Biocomplexity and Fractality in the Search of Biomarkers of Aging and Pathology: Mitochondrial DNA Profiling of Parkinson’s Disease


Abstract
:1. Introduction
2. Results
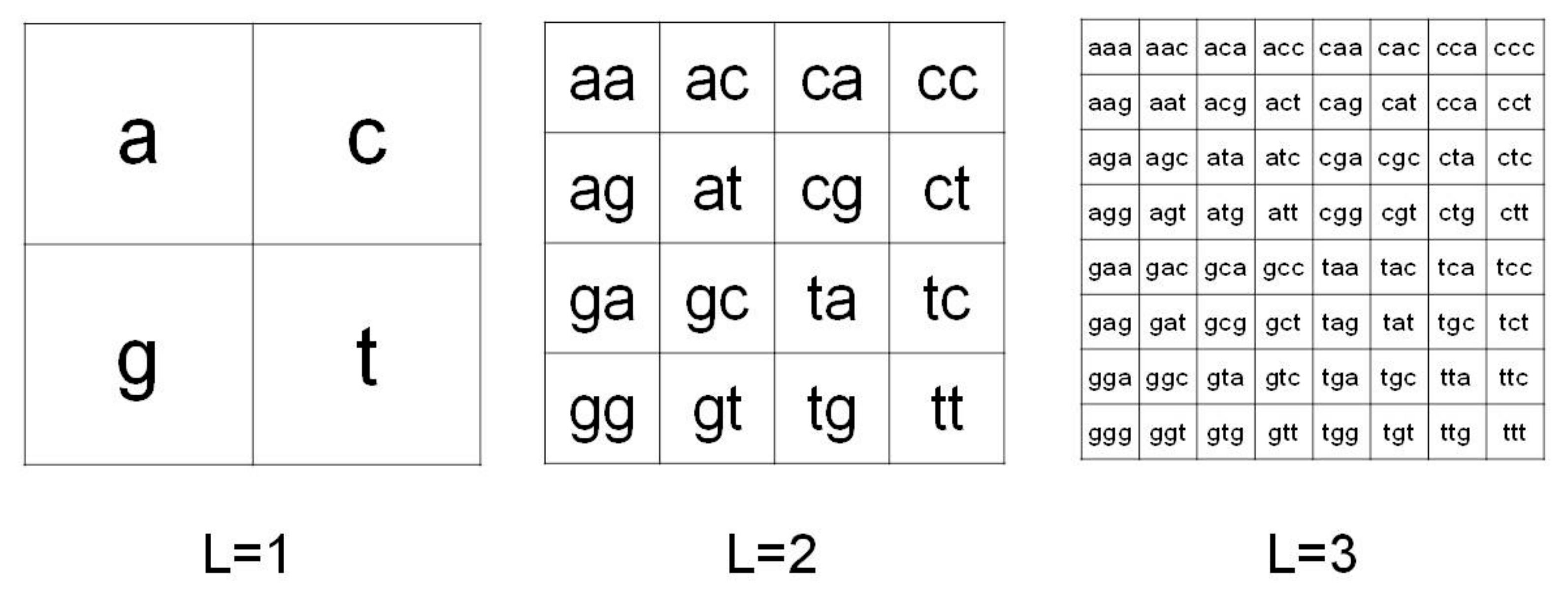
2.1. Chaos Game Representation
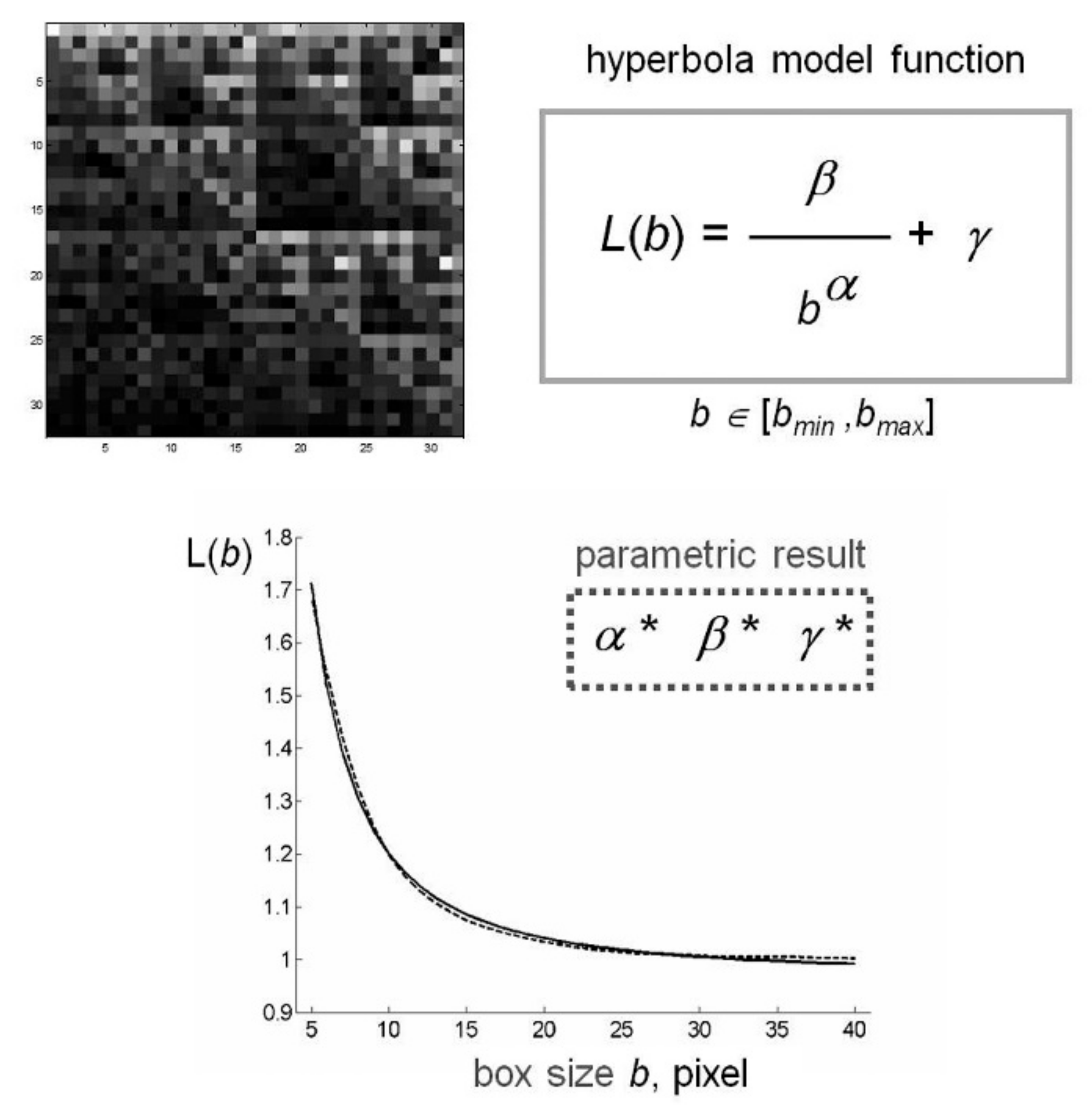
2.2. Fractal Analysis of mtDNA in Aging and Parkinson’s Disease
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. mtDNA Extraction and Resequencing
4.3. Chaos Game Representation of mtDNA
- initialize A to a zero matrix having 2L rows and 2L columns,
- for each substring s⸦S, increase by one the entry of A having position p(s).
- initialize A to a zero matrix having 2L rows and 2L columns,
- for each substring s⸦S, compute the number N of multiple strings generated by the undetermined symbols in s, and perform the following cycle,
- for n=1,2,…,N,
- i.
- generate the string sn from s,
- ii.
- increase by w=1/N the entry of A having position p(sn).
- end for.
4.4. Estimate of Lacunarity
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| ADL | Activities of Daily Living hierarchy scale |
| CGR | Chaos Game Representation |
| CI | Complex I |
| CPS | Cognitive Performance Scale |
| FD | Fractal Dimension |
| GBA | Gliding Box Algorithm |
| H&Y | Hoehn & Yahr stage |
| mtDNA | mitochondrial DNA |
| np | nucleotide position |
| PD | Parkinson’s Disease |
| rCRS ROS | revised Cambridge Reference Sequence Reactive Oxygen Species |
References
- de Rijk, M.C.; Breteler, M.M.; Graveland, G.A.; Ott, A.; Grobbee, D.E.; van der Meche, F.G.A.; Hofman, A. Prevalence of Parkinson’s disease in the elderly: The rotterdam study. Neurology 1995, 45, 2143–2146. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Lai, B.C.L.; Marion, S.A.; Teschke, K.; Tsui, J.K.C. Occupational and environmental risk factors for Parkinson’s disease. Parkinsonism Relat. Disord. 2002, 8, 297–309. [Google Scholar] [CrossRef]
- Gorell, J.M.; Johnson, C.C.; Rybicki, B.A.; Peterson, E.L.; Kortsha, G.X.; Brown, G.G.; Richardson, R.J. Occupational exposures to metals as risk factors for Parkinson’s disease. Neurology 1997, 48, 650–658. [Google Scholar] [CrossRef]
- Gorell, J.M.; Johnson, C.C.; Rybicki, B.A.; Peterson, E.L.; Richardson, R.J. The risk of Parkinson’s disease with exposure to pesticides, farming, well water, and rural living. Neurology 1998, 50, 1346–1350. [Google Scholar] [CrossRef]
- Zorzon, M.; Capus, L.; Pellegrino, A.; Cazzato, G.; Zivadinov, R. Familial and environmental risk factors in Parkinson’s disease: A case-control study in north-east Italy. Acta Neurol. Scand. 2002, 105, 77–82. [Google Scholar] [CrossRef]
- Calne, D.B.; Langston, J.W. Aetiology of Parkinson’s disease. Lancet 1983, 2, 1457–1459. [Google Scholar] [CrossRef]
- Huang, Z.; de la Fuente-Fernández, R.; Stoessl, A.J. Etiology of Parkinson’s disease. Can. J. Neurol. Sci. 2003, 30, S10–S18. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Goedert, M. Tau and Parkinson disease. JAMA 2001, 286, 2324–2326. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rüb, C.; Liu, Y.; Magrané, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. α-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St Lawrence, E.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s disease. Cell Stem. Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Khusnutdinova, E.; Gilyazova, I.; Ruiz-Pesini, E.; Derbeneva, O.; Khusainova, R.; Khidiyatova, I.; Magzhanov, R.; Wallace, D.C. A mitochondrial etiology of neurodegenerative diseases: Evidence from Parkinson’s disease. Ann. N. Y. Acad. Sci. 2008, 1147, 1–20. [Google Scholar] [CrossRef]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Why do we have a maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu. Rev. Biochem. 2007, 76, 781–821. [Google Scholar] [CrossRef] [Green Version]
- Franco-Iborra, S.; Vila, M.; Perier, C. The Parkinson disease mitochondrial hypothesis: Where are we at? Neuroscientist 2016, 22, 266–277. [Google Scholar] [CrossRef]
- Hahn, A.; Zuryn, S. The cellular mitochondrial genome landscape in disease. Trends Cell Biol. 2019, 29, 227–240. [Google Scholar] [CrossRef]
- Hudson, G.; Nalls, M.; Evans, J.R.; Breen, D.P.; Winder-Rhodes, S.; Morrison, K.E.; Morris, H.R.; Williams-Gray, C.H.; Barker, R.A.; Singleton, A.B.; et al. Two-stage association study and meta-analysis of mitochondrial DNA variants in Parkinson disease. Neurology 2013, 2013. 80, 2042–2048. [Google Scholar] [CrossRef]
- Ghezzi, D.; Marelli, C.; Achilli, A.; Goldwurm, S.; Pezzoli, G.; Barone, P.; Pellecchia, M.T.; Stanzione, P.; Brusa, L.; Bentivoglio, A.R.; et al. Mitochondrial DNA haplogroup K is associated with a lower risk of Parkinson’s disease in Italians. Eur. J. Hum. Genet. 2005, 13, 748–752. [Google Scholar] [CrossRef]
- Van Der Walt, J.M.; Nicodemus, K.K.; Martin, E.R.; Scott, W.K.; Nance, M.A.; Watts, R.L.; Hubble, J.P.; Haines, J.L.; Koller, W.C.; Lyons, K.; et al. Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. Am. J. Hum. Genet. 2003, 72, 804–811. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.K.; Pankratz, N.; Kissell, D.K.; Pauciulo, M.W.; Halter, C.A.; Rudolph, A.; Pfeiffer, R.F.; Nichols, W.C.; Foroud, T.; Parkinson Study Group-PROGENI Investigators. Maternal inheritance and mitochondrial DNA variants in familial Parkinson’s disease. BMC Med. Genet. 2010, 11, 53. [Google Scholar] [CrossRef] [PubMed]
- Andalib, S.; Vafaee, M.S.; Gjedde, A. Parkinson’s disease and mitochondrial gene variations: A review. J. Neurol. Sci. 2014, 346, 11–19. [Google Scholar] [CrossRef]
- Simon, D.K.; Matott, J.C.; Espinosa, J.; Abraham, N.A. Mitochondrial DNA mutations in Parkinson’s disease brain. Acta Neuropathol. Commun. 2017, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.M.N.; Schuck, P.F.; Wenk, G.L.; Ferreira, G.C. Metabolic disturbances in diseases with neurological involvement. Aging Dis. 2014, 5, 238–255. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Domingues, A.F.; Ferreira, I.L.; Januário, C.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial function in Parkinson’s disease cybrids containing an nt2 neuron-like nuclear background. Mitochondrion 2008, 8, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, J.G.; Hipp, M.J.; Montine, T.J.; Kennedy, S.R. Mitochondrial DNA mutations increase in early stage Alzheimer disease and are inconsistent with oxidative damage. Ann. Neurol. 2016, 80, 301–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casoli, T.; Spazzafumo, L.; Di Stefano, G.; Conti, F. Role of diffuse low-level heteroplasmy of mitochondrial DNA in Alzheimer’s disease neurodegeneration. Front. Aging Neurosci. 2015, 7, 142. [Google Scholar] [CrossRef] [Green Version]
- Borghammer, P.; van Den Bergeb, N. Brain-first versus gut-first Parkinson’s disease: A. hypothesis. J. Parkinsons Dis. 2019, 9, S281–S295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casoli, T.; Lisa, R.; Fabbietti, P.; Conti, F. Analysis of mitochondrial DNA allelic changes in Parkinson’s disease: A preliminary study. Aging Clin. Exp. Res. 2020, 32, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Zaia, A.; Maponi, P.; Di Stefano, G.; Casoli, T. Biocomplexity and fractality in the search of biomarkers of aging and pathology: Focus on mitochondrial DNA and Alzheimer’s disease. Aging Dis. 2017, 8, 44–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oiwa, N.N.; Glazier, J.A. Self-similar mitochondrial DNA. Cell. Biochem. Biophys. 2004, 41, 41–62. [Google Scholar] [CrossRef]
- Hao, B.L. Fractals from genomes—Exact solutions of a biology-inspired problem. Physica A 2000, 282, 225–246. [Google Scholar] [CrossRef] [Green Version]
- Kirilyuk, A.P. Complex-dynamical extension of the fractal paradigm and its applications in life sciences. In Fractals in Biology and Medicine; Losa, G.A., Merlini, D., Nonnenmacher, T.F., Weibel, E., Eds.; Birkhauser Press: Basel, Switzerland, 2004; pp. 233–244. [Google Scholar]
- Zhou, L.Q.; Yu, Z.G.; Deng, J.Q.; Anh, V.; Long, S.C. A fractal method to distinguish coding and non-coding sequences in a complete genome based on a number sequence representation. J. Theor. Biol. 2005, 232, 559–567. [Google Scholar] [CrossRef] [Green Version]
- Aldrich, P.R.; Horsley, R.K.; Turcic, S.M. Symmetry in the language of gene expression: A survey of gene promoter networks in multiple bacterial species and non-σ regulons. Symmetry 2011, 3, 750–766. [Google Scholar] [CrossRef] [Green Version]
- Cattani, C.; Pierro, G. On the fractal geometry of DNA by the binary image analysis. Bull. Math. Biol. 2013, 75, 1544–1570. [Google Scholar] [CrossRef]
- Piantanelli, L.; Rossolini, G.; Basso, A.; Piantanelli, A.; Malavolta, M.; Zaia, A. Use of mathematical models of survivorship in the study of biomarkers of aging: The role of heterogeneity. Mech. Ageing Dev. 2001, 122, 1461–1475. [Google Scholar] [CrossRef]
- Zaia, A. Osteoporosis and fracture risk: New perspectives for early diagnosis and treatment assessment. In Osteoporosis: Etiology, Diagnosis and Treatment; Mattingly, B.E., Pillare, A.C., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2009; pp. 267–290. [Google Scholar]
- Pettersson, M. Complexity and Evolution; Cambridge University Press: Cambridge, UK, 1996. [Google Scholar]
- Grassberger, A.; Procaccia, I. Measuring the strangeness of strange attractors. Physica D 1983, 9, 189–208. [Google Scholar] [CrossRef]
- Goldberger, A.L.; Rigney, D.R.; West, B.J. Chaos and fractals in human physiology. Sci. Am. 1990, 262, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Goldberger, A.L. Non-linear dynamics for clinicians: Chaos theory, fractals, and complexity at the bedside. Lancet 1996, 347, 1312–1314. [Google Scholar] [CrossRef]
- Nonnenmacher, T.F.; Baumann, G.; Losa, G.A. Self-organization and fractal scaling patterns in biological systems. In Trends in Biological Cybernetics; Menon, J., Ed.; Publication Manager, Research Trends, Council of Scientific Research Integration: Trivandrum, India, 1990; pp. 65–73. [Google Scholar]
- Weibel, E.R. Fractal geometry: A design principle for living organisms. Am. J. Physiol. 1991, 261, L361–L369. [Google Scholar] [CrossRef] [PubMed]
- Goldberger, L.A.; Peng, C.K.; Lipsitz, L.A. What is physiologic complexity and how does it change with aging and disease? Neurobiol. Aging 2002, 23, 23–26. [Google Scholar] [CrossRef]
- Piantanelli, A.; Serresi, S.; Ricotti, G.; Rossolini, G.; Zaia, A.; Basso, A.; Piantanelli, L. Color-based method for fractal dimension estimation of pigmented skin lesion contour. In Fractals in Biology and Medicine; Losa, G.A., Ed.; Birkhauser Press: Basel, Switzerland, 2002; pp. 127–136. [Google Scholar]
- Vaillancourt, D.E.; Newell, K.M. Changing complexity in human behaviour and physiology through aging and disease. Neurobiol. Aging 2002, 23, 1–11. [Google Scholar] [CrossRef]
- Lipsitz, L.A. Physiological complexity, aging, and the path to frailty. Sci. Aging Knowl. Environ. 2004, 16, pe16. [Google Scholar] [CrossRef]
- Doubal, F.N.; MacGillivray, T.J.; Patton, N.; Dhillon, B.; Dennis, M.S.; Wardlaw, J.M. Fractal analysis of retinal vessels suggests that a distinct vasculopathy causes lacunar stroke. Neurology 2010, 74, 1102–1107. [Google Scholar] [CrossRef] [Green Version]
- Fiz, J.A.; Monte-Moreno, E.; Andreo, F.; Auteri, S.J.; Sanz-Santos, J.; Serra, P.; Bonet, G.; Castellà, E.; Manzano, J.R. Fractal dimension analysis of malignant and benign endobronchial ultrasound nodes. BMC Med. Imaging 2014, 14, 22. [Google Scholar] [CrossRef] [Green Version]
- Captur, G.; Karperien, A.L.; Li, C.; Zemrak, F.; Tobon-Gomez, C.; Gao, X.; Bluemke, D.A.; Elliott, P.M.; Petersen, S.E.; Moon, J.C. Fractal frontiers in cardiovascular magnetic resonance: Towards clinical implementation. J. Cardiovasc. Magn. Reson. 2015, 17, 80. [Google Scholar] [CrossRef] [Green Version]
- Mandelbrot, B.B. A Fractal’s Lacunarity, and how it can be Tuned and Measured. In Fractals in Biology and Medicine; Nonnenmacher, T.F., Losa, G.A., Weibel, E.R., Eds.; Birkhauser Press: Basel, Switzerland, 1993; pp. 8–21. [Google Scholar]
- Plotnick, R.E.; Gardner, R.H.; Hargrove, W.W.; Prestegard, K.; Perlmutter, M. Lacunarity analysis: A general technique for the analysis of spatial patterns. Phys. Rev. E 1996, 53, 5461–5468. [Google Scholar] [CrossRef] [Green Version]
- Allain, C.; Cloitre, M. Characterizing the lacunarity of random and deterministic fractal sets. Phy. Rev. A 1991, 44, 3552–3558. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, H.J. Chaos game representation of gene structure. Nucleic Acids Res. 1990, 18, 2163–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deschavanne, P.J.; Giron, A.; Vilain, J.; Fagot, G.; Fertil, B. Genomic signature: Characterization and classification of species assessed by chaos game representation of sequences. Mol. Biol. Evol. 1999, 16, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Wang, Y.; Lu, D. Multifractal analysis of genomic sequences CGR images. In Proceedings of the 2005 IEEE Engineering in Medicine and Biology 27th Annual Conference, Shanghai, China, 17–18 January 2006; Volume 5, pp. 4783–4786. [Google Scholar] [CrossRef]
- Stan, C.; Cristescu, C.P.; Scarlat, E.I. Similarity analysis for DNA sequences based on chaos game representation. Case study: The albumin. J. Theor. Biol. 2010, 267, 513–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaia, A.; Eleonori, R.; Maponi, P.; Rossi, R.; Murri, R. Medical imaging and osteoporosis: Fractal’s lacunarity analysis of trabecular bone in MR images. In Proceedings of the Eighteenth IEEE Symposium on Computer-Based Medical Systems—CBMS 2005, Dublin, Ireland, 23–24 June 2005; pp. 3–8. [Google Scholar] [CrossRef]
- Zaia, A.; Eleonori, R.; Maponi, P.; Rossi, R.; Murri, R. MR imaging and osteoporosis: Fractal lacunarity analysis of trabecular Bone. IEEE Trans. Inf. Technol. Biomed. 2006, 10, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Zaia, A.; Rossi, R.; Egidi, N.; Maponi, P. Fractal’s lacunarity analysis of trabecular bone in MR images. In Computational Vision and Medical Image Processing; Tavares, J., Jorge, N., Eds.; CRC Press: Boca Raton, FL, USA, 2010; pp. 421–426. [Google Scholar]
- Zaia, A. Fractal lacunarity of trabecular bone and magnetic resonance imaging: New perspectives for osteoporotic fracture risk assessment. World J. Orthop. 2015, 6, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hill, K.; Singh, S.; Kari, L. The spectrum of genomic signatures: From dinucleotides to chaos game representation. Gene 2005, 346, 173–185. [Google Scholar] [CrossRef]
- Coskun, P.; Wyrembak, J.; Schriner, S.E.; Chen, H.W.; Marciniack, C.; Laferla, F.; Wallace, D.C. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim. Biophys. Acta 2012, 1820, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, V.S.; Fenech, M. Mutations that affect mitochondrial functions and their association with neurodegenerative diseases. Mutat. Res. Rev. Mutat. Res. 2014, 759, 1–13. [Google Scholar] [CrossRef]
- Smigrodzki, R.; Parks, J.; Parker, W.D. High frequency of mitochondrial complex I mutations in Parkinson’s disease and aging. Neurobiol. Aging 2004, 25, 1273–1281. [Google Scholar] [CrossRef]
- Ikebe, S.; Tanaka, M.; Ohno, K.; Sato, W.; Hattori, K.; Kondo, T.; Mizuno, Y.; Ozawa, T. Increase of deleted mitochondrial DNA in the striatum in Parkinson’s disease and senescence. Biochem. Biophys. Res. Commun. 1990, 170, 1044–1048. [Google Scholar] [CrossRef]
- Schapira, A.H.; Holt, I.J.; Sweeney, M.; Harding, A.E.; Jenner, P.; Marsden, C.D. Mitochondrial DNA analysis in Parkinson’s disease. Mov. Disord. 1990, 5, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Lestienne, P.; Nelson, I.; Riederer, P.; Reichmann, H.; Jellinger, K. Mitochondrial DNA in postmortem brain from patients with Parkinson’s disease. J. Neurochem. 1991, 56, 1819. [Google Scholar] [CrossRef] [PubMed]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T.; Tanaka, M.; Ino, H.; Ohno, K.; Sano, T.; Wada, Y.; Yoneda, M.; Tanno, Y.; Miyatake, T.; Tanaka, T.; et al. Distinct clustering of point mutations in mitochondrial DNA among patients with mitochondrial encephalomyopathies and with Parkinson’s disease. Biochem. Biophys. Res. Commun. 1991, 176, 938–946. [Google Scholar] [CrossRef]
- Ikebe, S.; Tanaka, M.; Ozawa, T. Point mutations of mitochondrial genome in Parkinson’s disease. Brain Res. Mol. Brain Res. 1995, 28, 281–295. [Google Scholar] [CrossRef]
- Shoffner, J.M.; Brown, M.D.; Torroni, A.; Lott, M.T.; Cabell, M.F.; Mirra, S.S.; Beal, M.F.; Yang, C.C.; Gearing, M.; Salvo, R.; et al. Mitochondrial DNA variants observed in Alzheimer disease and Parkinson disease patients. Genomics 1993, 17, 171–184. [Google Scholar] [CrossRef]
- Lucking, C.B.; Kosel, S.; Mehraein, P.; Graeber, M.B. Absence of the mitochondrial A7237T mutation in Parkinson’s disease. Biochem. Biophys. Res. Commun. 1995, 211, 700–704. [Google Scholar] [CrossRef]
- Kosel, S.; Grasbon-Frodl, E.M.; Hagenah, J.M.; Graeber, M.B.; Vieregge, P. Parkinson disease: Analysis of mitochondrial DNA in monozygotic twins. Neurogenetics 2000, 2, 227–230. [Google Scholar] [CrossRef]
- Richter, G.; Sonnenschein, A.; Grunewald, T.; Reichmann, H.; Janetzky, B. Novel mitochondrial DNA mutations in Parkinson’s disease. J. Neural. Transm. 2002, 109, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Vives-Bauza, C.; Andreu, A.L.; Manfredi, G.; Beal, M.F.; Janetzky, B.; Gruenewald, T.H.; Lin, M.T. Sequence analysis of the entire mitochondrial genome in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2002, 290, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- Hua, F.; Zhang, X.; Hou, B.; Xue, L.; Xie, A. Relationship between mitochondrial DNA A10398G polymorphism and Parkinson’s disease: A meta-analysis. Oncotarget 2017, 8, 78023–78030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coxhead, J.; Kurzawa-Akanbi, M.; Hussain, R.; Pyle, A.; Chinnery, P.; Hudson, G. Somatic mtDNA variation is an important component of Parkinson’s disease. Neurobiol. Aging 2016, 38, 217e1–217e6. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.T.; Cantuti-Castelvetri, I.; Zheng, K.; Jackson, K.E.; Tan, Y.B.; Arzberger, T.; Lees, A.J.; Betensky, R.A.; Beal, M.F.; Simon, D.K. Somatic mitochondrial DNA mutations in early Parkinson and incidental Lewy body disease. Ann. Neurol. 2012, 71, 850–854. [Google Scholar] [CrossRef] [Green Version]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Broderick, P.A.; Wenning, L. Neuromolecular imaging in Parkinson’s disease. In Compendium on Parkinson’s Disease; Preedy, V.R., Ed.; Elsevier Press: London, UK, 2020; Chapter 16. [Google Scholar]
- Bustacchini, S.; Abbatecola, A.M.; Bonfigli, A.R.; Chiatti, C.; Corsonello, A.; Di Stefano, G.; Galeazzi, R.; Fabbietti, P.; Lisa, R.; Guffanti, E.E.; et al. The Report-AGE project: A permanent epidemiological observatory to identify clinical and biological markers of health outcomes in elderly hospitalized patients in Italy. Aging Clin. Exp. Res. 2015, 27, 893–901. [Google Scholar] [CrossRef]
- Carpenter, G.I.; Hastie, C.L.; Morris, J.N.; Fries, B.E.; Ankri, J. Measuring change in activities of daily living in nursing home residents with moderate to severe cognitive impairment. BMC Geriatr. 2006, 3, 7. [Google Scholar] [CrossRef]
- Mithani, S.K.; Shao, C.; Tan, M.; Smith, I.M.; Califano, J.A.; El-Naggar, A.K.; Ha, P.K. Mitochondrial mutations in adenoid cystic carcinoma of the salivary glands. PLoS ONE 2009, 4, e8493. [Google Scholar] [CrossRef] [Green Version]
- Coon, K.D.; Valla, J.; Szelinger, S.; Schneider, L.E.; Niedzielko, T.L.; Brown, K.M.; Pearson, J.V.; Halperin, R.; Dunckley, T.; Papassotiropoulos, A.; et al. Quantitation of heteroplasmy of mtDNA sequence variants identified in a population of AD patients and controls by array-based resequencing. Mitochondrion 2006, 6, 194–210. [Google Scholar] [CrossRef]
- Lévêque, M.; Marlin, S.; Jonard, L.; Procaccio, V.; Reynier, P.; Amati-Bonneau, P.; Baulande, S.; Pierron, D.; Lacombe, D.; Duriez, F.; et al. Whole mitochondrial genome screening in maternally inherited non-syndromic hearing impairment using a microarray resequencing mitochondrial DNA chip. Eur. J. Hum. Genet. 2007, 15, 1145–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannwarth, S.; Procaccio, V.; Lebre, A.S.; Jardel, C.; Chaussenot, A.; Hoarau, C.; Maoulida, H.; Charrier, N.; Gai, X.; Xie, H.M.; et al. Prevalence of rare mitochondrial DNA mutations in mitochondrial disorders. J. Med. Genet. 2013, 50, 704–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, H.M.; Perin, J.C.; Schurr, T.G.; Dulik, M.C.; Zhadanov, S.I.; Baur, J.A.; King, M.P.; Place, E.; Clarke, C.; Grauer, M.; et al. Mitochondrial genome sequence analysis: A custom bioinformatics pipeline substantially improves Affymetrix MitoChip v2.0 call rate and accuracy. BMC Bioinform. 2011, 12, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamzami, M.A.; Price, G.R.; Taylor, R.W.; Blakely, E.L.; Oancea, I.; Bowling, F.; Duley, J.A. Insights into N-calls of mitochondrial DNA sequencing using MitoChip v2.0. BMC Res. Notes 2011, 4, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Vinga, S.; Carvalho, A.M.; Francisco, A.P.; Russo, L.M.; Almeida, J.S. Pattern matching through chaos game representation: Bridging numerical and discrete data structures for biological sequence analysis. Algorithms Mol. Biol. 2012, 7, 10. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PD Patients | Controls | |
|---|---|---|
| Number (M/F) | 15 (7/8) | 15 (6/9) |
| Age (year) | 78.8 ± 6.0 | 80.8 ± 5.1 |
| H&Y | 2.5 (2.0-3.0) | |
| CPS | 2.0 (0.5-3.0) | 2.0 (0.5-2.0) |
| ADL | 1.0 (0.0-2.0) | 0.0 (0.0-3.0) |
| Number | rCRS | PD patients | Controls | p Value |
|---|---|---|---|---|
| Subjects | 15 | 15 | ||
| Adenine | 5117 | 4884 ± 252 | 4912 ± 110 | 0.359 |
| Cytosine | 5175 | 4658 ± 206 | 4618 ± 168 | 0.301 |
| Guanine | 2163 | 2102 ± 92 | 2108 ± 49 | 0.421 |
| Thymine | 4089 | 3880 ± 239 | 3916 ± 101 | 0.311 |
| No-call | - | 1009 ± 765 | 980 ± 390 | 0.453 |
| Homoplasmy | - | 18 ± 8 | 21 ± 8 | 0.196 |
| Heteroplasmy | - | 12 ± 13 | 10 ± 6 | 0.332 |
| mtDNA | whole | frame1 | ||
|---|---|---|---|---|
| α | β | α | β | |
| rCRS | 1.6375 | 0.0053 | 1.3224 | 0.1179 |
| PD patients | 1.6405 ± 0.0005 | 0.0011 ± 0.0004 | 1.3106 ± 0.060 | 0.1019 ± 0.061 |
| Controls | 1.6403 ± 0.0007 | 0.0009 ± 0.0013 | 1.3150 ± 0.073 | 0.0910 ± 0.061 |
| p Value | 0.179 | 0.151 | 0.433 | 0.028 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaia, A.; Maponi, P.; Zannotti, M.; Casoli, T. Biocomplexity and Fractality in the Search of Biomarkers of Aging and Pathology: Mitochondrial DNA Profiling of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1758. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051758
Zaia A, Maponi P, Zannotti M, Casoli T. Biocomplexity and Fractality in the Search of Biomarkers of Aging and Pathology: Mitochondrial DNA Profiling of Parkinson’s Disease. International Journal of Molecular Sciences. 2020; 21(5):1758. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051758
Chicago/Turabian StyleZaia, Annamaria, Pierluigi Maponi, Martina Zannotti, and Tiziana Casoli. 2020. "Biocomplexity and Fractality in the Search of Biomarkers of Aging and Pathology: Mitochondrial DNA Profiling of Parkinson’s Disease" International Journal of Molecular Sciences 21, no. 5: 1758. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051758