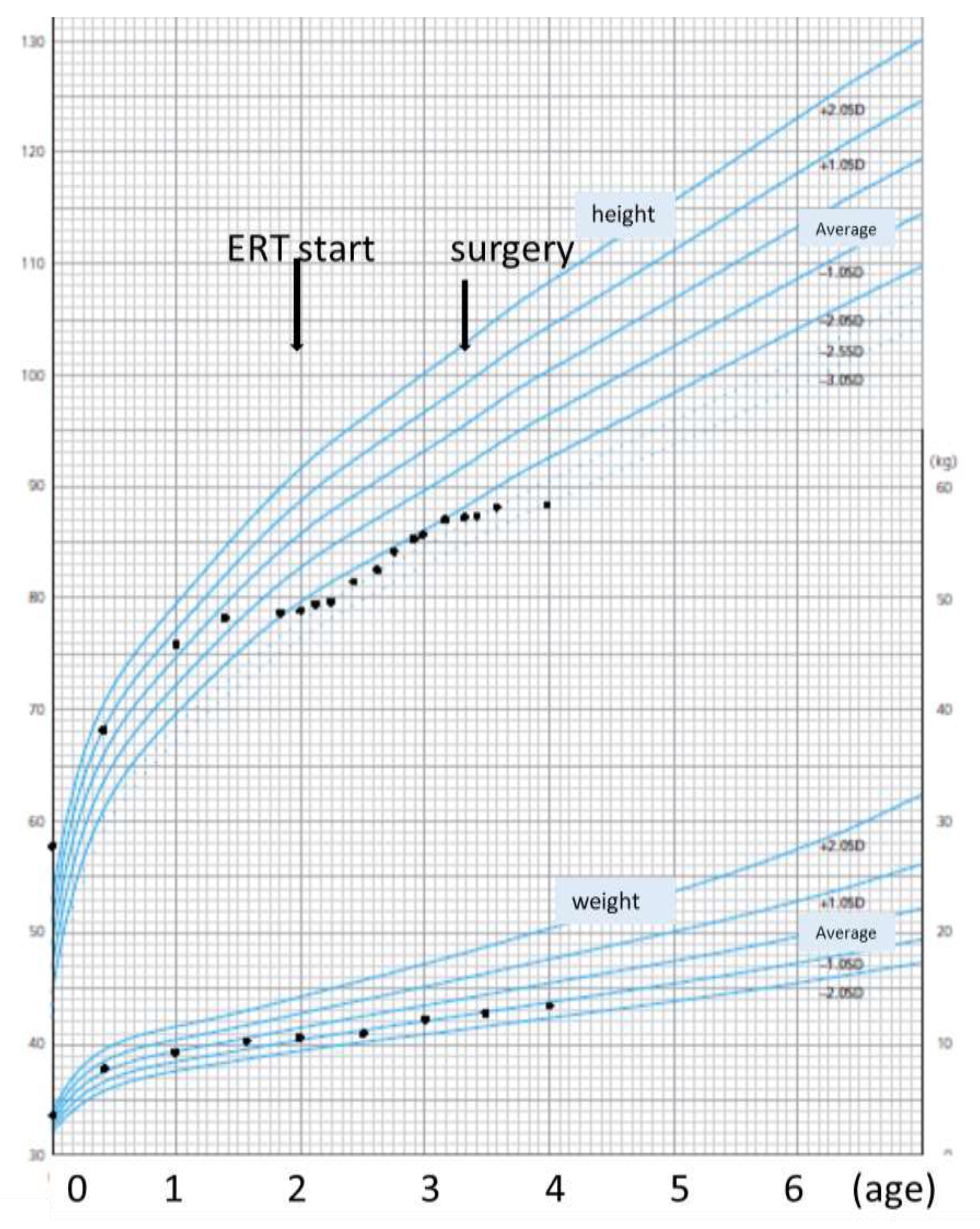
6.2. Body Height and Weight
6.3. Physical Activity
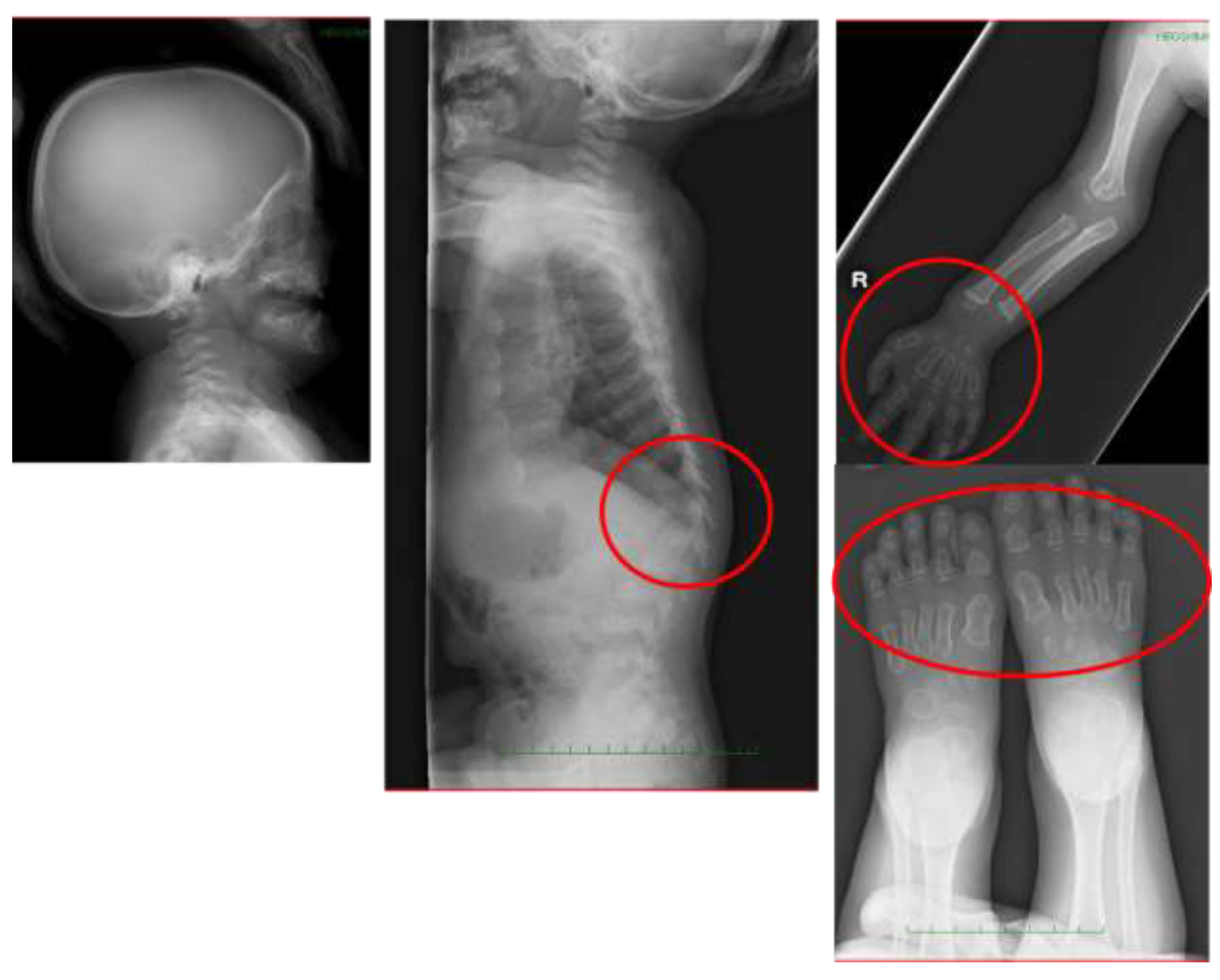
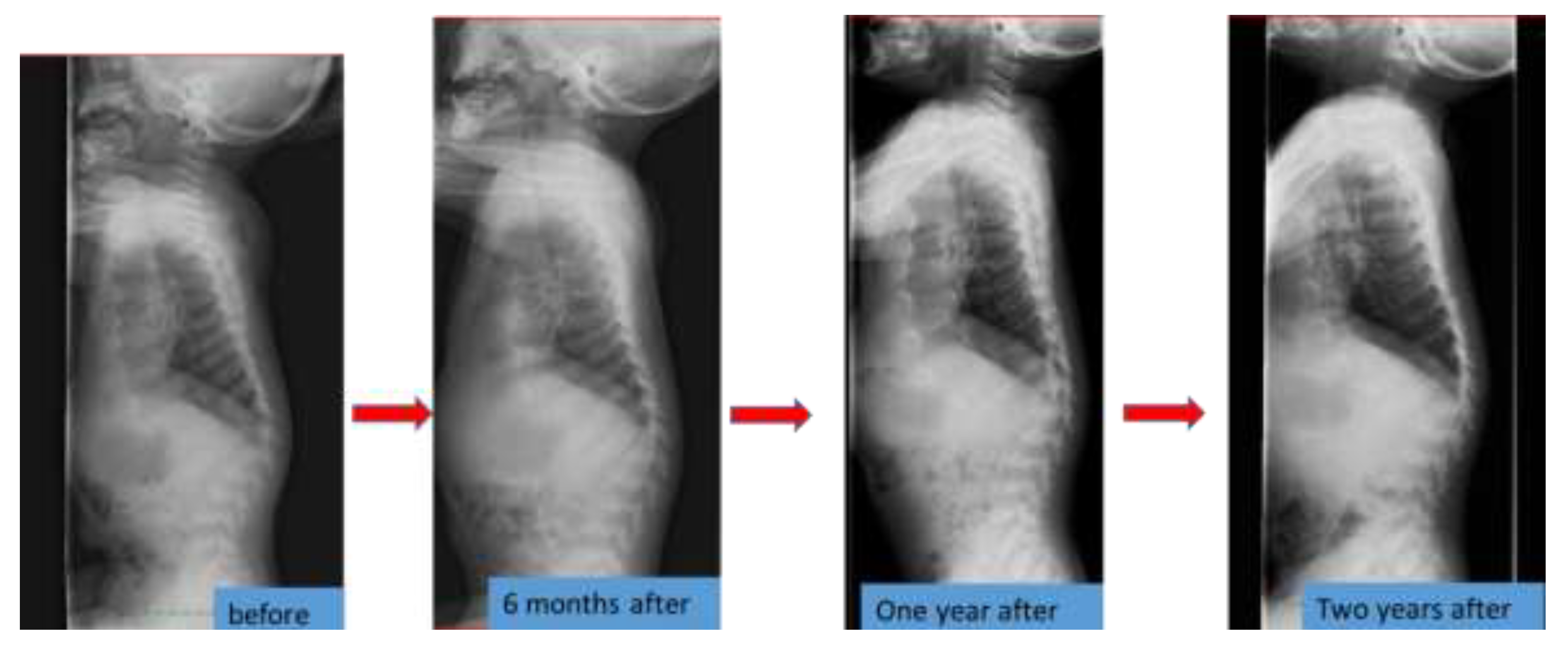
6.4. Spinal Lesions
6.5. Echography and Management in the Other Departments
7. Discussion
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| N-acetylgalactosamine-6-sulfatase | GalNac6S |
| MPS | mucopolysaccharidosis |
| GAG | glycosaminoglycan |
| KS | keratan sulfate |
| ERT | enzyme replacement therapy |
| SD | standard deviation |
| MRI | magnetic resonance imaging |
| GH | growth hormone |
References
- Khan, S.; Alméciga-Díaz, C.J.; Sawamoto, K.; Mackenzie, W.G.; Theroux, M.C.; Pizarro, C.; Mason, R.W.; Orii, T.; Tomatsu, S. Mucopolysaccaridosis ⅣA and glycosaminoglycans. Mol. Genet. Metab. 2017, 120, 78–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentili, C.; Cancedda, R. Cartilage and bone extracellular matrix. Curr. Pharm. Des. 2009, 15, 1334–1348. [Google Scholar] [CrossRef] [PubMed]
- Perache, H.; Sawamoto, K.; Averill, L.; Kecskemethy, H.; Therou, M.; Thacker, M.; Nagao, K.; Pizarro, C.; Mackenzie, W.; Kobayashi, H.; et al. Molecular genetics and metabolism special edition: Diagnosis and prognosis of mucopolysaccharidosis IVA. Mol. Genet. Metab. 2018, 125, 18–37. [Google Scholar] [CrossRef] [PubMed]
- Aliiston, T. Chondroitin sulfate and growth factor signaling in the skeleton: Possible link to MPS Ⅵ. J. Pediatr. Rehabil. Med. 2010, 3, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhlebach, M.S.; Wooten, W.; Muenzer, J. Respiratory manifestations in mucopolysaccharidoses. Pediatr. Respir. 2011, 12, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Averill, L.W.; Sawamoto, K.; Mackenzie, W.G.; Bober, M.B.; Pizarro, C.; Goff, C.J.; Xie, L.; Orii, T.; Theroux, M. Obstructive airway in Morquio A syndrome, the past, the present and the future. Mol. Genet. Metab. 2016, 117, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Bank, R.A.; Groener, J.E.; van Gemund, J.J.; Maaswinkel, P.D.; Hoeben, K.A.; Schut, H.A.; Everts, V. Deficiency in N-acetylgalactosamine-6-sulfate sulfatase results in collagen perturbations in cartilage of Morquio syndrome A patients. Mol. Genet. Metab. 2009, 97, 196–201. [Google Scholar] [CrossRef]
- Tomatsu, S.; Mackenzie, W.G.; Theroux, M.C.; Mason, R.W.; Thacker, M.M.; Shaffer, T.H.; Montaño, A.M.; Rowan, D.; Sly, W.; Alméciga-Díaz, C.J.; et al. Current and emerging treatments and surgical interventions for Morquio A syndrome: A review. Res. Rep. Endocr. Disord. 2012, 12, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Tomatsu, S.; Mackenzie, W.G.; Theroux, M.C.; Mason, R.W.; Thacker, M.M.; Shaffer, T.H.; Montaño, A.M.; Rowan., D.; Sly, W.; Alméciga-Díaz, C.J.; et al. Mucopolysaccharidosis type IVA (Morquio A disease): Clinical review and current treatment. Curr. Pharm. Biotechnol. 2011, 12, 931–945. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Nishioka., T.; Gutierrez, M.A.; Peña, O.M.; Tranda Firescu, G.G.; Lopez, P.; Yamaguchi, S.; Noguchi, A.; Orii, T. Mutation and polymorphism spectrum of the GALNS gene in mucopolysaccharidosis 4A. Hum. Mutat. 2005, 26, 500–521. [Google Scholar] [CrossRef]
- Hiramatsu, M.; Nakamura, K. Elosulfase alfa enzyme replacement therapy attenuates disease progression in a non-ambulatory Japanese patient with Morquio A syndrome (case report). Mol. Genet. Metab. Rep. 2017, 13, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Bialer, M.; Parini, R.; Martin, K.; Wang, H.; Yang, K.; Shaywitz, A.J.; Harmatz, P. Safety and clinical activity of elosulfase alfa in pediatric patients with Morquio A syndrome (mucopolysaccharidosis IVA) less than 5 y. Pediatr. Res. 2015, 78, 718–722. [Google Scholar] [CrossRef] [PubMed]
- Do Cao, J.; Wiedemann, A.; Quinaux, T.; Battaglia-Hsu, S.F.; Mainard, L.; Froissart, R.; Bonnemains, C.; Ragot, S.; Leheup, B.; Journeau, P.; et al. 30 months follow-up of an early enzyme replacement therapy in a severe Morquio A patient: About one case. Mol. Genet. Metab. Rep. 2016, 9, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Doherty, C.; Stapleton, M.; Piechnik, M.; Mason, R.W.; Mackenzie, W.G.; Yamaguchi, S.; Kobayashi, H.; Suzuki, Y.; Tomatsu, S. Effect of enzyme replacement therapy on the growth of patients with Morquio A. J. Hum. Genet. 2019, 64, 625–635. [Google Scholar] [CrossRef]
- Kosuga, M.; Mashima, R.; Hirakiyama, A.; Fuji, N.; Kumagai, T.; Seo, J.H.; Nikaido, M.; Saito, S.; Ohno, K.; Sakuraba, H.; et al. Molecular diagnosis of 65 families with mucopolysaccharidosis type II (Hunter syndrome) characterized by 16 novel mutations in the IDS gene: Genetic, pathological, and structural studies on iduronate-2-sulfatase. Mol. Genet. Metab. 2016, 118, 190–197. [Google Scholar] [CrossRef]
- Montano, A.M.; Tomatsu, S.; Brusius, A.; Smith, M.; Orii, T. Growth charts for patients affected with Morquio A disease. Am. J. Med. Genet. Part A 2008, 146, 1286–1295. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.; Oikawa, H.; Giugliani, R.; Harmatz, P.; Smith, M.; Suzuki, Y.; Orii, T. Impairment of Body Growth in Mucopolysaccharidoses. In Handbook of Growth and Growth Monitoring in Health and Disease; Preedy, V.R., Ed.; Springer Science & Business Media, LLC: New York, NY, USA, 2012; pp. 2091–2116. [Google Scholar] [CrossRef]
- Kato, N.; Isojima, G.; Murata, M. Growth standard charts for Japanese children with mean and standard deviation (SD) values based on the year 2000 national survey. Clinic Pediatr. Endocrinol. 2016, 25, 71–76. [Google Scholar] [CrossRef]
- Charrow, J.; Alden, T.D.; Breathnach, C.A.; Frawley, G.P.; Hendriksz, C.J.; Link, B.; Mackenzie, W.G.; Manara, R.; Offiah, A.C.; Solano, M.L.; et al. Diagnostic evaluation, monitoring, and perioperative management of spinal cord compression in patients with Morquio syndrome. Mol. Genet. Metab. 2015, 114, 11–18. [Google Scholar] [CrossRef]
- Simonaro, C.M.; D’Angelo, M.; He, X.; Eliyahu, E.; Shtraizent, N.; Haskins, M.E.; Schuchman, E.H. Mechanism of glycosaminoglycan-mediated bone and joint disease; implications for the mucopolysaccharidoses and other connective tissue disease. Am. J. Pathol. 2008, 172, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Matalon, R.; Arbogast, B.; Justice, P.; Brandt, I.K.; Dorfman, A. Morquio’s syndrome: Deficiency of a chondroitin sulfate N-acetylhexosamine sulfate sulfatase. Biochem. Biophys. Res. Commun. 1974, 61, 759–765. [Google Scholar] [CrossRef]
- Doherty, C.; Averill, L.W.; Theroux, M.; Mackenzie, W.G.; Pizarro, C.; Mason, R.W.; Tomatsu, S. Natural history of Morquio A patient with tracheal obstruction from birth to death. Mol. Genet. Metab. Reports. 2018, 1, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Melbouc, M. Growth impairment in mucopolysaccharidoses. Mol. Genet. Metab. 2018, 124, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Simonaro, C.M.; Ge, Y.; Eliyahu, E.; He, X.; Jepsen, K.J.; Schuchman, E.H. Involvement of the Toll-like receptor 4 pathway and use of TNF-alpha antagonists for treatment of the mucopolysaccharidoses. Proc. Natl. Acad. Sci. USA 2010, 107, 222–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattoni, A.; Motta, S.; Masera, N.; Gasperini, S.; Rovelli, A.; Parini, R. The use of recombinant human growth hormone in patients with Mucopolysaccharidosis and growth hormone deficiency: A case series. Ital. J. Pediatrics. 2019, 45, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuchman, E.H.; Ge, Y.; Lai, A.; Borisov, Y.; Faillace, M.; Eliyahu, E.; He, X.; Iatridis, J.; Vlassara, H.; Striker, G.; et al. Pentosan polysulfate: A novel therapy for the mucopolysaccharidoses. PLoS ONE 2013, 8, e54459. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, J.; Bravo, S.B.; García-Vence, M.; De Castro, M.J.; Luzardo, A.; Colón, C.; Tomatsu, S.; Otero-Espinar, F.J.; Couce, M. Proteomic analysis in Morquio A cells treated with immobilized enzymatic replacement therapy on nanostructured lipid systems. Int. J. Mol. Sci. 2019, 20, 4610. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | References | |
|---|---|---|
| White blood cell count(/µL) | 7680 | 3.3~8.6 × 10^3 |
| Red Blood cell count(/µL) | 4.58 × 10^6 | 4.35~5.55 × 10^6 |
| Hb (g/dL) | 11.6 | 13.7~16.8 |
| PLT(/µL) | 327 × 10^3 | 158~348 × 10^3 |
| AST(IU/L) | 30 | 13~30 |
| ALT(IU/L) | 15 | 10~42 |
| LDH(IU/L) | 239 | 124~222 |
| BUN(mg/dL) | 17.3 | 8~20 |
| Cr,(mg/dL) | 0.20 | 0.65~1.07 |
| Na(mEq/L) | 139 | 138~145 |
| K(mEq/L) | 4.4 | 3.6~4.8 |
| Cl(mEq/L) | 107 | 101~108 |
| Ca(mg/dL) | 5.0 | 4.3~5.2 |
| P(mg/dL) | 4.9 | 2.5~4.7 |
| CRP(mg/dL) | 0.02 | <0.3 |
| ph (vein) | 7.484 | 7.35~7.45 |
| pCO2 | 27.4 | 35~45 |
| HCO3- | 20.3 | 22~24 |
| BE | −0.7 | −3.0~3.0 |
| Leukocyte GalNac6S Activity(nmol/mg protein/17 h) | <1.3 | 104.6 |
| Urine uronic acid (mg/g·creatinine) | 166 | 29.7 ± 13.3 |
| Fraction of KS (%) | 13 | Not detected |
| Before | 6 Months after ERT | 12 Months after ERT | 25 Months after ERT | |
|---|---|---|---|---|
| Body height | 79 cm (−2.3 SD) | 81 cm (−2.5 SD) | 85.5 cm (−2 SD) | 88 cm (−2.9 SD) |
| Urine KS(µg/g·creatinine) | 66.24 | - | 41.71 | - |
| Walk | 4~5 steps | Walk longer and Trot | 50 m for 6 min | By walker |
| Posture at sitting | A forward-bend posture | Up and Better posture | Keep better posture | Slight forward tilting posture |
| Kyphosis angle (degree) | 126 | 131 | 141 | 138 |
| Shoulder joint mobility | Disability of elevation | Improve of elevation (ball playing) | Keep elevation (shoulder backpack) | Keep elevation |
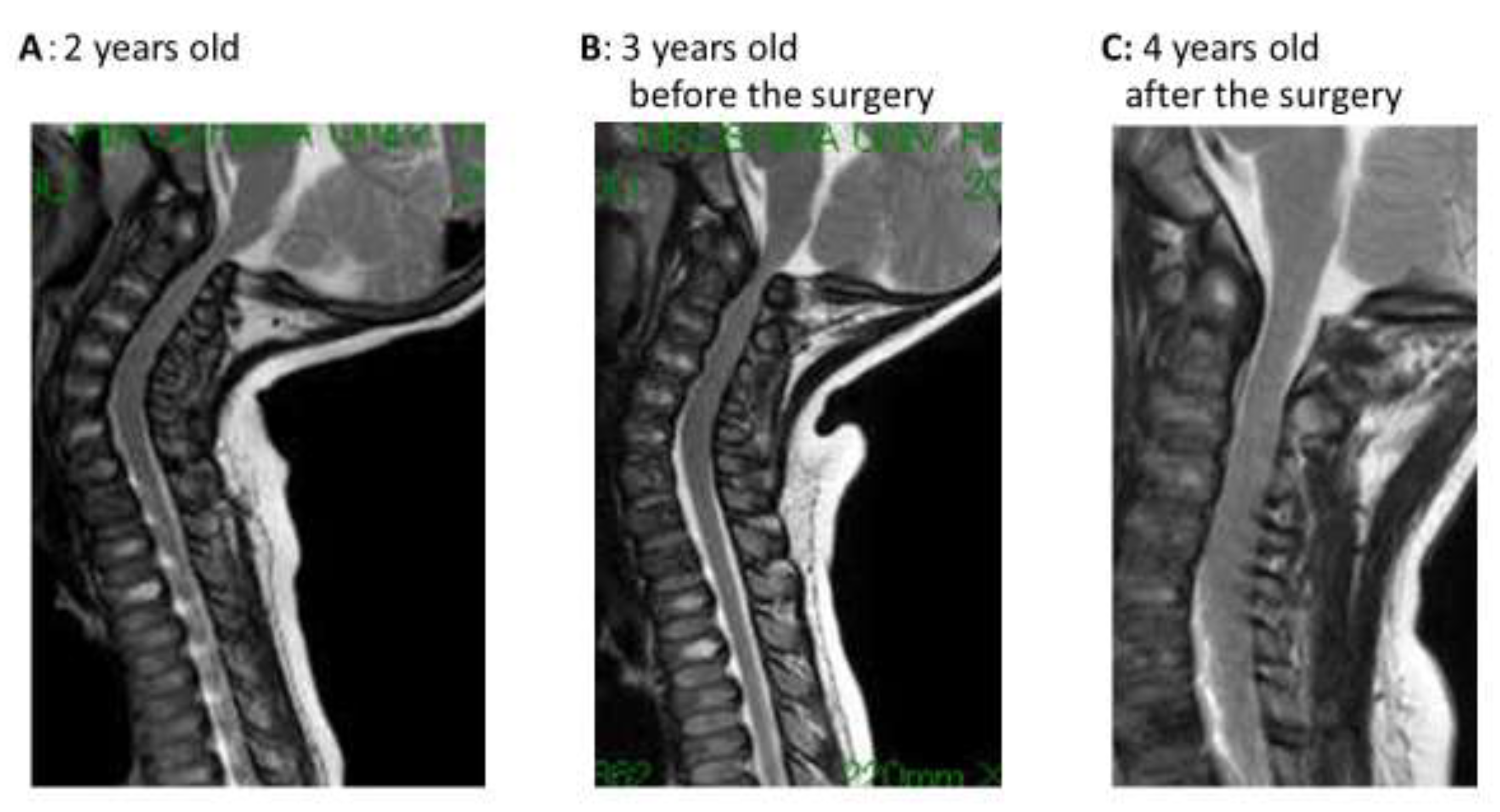
| Spinal compression at foramen magnum | Compression by magnetic resonance imaging (MRI) No symptom | - | Compression by MRI No symptom | Improvement after decompression surgery |
| Echocardiography | MR trivial | MR trivial | MR trivial | MR trivial |
| Abdominal echography | Hepatosplenomegaly- | - | - | - |
| Otolaryngeography | Otitis media + Mild deafness(40Db) | Otitis media+ | Otitis media- Improved | No medication Hearing test: normal(25Db) |
| Opthalmology | Not particular | - | Not particular | Not particular |
| Communication level | Several words | Short talk | Talk well | Communicate well |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura-Utsunomiya, A.; Nakamae, T.; Kagawa, R.; Karakawa, S.; Sakata, S.; Sakura, F.; Tani, C.; Matsubara, Y.; Ishino, T.; Tajima, G.; et al. A Case Report of a Japanese Boy with Morquio A Syndrome: Effects of Enzyme Replacement Therapy Initiated at the Age of 24 Months. Int. J. Mol. Sci. 2020, 21, 989. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030989
Nakamura-Utsunomiya A, Nakamae T, Kagawa R, Karakawa S, Sakata S, Sakura F, Tani C, Matsubara Y, Ishino T, Tajima G, et al. A Case Report of a Japanese Boy with Morquio A Syndrome: Effects of Enzyme Replacement Therapy Initiated at the Age of 24 Months. International Journal of Molecular Sciences. 2020; 21(3):989. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030989
Chicago/Turabian StyleNakamura-Utsunomiya, Akari, Toshio Nakamae, Reiko Kagawa, Shuhei Karakawa, Sonoko Sakata, Fumiaki Sakura, Chihiro Tani, Yoshiko Matsubara, Takashi Ishino, Go Tajima, and et al. 2020. "A Case Report of a Japanese Boy with Morquio A Syndrome: Effects of Enzyme Replacement Therapy Initiated at the Age of 24 Months" International Journal of Molecular Sciences 21, no. 3: 989. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030989