Benzylaminoethyureido-Tailed Benzenesulfonamides: Design, Synthesis, Kinetic and X-ray Investigations on Human Carbonic Anhydrases

 , , , , ,
, , , , ,
Abstract
:
1. Introduction
2. Results and Discussion
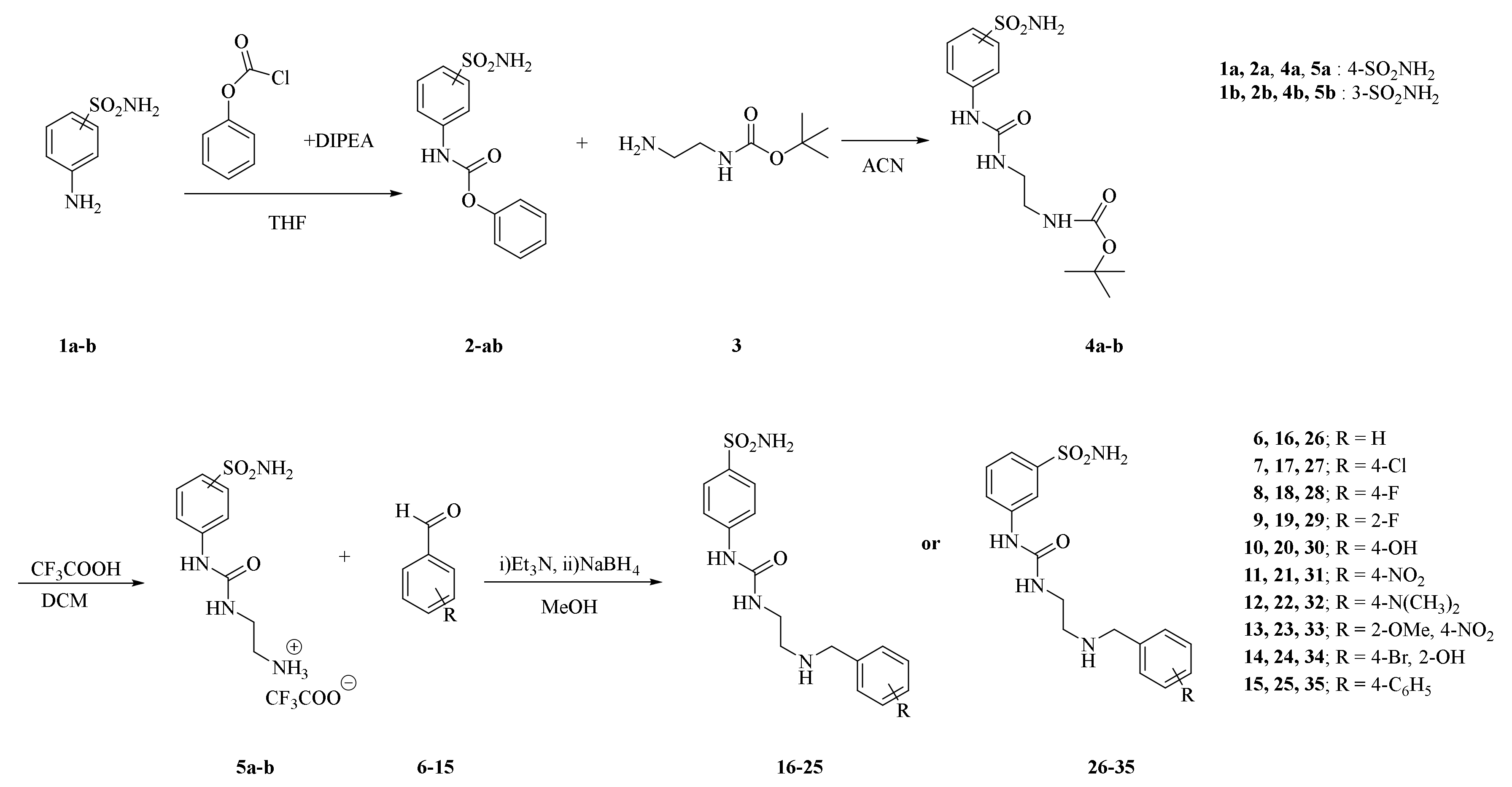
2.1. Chemistry
2.2. Carbonic Anhydrase Inhibition
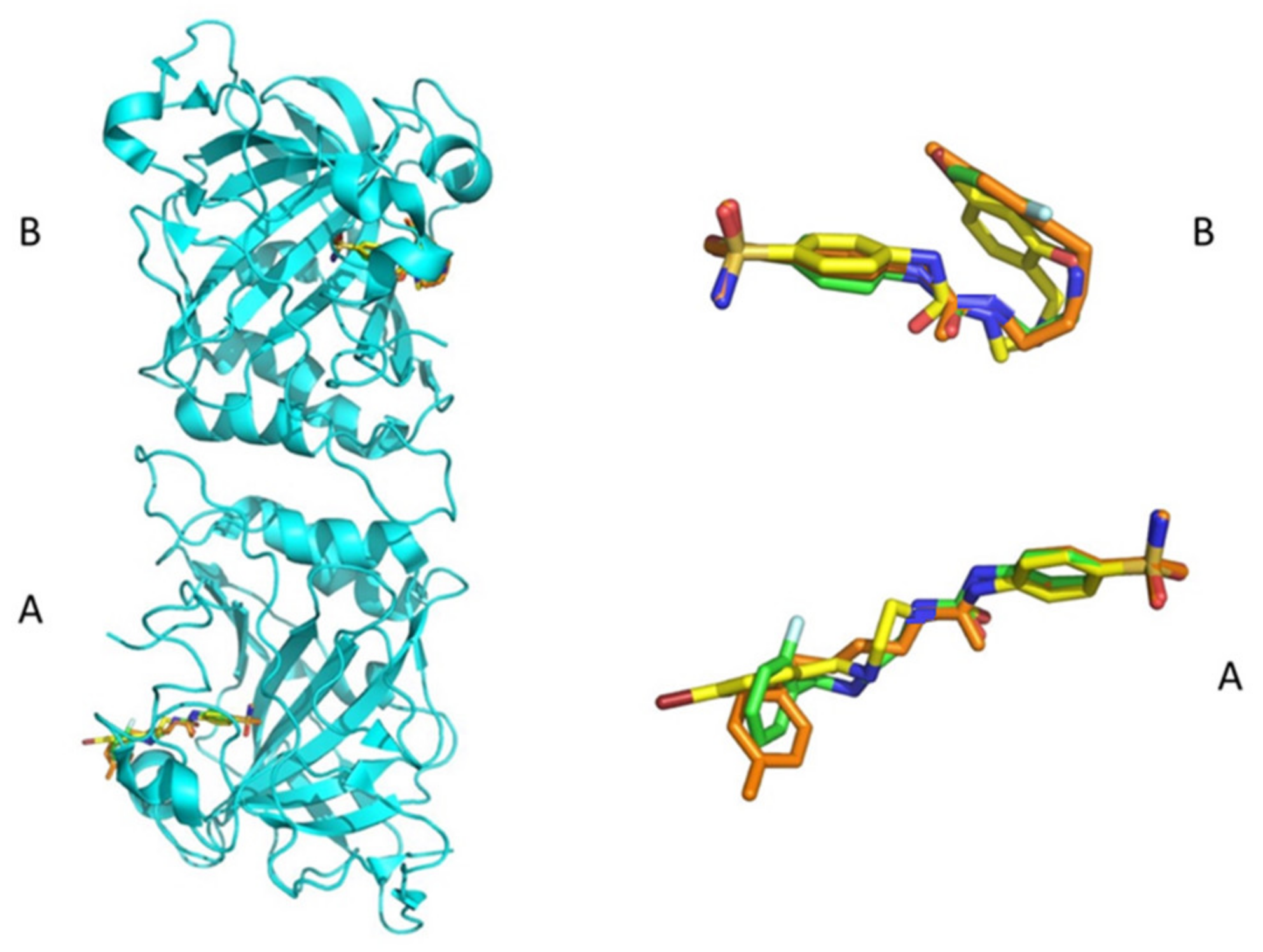
- hCA I was inhibited by all these sulfonamides with a variety of potencies. The most potent inhibitor was the 4-F substituted SA derivative 18 with a KI of 24.6 nM. Followed by 4-Br, the 2-OH substituted SA derivative 24 showed medium potency with a KI of 138.4 nM. The remaining SA derivatives 16–18, 20–23 and 25 were weak inhibitors with KIs spanning between 526.9 and 1506 nM. The MA derivatives of the series 26–35 were poor to ineffective inhibitors of the cytosolic widespread hCA I. 26–32 and 34 were weak inhibitors with KIs spanning between 653.8 and 6250 nM. 33 and 35 showed ineffective inhibition of hCA I whereas clinically used drug AAZ was medium potency inhibitor with a KI of 250 nM (Table 1).
- The series of sulfonamides 16–35 showed nanomolar range inhibitory potencies on the physiologically dominant cytosolic isoform hCA II with KIs spanning between 2.8 and 564.9 nM. The compounds 18–19, 24–25, 27–29 and 34 were effective inhibitors with KIs in the range of 2.8–63 nM. The most effective inhibitor of the series was 4-Br, 2-OH substituted MA derivative 34 with a KI of 2.8 nM, also its SA derivative 24 was the third most effective inhibitor reported here with a KI of 5.3 nM. 2-F derivative of SA 19 was the second most potent inhibitor with a KI of 3.9 nM and its MA derivative 29 inhibited this isoform effectively with a KI of 53.8 (8.6-fold less potent). When the fluorine substitution pattern of 19 shifted from the ortho- to para- position to afford 18 resulted as a slight reduction of the inhibition constant (0.4-fold) to 9.2 nM. MA derivative 28 was a less effective inhibitor compared to its SA counterpart 18 with a KI of 53.8 nM (5.8-fold less potent). The last high potency inhibitor of this isoform was 4-Cl substituted MA derivative 27 with a KI of 36.9 nM whereas its SA warhead possessing derivative 17 showed medium potency with 3.4-fold decrease compared to 27 with a KI of 126.9 nM. SA derivatives 21–23, 25 and MA derivatives 26, 33 were medium potency inhibitors with KIs spanning between 63.2–96.1 nM and 76.3–90.2 nM, respectively. The remaining SA derivatives 16, 20, and MA derivatives 30–32, 35 were weak to ineffective inhibitors of hCA II with KIs of 267–350 nM and 166.4–564.9 nM, respectively.
- The tumor associated transmembrane isoform hCA IX was effectively inhibited only by two SA derivatives with fluoro substitutions, 18 in–para and 19 in–ortho positions with KIs of 30.1 and 20.3 nM, respectively. Their inhibition levels are in comparison with clinically used drug AAZ (KI 25.8 nM). Compounds 17 and 21–24 were medium potency inhibitors with KIs spanning between 115.6 and 458.3 nM. The remaining ones 16, 20, 25, 27, 28 and 30–34 were low micromolar KI values obtained and comprised between 0.97 and 4.21 µM. The remaining of the series 26, 29 and 35 were not inhibitors of the hCA IX.
- The second extracellular tumor associated isoform hCA XII was inhibited by 16-35 more effectively (except 18 and 19) compared to the other tumor associated isoform hCA IX investigated here. The 4-bromo-2-hydroxyphenyl substituted derivative 35 was the most potent inhibitor of this isoform with a KI of 7.2 nM. 35 showed a comparable KI with AAZ (KI of 5.7 nM). Compounds 16-21, 23–25, 28, 31 and 32–33 were potent inhibitors with KIs between 32 and 97.6 nM. Compounds 22, 27 and 30 showed medium potency inhibition of this isoform with KIs spanning between 228.1 and 317.1 nM whereas compounds 26, 29 and 35 showed lower inhibitory effects with inhibition constants between 478.8 and 772.1 nM. The comparison of two subset reported here showed us the derivatives of SA were more potent inhibitors versus MA derivatives. Whereas 4-nitro substituted 31, 4-dimethylamino substituted 32 and 4-bromo-2-hydroxy substituted 34 were more potent compare to their SA derivatives (1.3, 3.4 and 13.6-folds, respectively). 4-nitro-2-methoxy substituted derivatives 23 and 33 showed very similar inhibition activities on this isoform with KIs of 53.6 and 53.1 nM, respectively.
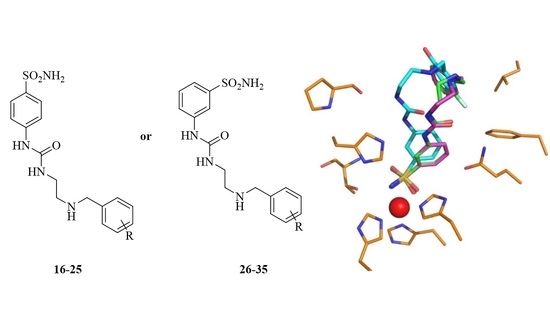
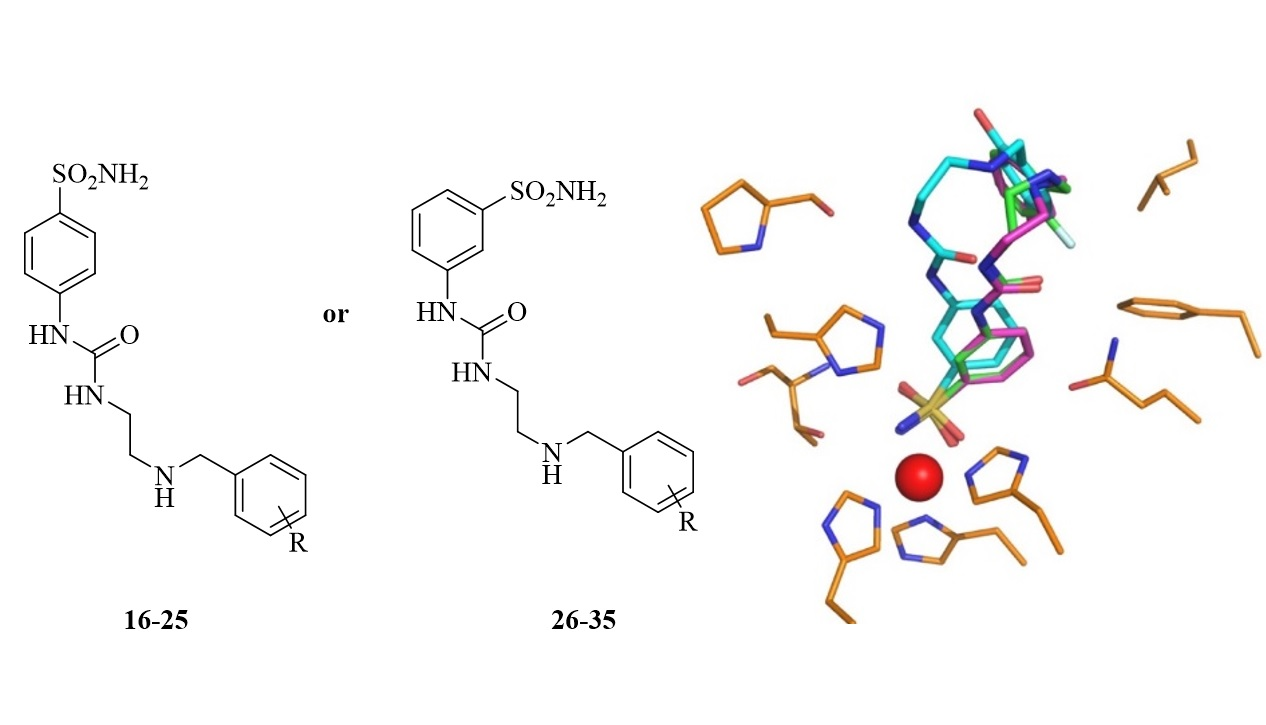
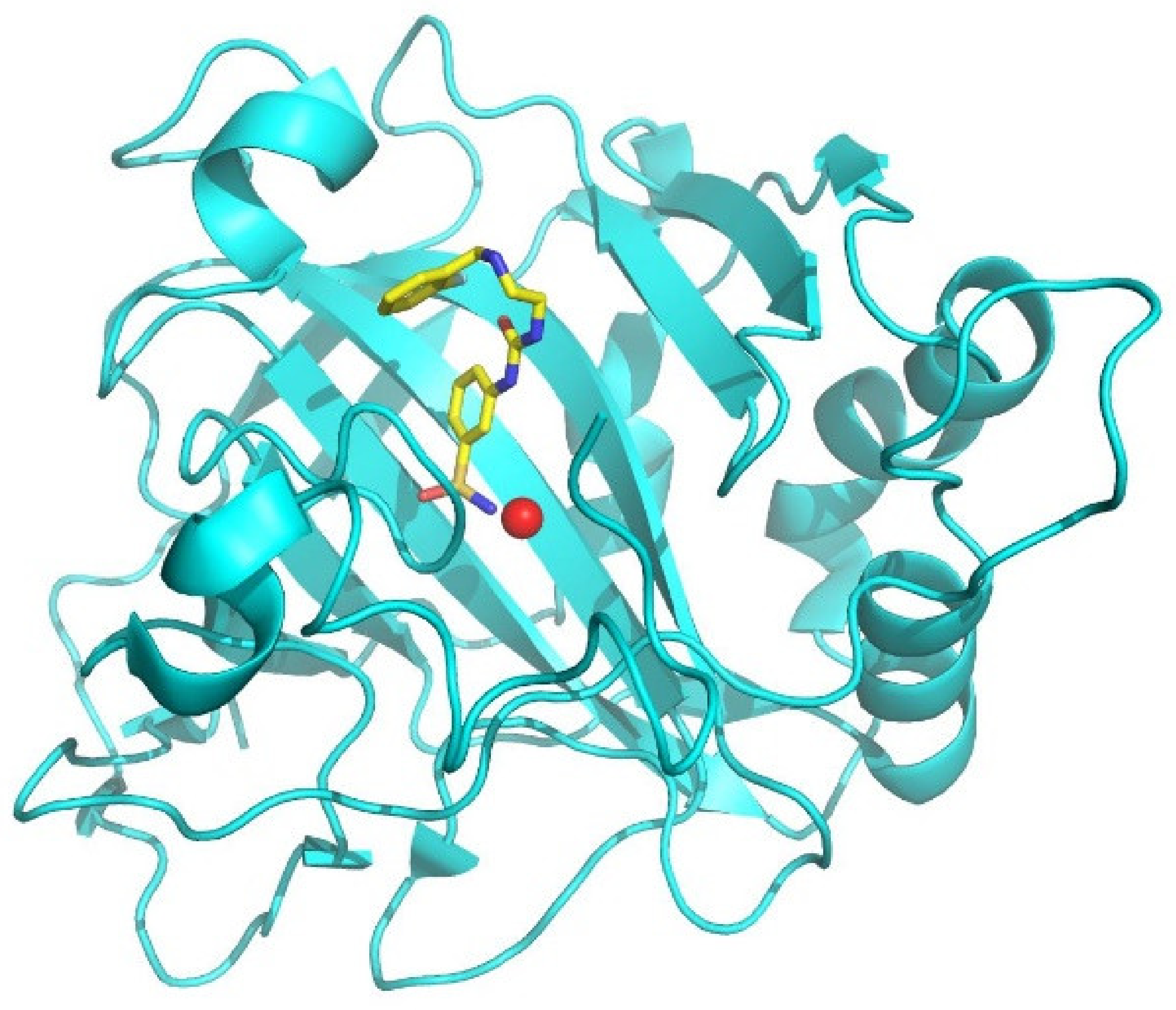
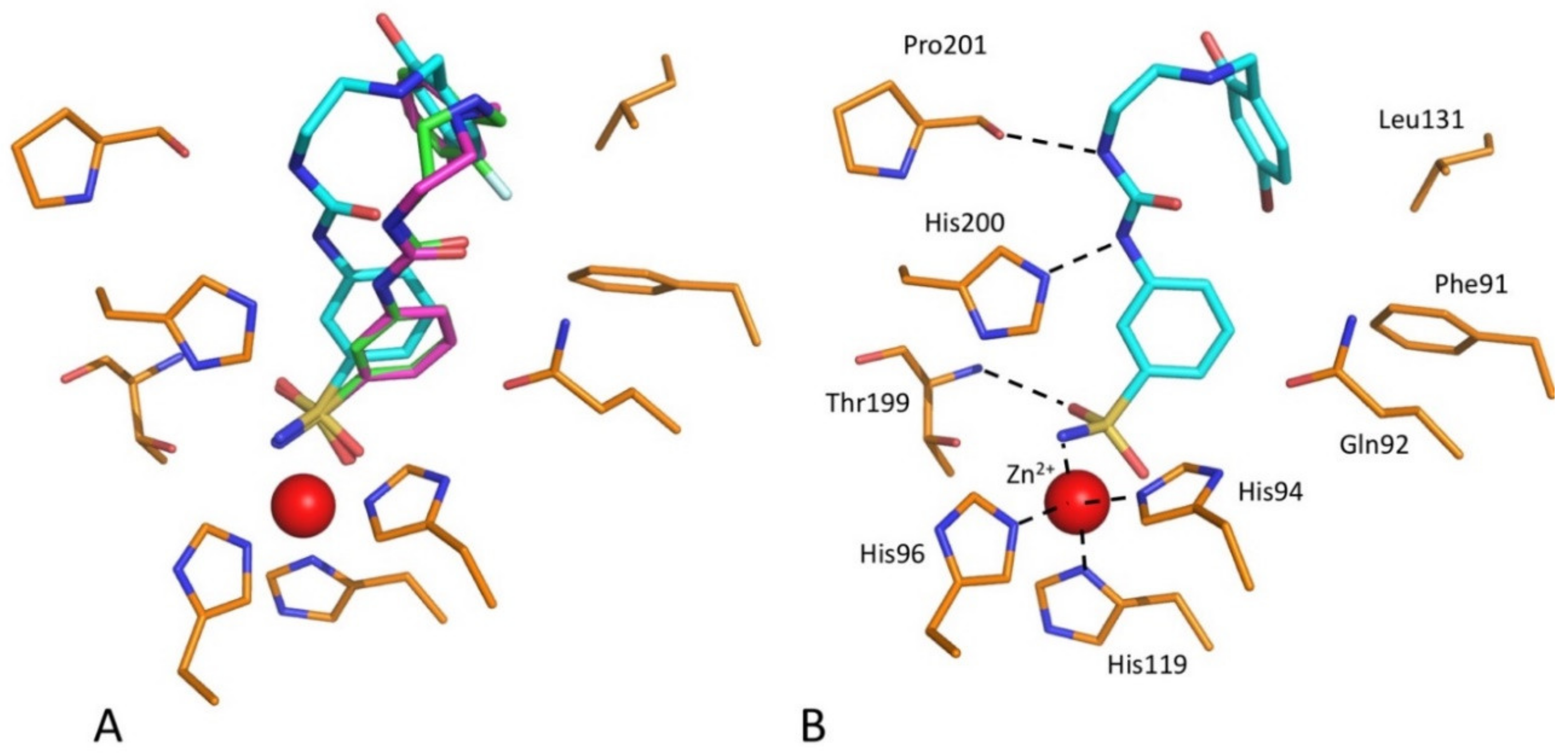
2.3. Structure of hCA I/Inhibitor Complexes
3. Conclusions
4. Experimental Protocols
4.1. Chemistry
4.2. CA Inhibition
4.3. Crystallization, Data Collection and Structure Determination
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nocentini, A.; Supuran, C.T. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin Drug Discov. 2019, 14, 1175–1197. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrases. Bioorg. Med. Chem. 2013, 21, 1377–1378. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scozzafava, A.; Supuran, C.T. Glaucoma and the applications of carbonic anhydrase inhibitors. Subcell. Biochem. 2014, 75, 349–359. [Google Scholar] [PubMed]
- A Study of SLC-0111 and Gemcitabine for Metastatic Pancreatic Ductal Cancer in Subjects Positive for CAIX-Full Text View-ClinicalTrials. Available online: https://clinicaltrials.gov/ct2/show/NCT03450018 (accessed on 31 March 2020).
- Pacchiano, F.; Aggarwal, M.; Avvaru, B.S.; Robbins, A.H.; Scozzafava, A.; McKenna, R.; Supuran, C.T. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem. Commun. (Camb.) 2010, 46, 8371–8373. [Google Scholar] [CrossRef]
- Pacchiano, F.; Carta, F.; McDonald, P.C.; Lou, Y.; Vullo, D.; Scozzafava, A.; Dedhar, S.; Supuran, C.T. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J. Med. Chem. 2011, 54, 1896–1902. [Google Scholar] [CrossRef] [Green Version]
- Lou, Y.; McDonald, P.C.; Oloumi, A.; Chia, S.; Ostlund, C.; Ahmadi, A.; Kyle, A.; Auf dem Keller, U.; Leung, S.; Huntsman, D.; et al. Targeting tumor hypoxia: Suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res. 2011, 71, 3364–3376. [Google Scholar] [CrossRef] [Green Version]
- Supuran, C.T. Carbonic anhydrase inhibition and the management of neuropathic pain. Expert Rev. Neurother 2016, 16, 961–968. [Google Scholar] [CrossRef]
- Carta, F.; Di Cesare Mannelli, L.; Pinard, M.; Ghelardini, C.; Scozzafava, A.; McKenna, R.; Supuran, C.T. A class of sulfonamide carbonic anhydrase inhibitors with neuropathic pain modulating effects. Bioorg. Med. Chem. 2015, 23, 1828–1840. [Google Scholar] [CrossRef] [PubMed]
- Angeli, A.; Di Cesare Mannelli, L.; Lucarini, E.; Peat, T.S.; Ghelardini, C.; Supuran, C.T. Design, synthesis and X-ray crystallography of selenides bearing benzenesulfonamide moiety with neuropathic pain modulating effects. Eur. J. Med. Chem 2018, 154, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Bozdag, M.; Poli, G.; Angeli, A.; Lucarini, E.; Tuccinardi, T.; Di Cesare Mannelli, L.; Selleri, S.; Ghelardini, C.; Winum, J.-Y.; Carta, F.; et al. N-aryl-N’-ureido-O-sulfamates: Potent and selective inhibitors of the human Carbonic Anhydrase VII isoform with neuropathic pain relieving properties. Bioorganic. Chem. 2019, 89, 103033. [Google Scholar] [CrossRef] [PubMed]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple binding modes of inhibitors to carbonic anhydrases: How to design specific drugs targeting 15 different isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbate, F.; Winum, J.-Y.; Potter, B.V.L.; Casini, A.; Montero, J.-L.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors: X-ray crystallographic structure of the adduct of human isozyme II with EMATE, a dual inhibitor of carbonic anhydrases and steroid sulfatase. Bioorg. Med. Chem. Lett. 2004, 14, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. X-Ray Crystallography of Carbonic Anhydrase Inhibitors and Its Importance in Drug Design; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Casini, A.; Antel, J.; Abbate, F.; Scozzafava, A.; David, S.; Waldeck, H.; Schäfer, S.; Supuran, C.T. Carbonic anhydrase inhibitors: SAR and X-ray crystallographic study for the interaction of sugar sulfamates/sulfamides with isozymes I, II and IV. Bioorg. Med. Chem. Lett. 2003, 13, 841–845. [Google Scholar] [CrossRef]
- De Simone, G.; Di Fiore, A.; Menchise, V.; Pedone, C.; Antel, J.; Casini, A.; Scozzafava, A.; Wurl, M.; Supuran, C.T. Carbonic anhydrase inhibitors. Zonisamide is an effective inhibitor of the cytosolic isozyme II and mitochondrial isozyme V: Solution and X-ray crystallographic studies. Bioorg. Med. Chem. Lett. 2005, 15, 2315–2320. [Google Scholar] [CrossRef]
- Temperini, C.; Innocenti, A.; Guerri, A.; Scozzafava, A.; Rusconi, S.; Supuran, C.T. Phosph(on)ate as a zinc-binding group in metalloenzyme inhibitors: X-ray crystal structure of the antiviral drug foscarnet complexed to human carbonic anhydrase I. Bioorg. Med. Chem. Lett. 2007, 17, 2210–2215. [Google Scholar] [CrossRef]
- Petreni, A.; Bonardi, A.; Lomelino, C.; Osman, S.M.; ALOthman, Z.A.; Eldehna, W.M.; El-Haggar, R.; McKenna, R.; Nocentini, A.; Supuran, C.T. Inclusion of a 5-fluorouracil moiety in nitrogenous bases derivatives as human carbonic anhydrase IX and XII inhibitors produced a targeted action against MDA-MB-231 and T47D breast cancer cells. Eur. J. Med. Chem. 2020, 190, 112112. [Google Scholar] [CrossRef]
- Tanini, D.; Capperucci, A.; Scopelliti, M.; Milaneschi, A.; Angeli, A.; Supuran, C.T. Syntesis of thio- and seleno-acetamides bearing benzenesulfonamide as potent inhibitors of human carbonic anhydrase II and XII. Bioorg. Chem. 2019, 89, 102984. [Google Scholar] [CrossRef]
- Supuran, C.T. Structure-based drug discovery of carbonic anhydrase inhibitors. J. Enzyme Inhib. Med. Chem 2012, 27, 759–772. [Google Scholar] [CrossRef]
- Supuran, C.T. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017, 12, 61–88. [Google Scholar] [CrossRef]
- Vats, L.; Kumar, R.; Bua, S.; Nocentini, A.; Gratteri, P.; Supuran, C.T.; Sharma, P.K. Continued exploration and tail approach synthesis of benzenesulfonamides containing triazole and dual triazole moieties as carbonic anhydrase I, II, IV and IX inhibitors. Eur. J. Med. Chem. 2019, 183, 111698. [Google Scholar] [CrossRef]
- Tanpure, R.P.; Ren, B.; Peat, T.S.; Bornaghi, L.F.; Vullo, D.; Supuran, C.T.; Poulsen, S.-A. Carbonic anhydrase inhibitors with dual-tail moieties to match the hydrophobic and hydrophilic halves of the carbonic anhydrase active site. J. Med. Chem. 2015, 58, 1494–1501. [Google Scholar] [CrossRef]
- Ibrahim, H.S.; Allam, H.A.; Mahmoud, W.R.; Bonardi, A.; Nocentini, A.; Gratteri, P.; Ibrahim, E.S.; Abdel-Aziz, H.A.; Supuran, C.T. Dual-tail arylsulfone-based benzenesulfonamides differently match the hydrophobic and hydrophilic halves of human carbonic anhydrases active sites: Selective inhibitors for the tumor-associated hCA IX isoform. Eur. J. Med. Chem. 2018, 152, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Bozdag, M.; Carta, F.; Ceruso, M.; Ferraroni, M.; McDonald, P.C.; Dedhar, S.; Supuran, C.T. Discovery of 4-Hydroxy-3-(3-(phenylureido)benzenesulfonamides as SLC-0111 Analogues for the Treatment of Hypoxic Tumors Overexpressing Carbonic Anhydrase IX. J. Med. Chem. 2018, 61, 6328–6338. [Google Scholar] [CrossRef] [Green Version]
- Bozdag, M.; Ferraroni, M.; Ward, C.; Carta, F.; Bua, S.; Angeli, A.; Langdon, S.P.; Kunkler, I.H.; Al-Tamimi, A.-M.S.; Supuran, C.T. Carbonic anhydrase inhibitors based on sorafenib scaffold: Design, synthesis, crystallographic investigation and effects on primary breast cancer cells. Eur. J. Med. Chem. 2019, 182, 111600. [Google Scholar] [CrossRef]
- Nocentini, A.; Trallori, E.; Singh, S.; Lomelino, C.L.; Bartolucci, G.; Di Cesare Mannelli, L.; Ghelardini, C.; McKenna, R.; Gratteri, P.; Supuran, C.T. 4-Hydroxy-3-nitro-5-ureido-benzenesulfonamides Selectively Target the Tumor-Associated Carbonic Anhydrase Isoforms IX and XII Showing Hypoxia-Enhanced Antiproliferative Profiles. J. Med. Chem. 2018, 61, 10860–10874. [Google Scholar] [CrossRef]
- Ceruso, M.; Antel, S.; Vullo, D.; Scozzafava, A.; Supuran, C.T. Inhibition studies of new ureido-substituted sulfonamides incorporating a GABA moiety against human carbonic anhydrase isoforms I-XIV. Bioorg. Med. Chem. 2014, 22, 6768–6775. [Google Scholar] [CrossRef]
- Congiu, C.; Onnis, V.; Deplano, A.; Balboni, G.; Dedeoglu, N.; Supuran, C.T. Synthesis of sulfonamides incorporating piperazinyl-ureido moieties and their carbonic anhydrase I, II, IX and XII inhibitory activity. Bioorg. Med. Chem. Lett. 2015, 25, 3850–3853. [Google Scholar] [CrossRef]
- Gieling, R.G.; Babur, M.; Mamnani, L.; Burrows, N.; Telfer, B.A.; Carta, F.; Winum, J.-Y.; Scozzafava, A.; Supuran, C.T.; Williams, K.J. Antimetastatic effect of sulfamate carbonic anhydrase IX inhibitors in breast carcinoma xenografts. J. Med. Chem. 2012, 55, 5591–5600. [Google Scholar] [CrossRef]
- Carta, F.; Maresca, A.; Scozzafava, A.; Supuran, C.T. Novel coumarins and 2-thioxo-coumarins as inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg. Med. Chem. 2012, 20, 2266–2273. [Google Scholar] [CrossRef]
- Temperini, C.; Innocenti, A.; Scozzafava, A.; Supuran, C.T. N-hydroxyurea--a versatile zinc binding function in the design of metalloenzyme inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 4316–4320. [Google Scholar] [CrossRef]
- Bozdag, M.; Carta, F.; Angeli, A.; Osman, S.M.; Alasmary, F.A.S.; AlOthman, Z.; Supuran, C.T. Synthesis of N’-phenyl-N-hydroxyureas and investigation of their inhibitory activities on human carbonic anhydrases. Bioorg. Chem. 2018, 78, 1–6. [Google Scholar] [CrossRef]
- Ceruso, M.; Antel, S.; Scozzafava, A.; Supuran, C.T. Synthesis and inhibition potency of novel ureido benzenesulfonamides incorporating GABA as tumor-associated carbonic anhydrase IX and XII inhibitors. J. Enzyme Inhib Med. Chem 2016, 31, 205–211. [Google Scholar] [CrossRef]
- Khalifah, R.G. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar]
- Abdoli, M.; Angeli, A.; Bozdag, M.; Carta, F.; Kakanejadifard, A.; Saeidian, H.; Supuran, C.T. Synthesis and carbonic anhydrase I, II, VII, and IX inhibition studies with a series of benzo[d]thiazole-5- and 6-sulfonamides. J. Enzyme Inhib. Med. Chem 2017, 32, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Bozdag, M.; Ferraroni, M.; Nuti, E.; Vullo, D.; Rossello, A.; Carta, F.; Scozzafava, A.; Supuran, C.T. Combining the tail and the ring approaches for obtaining potent and isoform-selective carbonic anhydrase inhibitors: Solution and X-ray crystallographic studies. Bioorg. Med. Chem. 2014, 22, 334–340. [Google Scholar] [CrossRef]
- Bozdag, M.; Pinard, M.; Carta, F.; Masini, E.; Scozzafava, A.; McKenna, R.; Supuran, C.T. A Class of 4-Sulfamoylphenyl-ω-aminoalkyl Ethers with Effective Carbonic Anhydrase Inhibitory Action and Antiglaucoma Effects. J. Med. Chem. 2014, 57, 9673–9686. [Google Scholar] [CrossRef] [Green Version]
- Güzel, O.; Innocenti, A.; Scozzafava, A.; Salman, A.; Supuran, C.T. Carbonic anhydrase inhibitors. Aromatic/heterocyclic sulfonamides incorporating phenacetyl, pyridylacetyl and thienylacetyl tails act as potent inhibitors of human mitochondrial isoforms VA and VB. Bioorg. Med. Chem. 2009, 17, 4894–4899. [Google Scholar] [CrossRef]
- Akgul, O.; Di Cesare Mannelli, L.; Vullo, D.; Angeli, A.; Ghelardini, C.; Bartolucci, G.; Altamimi, A.S.A.; Scozzafava, A.; Supuran, C.T.; Carta, F. Discovery of Novel Nonsteroidal Anti-Inflammatory Drugs and Carbonic Anhydrase Inhibitors Hybrids (NSAIDs-CAIs) for the Management of Rheumatoid Arthritis. J. Med. Chem. 2018, 61, 4961–4977. [Google Scholar] [CrossRef] [PubMed]
- Küçükbay, H.; Buğday, N.; Küçükbay, F.Z.; Berrino, E.; Bartolucci, G.; Del Prete, S.; Capasso, C.; Supuran, C.T. Synthesis and carbonic anhydrase inhibitory properties of novel 4-(2-aminoethyl)benzenesulfonamide-dipeptide conjugates. Bioorg. Chem. 2019, 83, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Berrino, E.; Milazzo, L.; Micheli, L.; Vullo, D.; Angeli, A.; Bozdag, M.; Nocentini, A.; Menicatti, M.; Bartolucci, G.; Di Cesare Mannelli, L.; et al. Synthesis and Evaluation of Carbonic Anhydrase Inhibitors with Carbon Monoxide Releasing Properties for the Management of Rheumatoid Arthritis. J. Med. Chem. 2019, 62, 7233–7249. [Google Scholar] [CrossRef] [PubMed]
- Bua, S.; Berrino, E.; Del Prete, S.; Murthy, V.S.; Vijayakumar, V.; Tamboli, Y.; Capasso, C.; Cerbai, E.; Mugelli, A.; Carta, F.; et al. Synthesis of novel benzenesulfamide derivatives with inhibitory activity against human cytosolic carbonic anhydrase I and II and Vibrio cholerae α- and β-class enzymes. J. Enzyme Inhib. Med. Chem. 2018, 33, 1125–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salerno, S.; Barresi, E.; Amendola, G.; Berrino, E.; Milite, C.; Marini, A.M.; Da Settimo, F.; Novellino, E.; Supuran, C.T.; Cosconati, S.; et al. 4-Substituted Benzenesulfonamides Incorporating Bi/Tricyclic Moieties Act as Potent and Isoform-Selective Carbonic Anhydrase II/IX Inhibitors. J. Med. Chem. 2018, 61, 5765–5770. [Google Scholar] [CrossRef]
- Bozdag, M.; Alafeefy, A.M.; Carta, F.; Ceruso, M.; Al-Tamimi, A.-M.S.; Al-Kahtani, A.A.; Alasmary, F.A.S.; Supuran, C.T. Synthesis 4-[2-(2-mercapto-4-oxo-4H-quinazolin-3-yl)-ethyl]-benzenesulfonamides with subnanomolar carbonic anhydrase II and XII inhibitory properties. Bioorg. Med. Chem. 2016, 24, 4100–4107. [Google Scholar] [CrossRef]
- Bozdag, M.; Alafeefy, A.M.; Altamimi, A.M.; Carta, F.; Supuran, C.T.; Vullo, D. Synthesis of new 3-(2-mercapto-4-oxo-4H-quinazolin-3-yl)-benzenesulfonamides with strong inhibition properties against the tumor associated carbonic anhydrases IX and XII. Bioorg. Med. Chem. 2017, 25, 2782–2788. [Google Scholar] [CrossRef]
- Bozdag, M.; Alafeefy, A.M.; Vullo, D.; Carta, F.; Dedeoglu, N.; Al-Tamimi, A.-M.S.; Al-Jaber, N.A.; Scozzafava, A.; Supuran, C.T. Benzenesulfonamides incorporating bulky aromatic/heterocyclic tails with potent carbonic anhydrase inhibitory activity. Bioorg. Med. Chem. 2015, 23, 7751–7764. [Google Scholar] [CrossRef]
- Bertol, E.; Mari, F.; Orzalesi, G.; Volpato, I. Combustion products from various kinds of fibers: Toxicological hazards from smoke exposure. Forensic Sci. Int. 1983, 22, 111–116. [Google Scholar] [CrossRef]
- Bertol, E.; Argo, A.; Procaccianti, P.; Vaiano, F.; Di Milia, M.G.; Furlanetto, S.; Mari, F. Detection of gamma-hydroxybutyrate in hair: Validation of GC-MS and LC-MS/MS methods and application to a real case. J. Pharm Biomed. Anal. 2012, 70, 518–522. [Google Scholar] [CrossRef] [Green Version]
- Supuran, C.T.; Vullo, D.; Manole, G.; Casini, A.; Scozzafava, A. Designing of novel carbonic anhydrase inhibitors and activators. Curr Med. Chem. Cardiovasc. Hematol. Agents 2004, 2, 49–68. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Angeli, A.; Ferraroni, M.; Supuran, C.T. Famotidine, an Antiulcer Agent, Strongly Inhibits Helicobacter pylori and Human Carbonic Anhydrases. ACS Med. Chem. Lett. 2018, 9, 1035–1038. [Google Scholar] [CrossRef]
- Adams, P.D.; Baker, D.; Brunger, A.T.; Das, R.; DiMaio, F.; Read, R.J.; Richardson, D.C.; Richardson, J.S.; Terwilliger, T.C. Advances, interactions, and future developments in the CNS, Phenix, and Rosetta structural biology software systems. Ann. Rev. Biophys 2013, 42, 265–287. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KI (nM) * | |||||
|---|---|---|---|---|---|
| Compound | R | hCA I | hCA II | hCA IX | hCA XII |
| 16 | 552.4 | 350.1 | 1350 | 54.5 | |
| 17 | 4-Cl | 582.1 | 126.9 | 458.3 | 32 |
| 18 | 4-F | 724.7 | 9.2 | 30.1 | 53.2 |
| 19 | 2-F | 24.6 | 3.9 | 20.3 | 58.3 |
| 20 | 4-OH | 781 | 267 | 974.2 | 75.8 |
| 21 | 4-NO2 | 526.9 | 96.1 | 317.1 | 77.5 |
| 22 | 4-Me2N | 1506 | 73.2 | 342.8 | 264.1 |
| 23 | 4-NO2, 2-MeO | 788.4 | 82.4 | 290.6 | 53.6 |
| 24 | 4-Br, 2-OH | 138.4 | 5.3 | 115.6 | 97.6 |
| 25 | 4-C6H5 | 781.7 | 63.2 | 1150 | 63.7 |
| 26 | - | 928.3 | 76.3 | >10,000 | 772.1 |
| 27 | 4-Cl | 702.4 | 36.9 | 3378 | 228.1 |
| 28 | 4-F | 867.5 | 53.8 | 4212 | 71.3 |
| 29 | 2-F | 728 | 33.4 | >10,000 | 478.8 |
| 30 | 4-OH | 6250 | 166.4 | 3511 | 317.1 |
| 31 | 4-NO2 | 5851 | 168.4 | 2325 | 60.7 |
| 32 | 4-Me2N | 8906 | 564.9 | 1199 | 78 |
| 33 | 4-NO2, 2-MeO | >10,000 | 90.2 | 2606 | 53.1 |
| 34 | 4-Br, 2-OH | 653.8 | 2.8 | 1915 | 7.2 |
| 35 | 4-C6H5 | >10,000 | 456.3 | >10,000 | 688 |
| AAZ | 250 | 12.1 | 25.8 | 5.7 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, M.; Bozdag, M.; Farooq, U.; Angeli, A.; Carta, F.; Berto, P.; Zanotti, G.; Supuran, C.T. Benzylaminoethyureido-Tailed Benzenesulfonamides: Design, Synthesis, Kinetic and X-ray Investigations on Human Carbonic Anhydrases. Int. J. Mol. Sci. 2020, 21, 2560. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072560
Ali M, Bozdag M, Farooq U, Angeli A, Carta F, Berto P, Zanotti G, Supuran CT. Benzylaminoethyureido-Tailed Benzenesulfonamides: Design, Synthesis, Kinetic and X-ray Investigations on Human Carbonic Anhydrases. International Journal of Molecular Sciences. 2020; 21(7):2560. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072560
Chicago/Turabian StyleAli, Majid, Murat Bozdag, Umar Farooq, Andrea Angeli, Fabrizio Carta, Paola Berto, Giuseppe Zanotti, and Claudiu T. Supuran. 2020. "Benzylaminoethyureido-Tailed Benzenesulfonamides: Design, Synthesis, Kinetic and X-ray Investigations on Human Carbonic Anhydrases" International Journal of Molecular Sciences 21, no. 7: 2560. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072560