Interleukin-17 Reduces βENaC via MAPK Signaling in Vascular Smooth Muscle Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Cell Culture
2.2. Cell Treatment
2.3. Protein Isolation
2.4. Western Blot on A10 Protein Isolates
2.5. Live/Dead Viability/Cytotoxicity Assay
2.6. Statistical Analysis.
3. Results
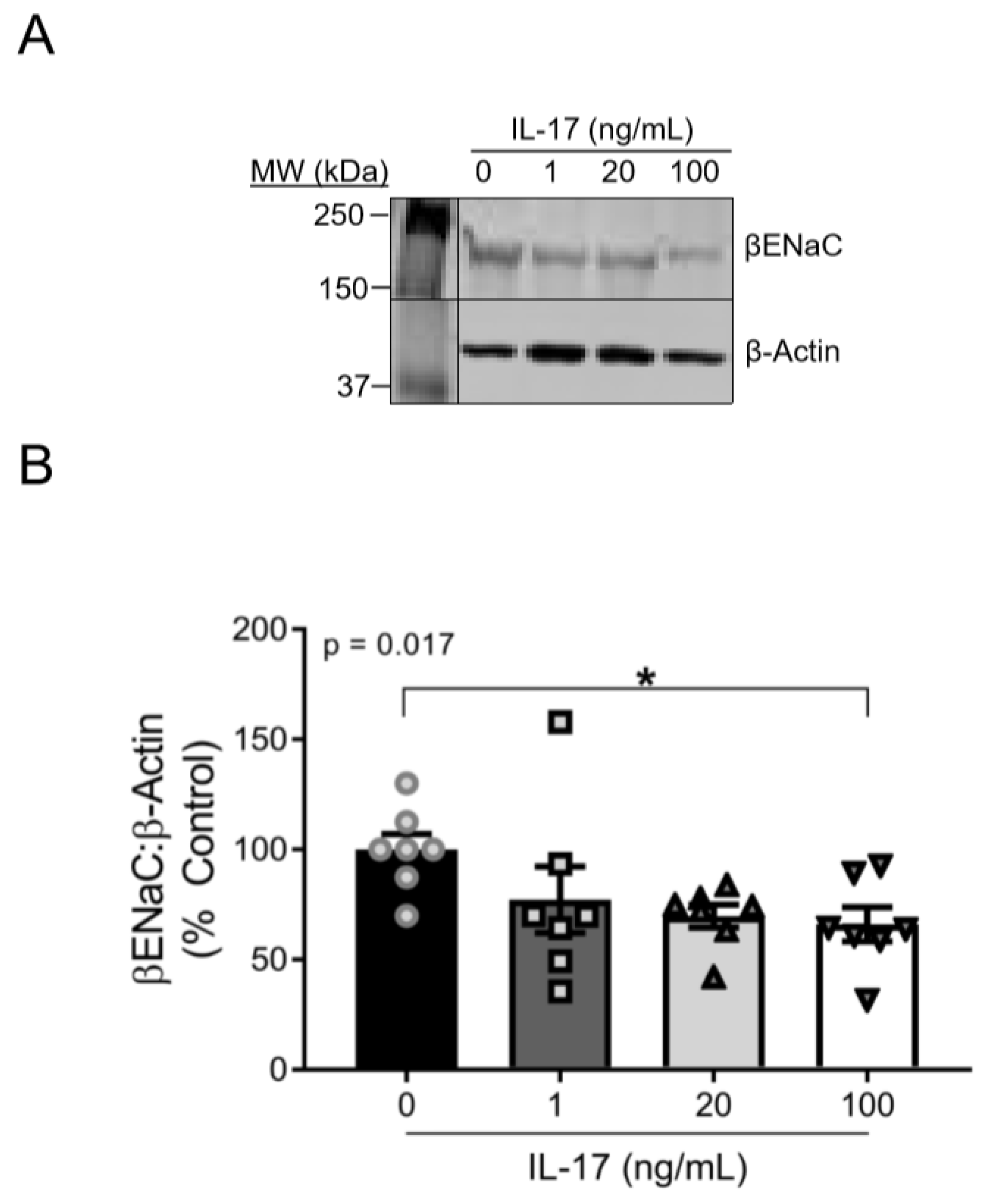
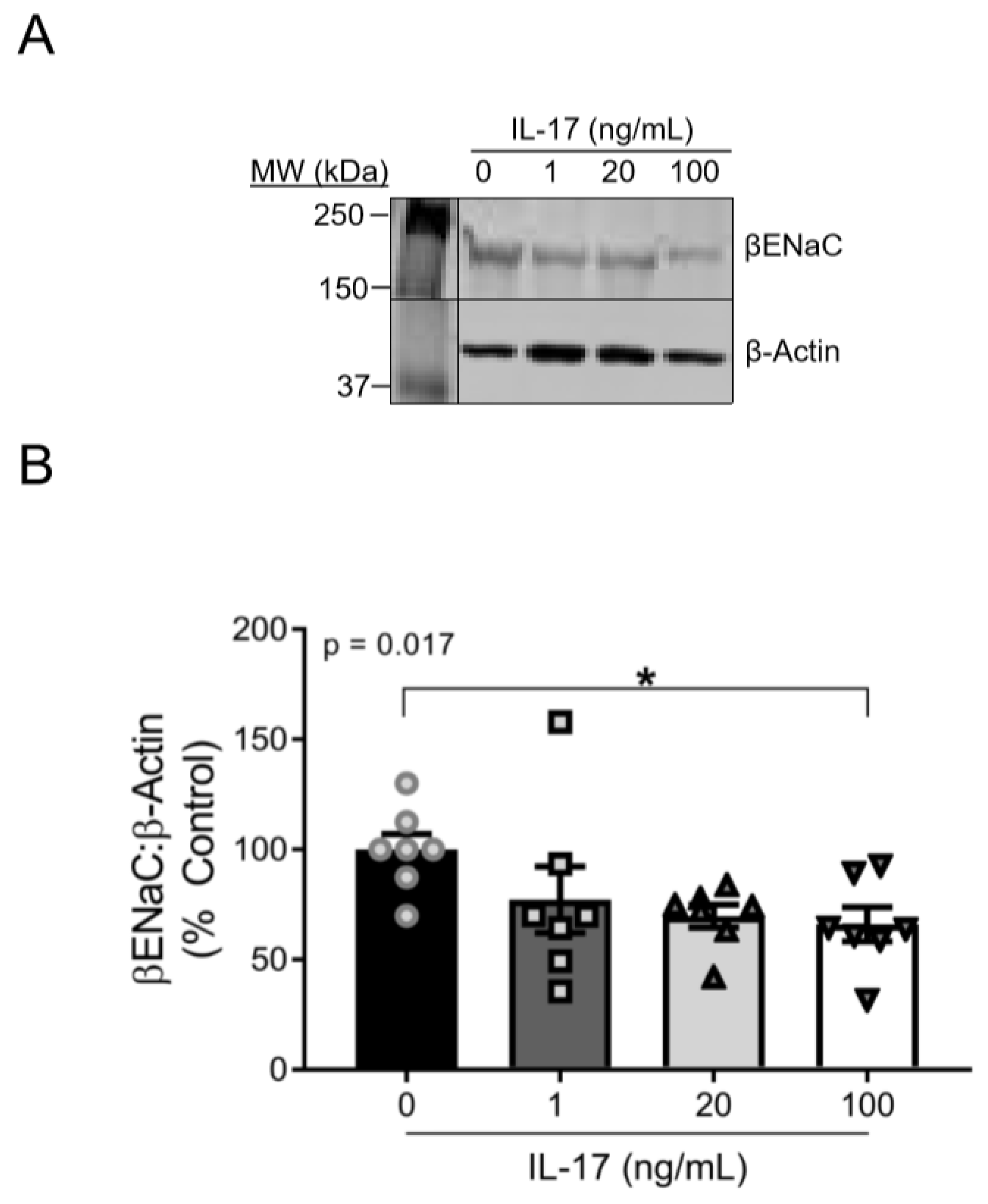
3.1. IL-17 Reduces βENaC Expression in a Concentration-Dependent Fashion in Cultured VSMCs
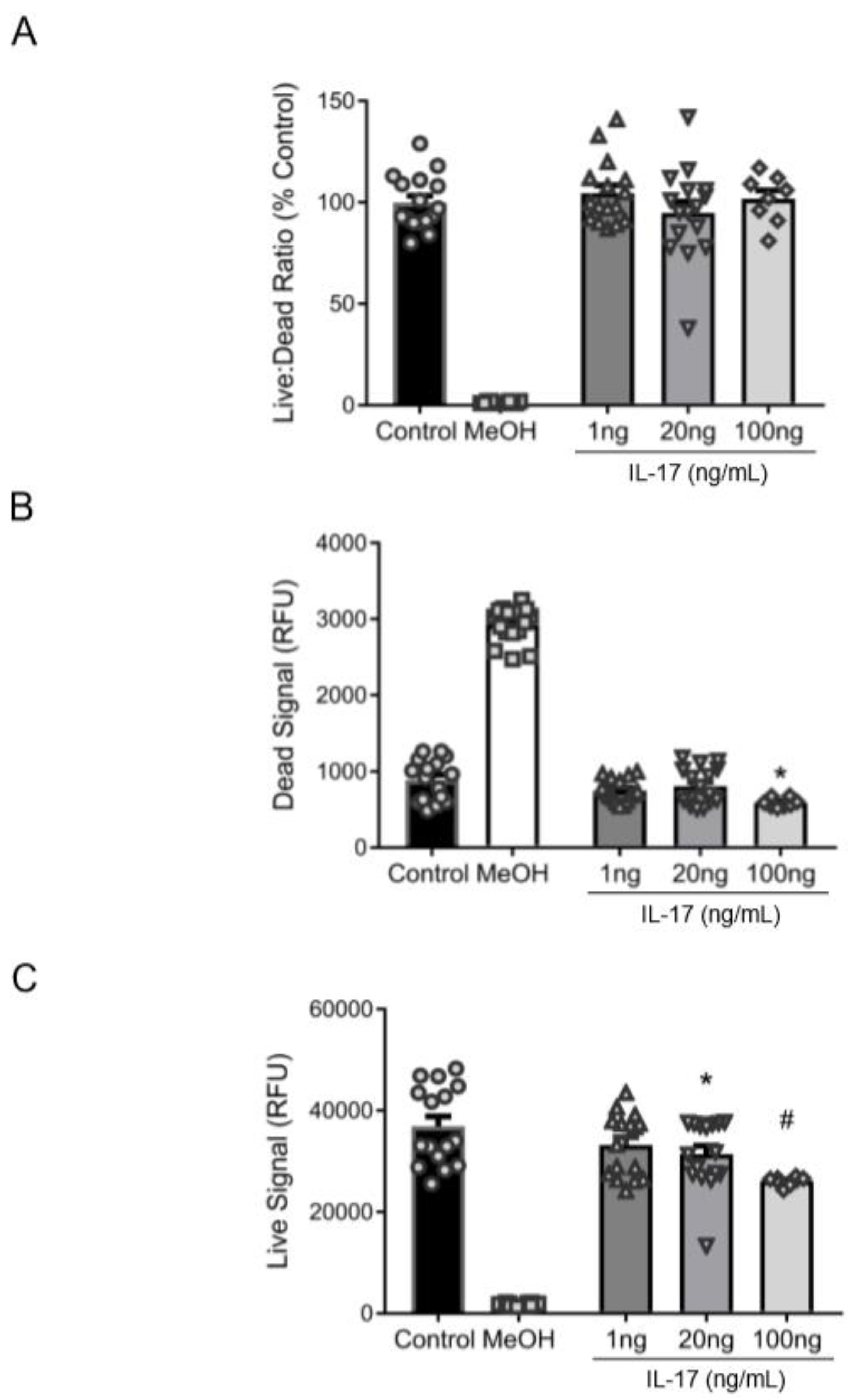
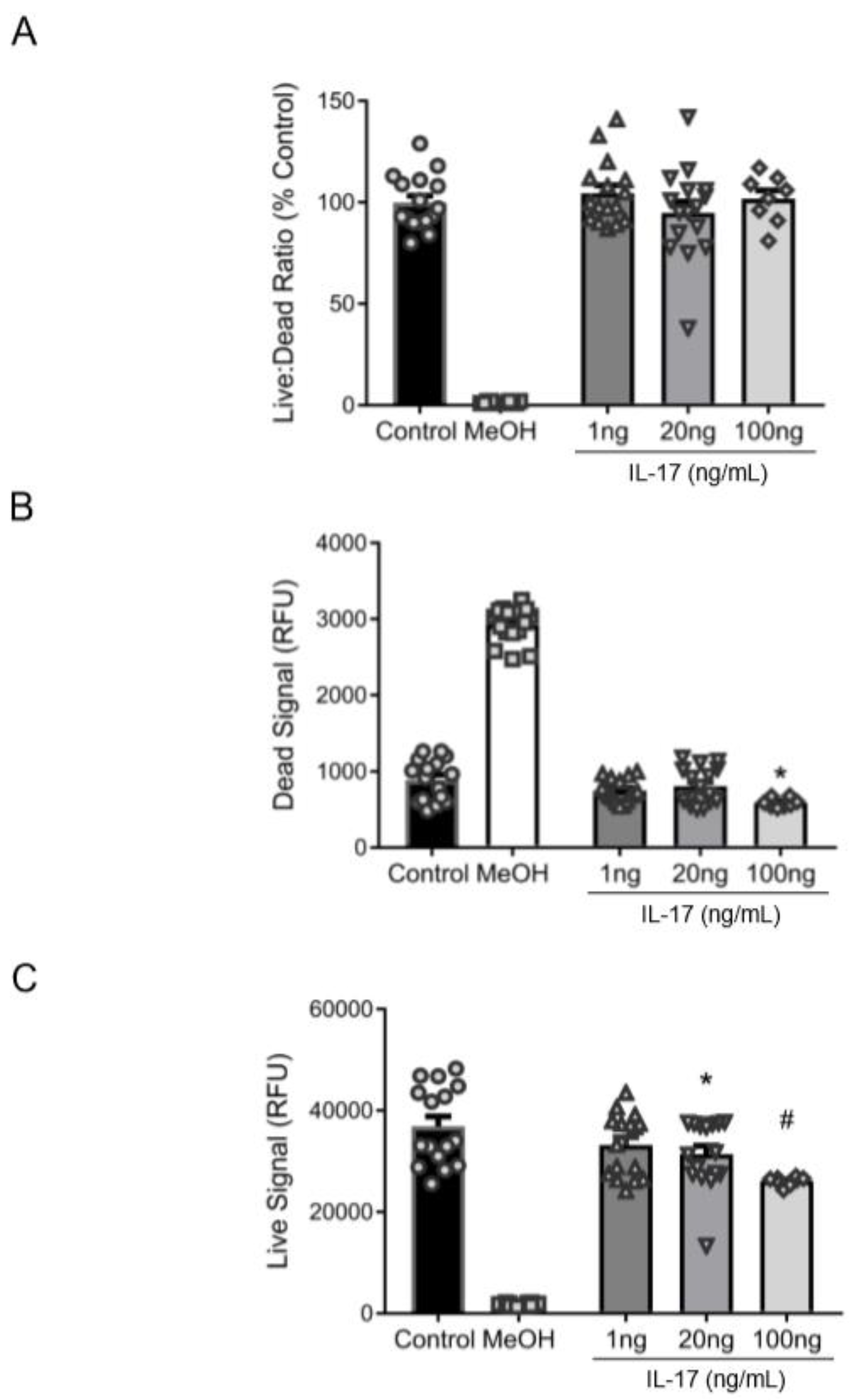
3.2. Reduction in βENaC by IL-17 Is Not Associated with Cell Death
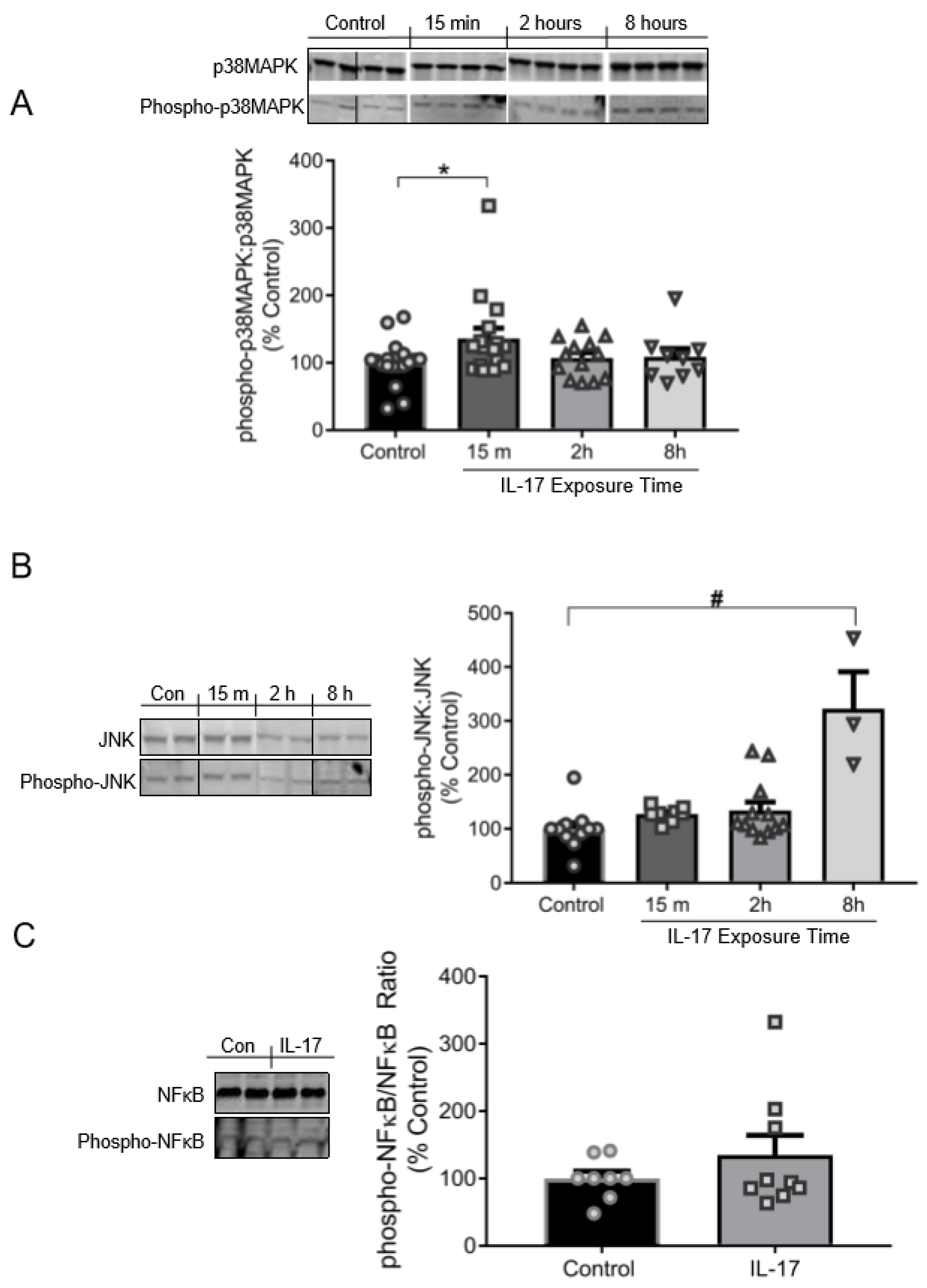
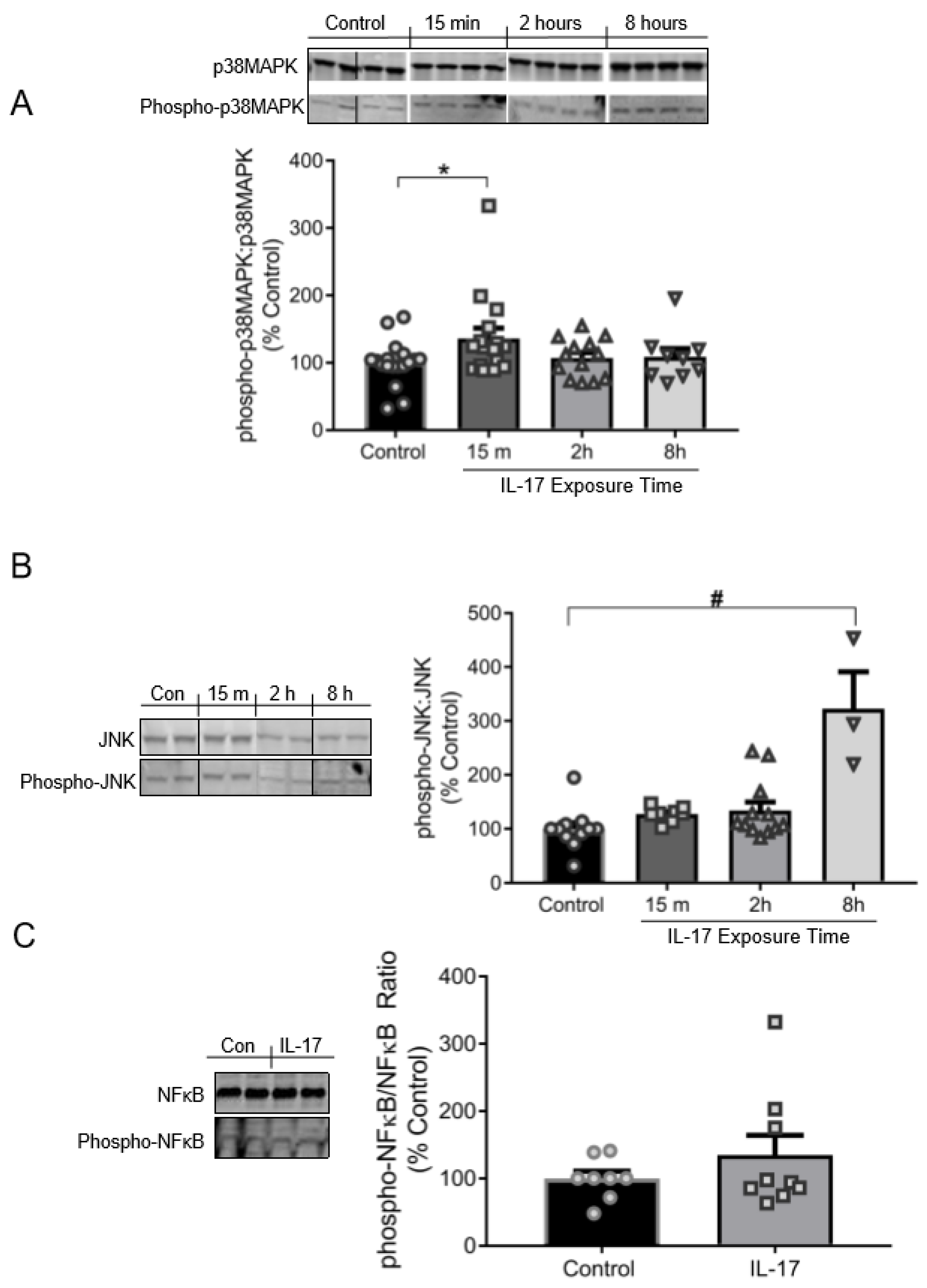
3.3. IL-17 Induces the Phosphorylation of p38MAPK and JNK, but Not NFκB
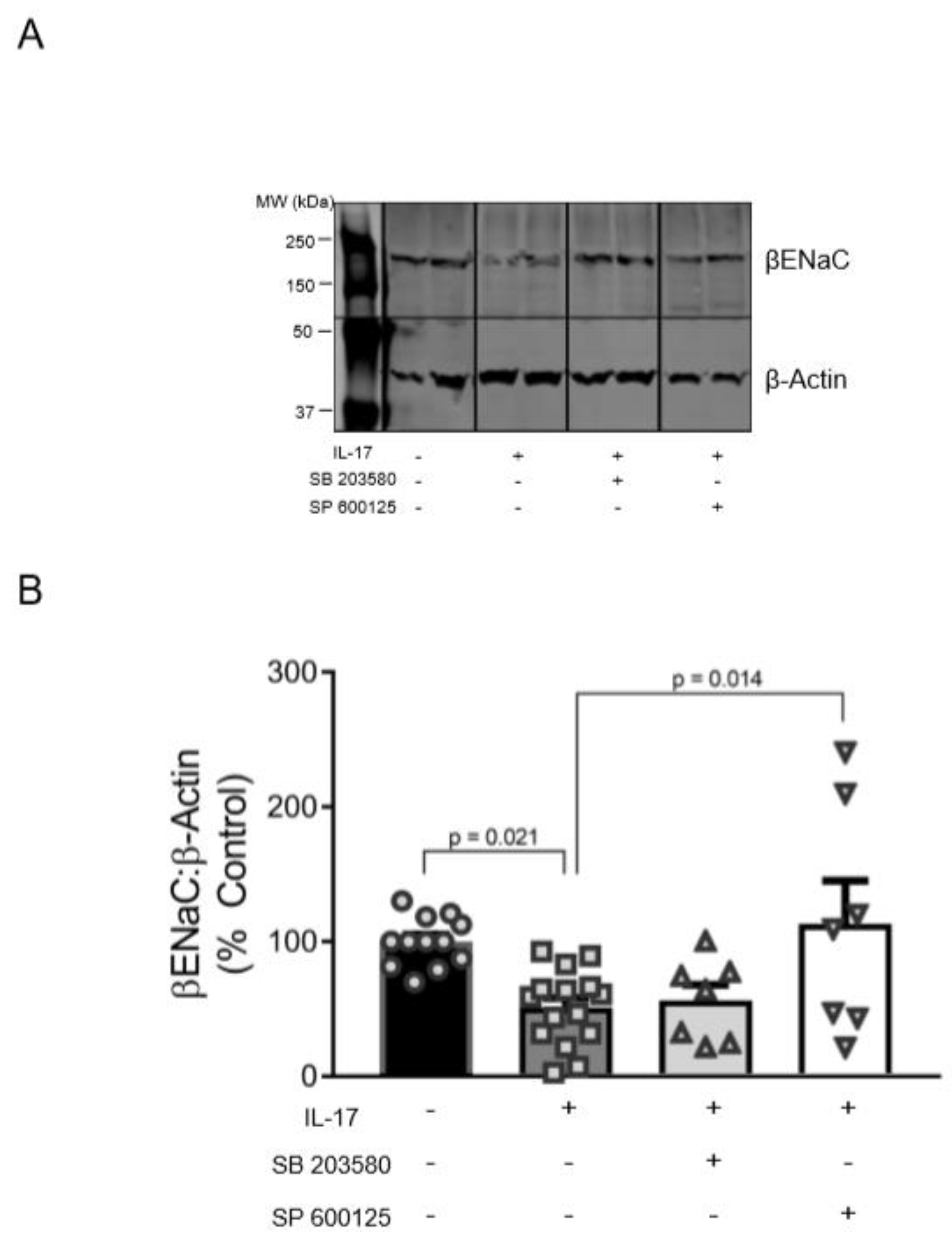
3.4. Role of p38MAPK and JNK in the Reduction of βENaC by IL-17
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Drummond, H.A.; Grifoni, S.C.; Jernigan, N.L. A new trick for an old dogma: Enac proteins as mechanotransducers in vascular smooth muscle. Physiology (Bethesda) 2008, 23, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimes, B.W.; Brandt, B.L. Characterization of two putative smooth muscle cell lines from rat thoracic aorta. Exp. Cell Res. 1976, 98, 349–366. [Google Scholar] [CrossRef]
- Ben-Shahar, Y. Sensory functions for degenerin/epithelial sodium channels (deg/enac). Adv. Genet. 2011, 76, 1–26. [Google Scholar] [PubMed] [Green Version]
- Drummond, H.A.; Gebremedhin, D.; Harder, D.R. Degenerin/epithelial Na+ channel proteins: Components of a vascular mechanosensor. Hypertension 2004, 44, 643–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jernigan, N.L.; Drummond, H.A. Vascular enac proteins are required for renal myogenic constriction. Am. J. Physiol. Renal Physiol. 2005, 289, F891–F901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, H.A. Betaenac is a molecular component of a vsmc mechanotransducer that contributes to renal blood flow regulation, protection from renal injury, and hypertension. Front. Physiol. 2012, 3, 341. [Google Scholar] [CrossRef] [Green Version]
- Drummond, H.A.; Stec, D.E. Betaenac acts as a mechanosensor in renal vascular smooth muscle cells that contributes to renal myogenic blood flow regulation, protection from renal injury and hypertension. J. Nephrol. Res. 2015, 1, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.; Gannon, K.; Gousset, M.; Liu, R.; Murphey, B.; Drummond, H.A. Impaired myogenic constriction of the renal afferent arteriole in a mouse model of reduced betaenac expression. Am. J. Physiol. Renal Physiol. 2012, 302, F1486–F1493. [Google Scholar] [CrossRef]
- Grifoni, S.C.; Chiposi, R.; McKey, S.E.; Ryan, M.J.; Drummond, H.A. Altered whole kidney blood flow autoregulation in a mouse model of reduced beta-enac. Am. J. Physiol. Renal Physiol. 2010, 298, F285–F292. [Google Scholar] [CrossRef] [Green Version]
- Guan, Z.; Pollock, J.S.; Cook, A.K.; Hobbs, J.L.; Inscho, E.W. Effect of epithelial sodium channel blockade on the myogenic response of rat juxtamedullary afferent arterioles. Hypertension 2009, 54, 1062–1069. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.C.; Ahn, D.S.; Yeon, S.I.; Lim, M.; Lee, Y.H. Epithelial Na+ channel proteins are mechanotransducers of myogenic constriction in rat posterior cerebral arteries. Exp. Physiol. 2012, 97, 544–555. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.C.; Choi, S.K.; Lim, M.; Yeon, S.I.; Lee, Y.H. Role of endogenous Enac and TRP channels in the myogenic response of rat posterior cerebral arteries. PLoS ONE 2013, 8, e84194. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, T.; Imig, J.D. Afferent arteriolar responses to beta, gamma-methylene atp and 20-hete are not blocked by enac inhibition. Physiol. Rep. 2013, 1, e00082. [Google Scholar] [CrossRef]
- Syntichaki, P.; Tavernarakis, N. Genetic models of mechanotransduction: The nematode Caenorhabditis elegans. Physiol. Rev. 2004, 84, 1097–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanLandingham, L.G.; Gannon, K.P.; Drummond, H.A. Pressure-induced constriction is inhibited in a mouse model of reduced betaenac. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R723–R728. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Takeya, K.; Aaronson, P.I.; Loutzenhiser, K.; Loutzenhiser, R. Effects of amiloride, benzamil, and alterations in extracellular Na+ on the rat afferent arteriole and its myogenic response. Am. J. Physiol. Renal Physiol. 2008, 295, F272–F282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grifoni, S.C.; Gannon, K.P.; Stec, D.E.; Drummond, H.A. Enac proteins contribute to VSMC migration. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H3076–H3086. [Google Scholar] [CrossRef] [Green Version]
- Benos, D.J.; Stanton, B.A. Functional domains within the degenerin/epithelial sodium channel (deg/enac) superfamily of ion channels. J. Physiol 1999, 520 Pt 3, 631–644. [Google Scholar] [CrossRef]
- Canessa, C.M.; Schild, L.; Buell, G.; Thorens, B.; Gautschi, I.; Horisberger, J.D.; Rossier, B.C. Amiloride-sensitive epithelial na+ channel is made of three homologous subunits. Nature 1994, 367, 463–467. [Google Scholar] [CrossRef]
- Kellenberger, S.; Schild, L. Epithelial sodium channel/degenerin family of ion channels: A variety of functions for a shared structure. Physiol. Rev. 2002, 82, 735–767. [Google Scholar] [CrossRef] [Green Version]
- Bonny, O.; Chraibi, A.; Loffing, J.; Jaeger, N.F.; Grunder, S.; Horisberger, J.D.; Rossier, B.C. Functional expression of a pseudohypoaldosteronism type i mutated epithelial Na+ channel lacking the pore-forming region of its alpha subunit. J. Clin. Invest. 1999, 104, 967–974. [Google Scholar] [CrossRef] [Green Version]
- Wesch, D.; Althaus, M.; Miranda, P.; Cruz-Muros, I.; Fronius, M.; Gonzalez-Hernandez, T.; Clauss, W.G.; Alvarez de la Rosa, D.; Giraldez, T. Differential n termini in epithelial Na+ channel delta-subunit isoforms modulate channel trafficking to the membrane. Am. J. Physiol. Cell Physiol. 2012, 302, C868–C879. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Choi, Y.S.; Kim, S.J.; Son, E.J.; Choi, H.S.; Yoon, J.H. Interleukin-1beta suppresses epithelial sodium channel beta-subunit expression and enac-dependent fluid absorption in human middle ear epithelial cells. Eur. J. Pharmacol. 2007, 567, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Dames, P.; Bergann, T.; Fromm, A.; Bucker, R.; Barmeyer, C.; Krug, S.M.; Fromm, M.; Schulzke, J.D. Interleukin-13 affects the epithelial sodium channel in the intestine by coordinated modulation of stat6 and p38 mapk activity. J. Physiol. 2015, 593, 5269–5282. [Google Scholar] [CrossRef] [Green Version]
- Wynne, B.M.; Zou, L.; Linck, V.; Hoover, R.S.; Ma, H.P.; Eaton, D.C. Regulation of lung epithelial sodium channels by cytokines and chemokines. Front. Immunol. 2017, 8, 766. [Google Scholar] [CrossRef] [Green Version]
- Dagenais, A.; Frechette, R.; Yamagata, Y.; Yamagata, T.; Carmel, J.F.; Clermont, M.E.; Brochiero, E.; Masse, C.; Berthiaume, Y. Downregulation of enac activity and expression by TNF-alpha in alveolar epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 286, L301–L311. [Google Scholar] [CrossRef] [PubMed]
- Galietta, L.J.; Pagesy, P.; Folli, C.; Caci, E.; Romio, L.; Costes, B.; Nicolis, E.; Cabrini, G.; Goossens, M.; Ravazzolo, R.; et al. IL-4 is a potent modulator of ion transport in the human bronchial epithelium in vitro. J. Immunol. 2002, 168, 839–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, J.; Kawakatsu, H.; Gartland, B.; Pespeni, M.; Sheppard, D.; Matthay, M.A.; Canessa, C.M.; Pittet, J.F. Interleukin-1beta decreases expression of the epithelial sodium channel alpha-subunit in alveolar epithelial cells via a p38 mapk-dependent signaling pathway. J. Biol. Chem. 2005, 280, 18579–18589. [Google Scholar] [CrossRef] [Green Version]
- Amatya, N.; Garg, A.V.; Gaffen, S.L. IL-17 signaling: The yin and the yang. Trends Immunol. 2017, 38, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Gaffen, S.L. An overview of IL-17 function and signaling. Cytokine 2008, 43, 402–407. [Google Scholar] [CrossRef] [Green Version]
- Senaud, J.; Vendrely, R.; Tronche, P. On the nature of the toxic substance of the sarcosporidic cysts of the sheep (Toxoplasmea), active on rabbits. C. R. Acad. Hebd. Seances Acad. Sci. D 1968, 266, 1137–1138. [Google Scholar] [PubMed]
- Matsuda, N.; Hattori, Y. Vascular biology in sepsis: Pathophysiological and therapeutic significance of vascular dysfunction. J. Smooth Muscle Res. 2007, 43, 117–137. [Google Scholar] [CrossRef] [Green Version]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.V.; Shelling, M.L.; Prodanovich, S.; Federman, D.G.; Kirsner, R.S. Psoriasis and vascular disease-risk factors and outcomes: A systematic review of the literature. J. Gen. Intern. Med. 2011, 26, 1036–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, S.P.; Handa, R.; Gulati, G.S.; Sharma, S.; Pandey, R.M.; Aggarwal, P.; Ramakrishnan, L.; Shankar, S. Peripheral vascular disease in systemic lupus erythematosus. Lupus 2007, 16, 720–723. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, C.; Rudemiller, N.P.; Abais, J.M.; Mattson, D.L. Inflammation and hypertension: New understandings and potential therapeutic targets. Curr. Hypertens. Rep. 2015, 17, 507. [Google Scholar] [CrossRef] [Green Version]
- Raines, E.W.; Ferri, N. Thematic review series: The immune system and atherogenesis. Cytokines affecting endothelial and smooth muscle cells in vascular disease. J. Lipid Res. 2005, 46, 1081–1092. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, R.; Fusco, R.; Gugliandolo, E.; Cordaro, M.; Siracusa, R.; Impellizzeri, D.; Peritore, A.F.; Crupi, R.; Cuzzocrea, S.; Di Paola, R. Effects of a new compound containing Palmitoylethanolamide and baicalein in myocardial ischaemia/reperfusion injury in vivo. Phytomedicine 2019, 54, 27–42. [Google Scholar] [CrossRef]
- Impellizzeri, D.; Siracusa, R.; Cordaro, M.; Crupi, R.; Peritore, A.F.; Gugliandolo, E.; D’Amico, R.; Petrosino, S.; Evangelista, M.; Di Paola, R.; et al. N-palmitoylethanolamine-oxazoline (Pea-oxa): A new therapeutic strategy to reduce neuroinflammation, oxidative stress associated to vascular dementia in an experimental model of repeated bilateral common carotid arteries occlusion. Neurobiol. Dis. 2019, 125, 77–91. [Google Scholar] [CrossRef]
- Karbach, S.; Croxford, A.L.; Oelze, M.; Schuler, R.; Minwegen, D.; Wegner, J.; Koukes, L.; Yogev, N.; Nikolaev, A.; Reissig, S.; et al. Interleukin 17 drives vascular inflammation, endothelial dysfunction, and arterial hypertension in psoriasis-like skin disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2658–2668. [Google Scholar] [CrossRef] [Green Version]
- Madhur, M.S.; Funt, S.A.; Li, L.; Vinh, A.; Chen, W.; Lob, H.E.; Iwakura, Y.; Blinder, Y.; Rahman, A.; Quyyumi, A.A.; et al. Role of interleukin 17 in inflammation, atherosclerosis, and vascular function in apolipoprotein e-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1565–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuler, R.; Efentakis, P.; Wild, J.; Lagrange, J.; Garlapati, V.; Molitor, M.; Kossmann, S.; Oelze, M.; Stamm, P.; Li, H.; et al. T cell-derived il-17a induces vascular dysfunction via perivascular fibrosis formation and dysregulation of (.)no/cgmp signaling. Oxid Med. Cell Longev. 2019, 2019, 6721531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garty, H.; Palmer, L.G. Epithelial sodium channels: Function, structure, and regulation. Physiol. Rev. 1997, 77, 359–396. [Google Scholar] [CrossRef] [PubMed]
- Kusche-Vihrog, K.; Tarjus, A.; Fels, J.; Jaisser, F. The epithelial Na+ channel: A new player in the vasculature. Curr. Opin. Nephrol. Hypertens. 2014, 23, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.S.; Weissman, J.L.; Farley, J.; Drummond, H.A. Betaenac is required for whole cell mechanically gated currents in renal vascular smooth muscle cells. Am. J. Physiol. Renal Physiol. 2013, 304, F1428–F1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, H.A.; Grifoni, S.C.; Abu-Zaid, A.; Gousset, M.; Chiposi, R.; Barnard, J.M.; Murphey, B.; Stec, D.E. Renal inflammation and elevated blood pressure in a mouse model of reduced {beta}-Enac. Am. J. Physiol. Renal Physiol. 2011, 301, F443–F449. [Google Scholar] [CrossRef] [Green Version]
- Schramm, P.; Klein, K.U.; Falkenberg, L.; Berres, M.; Closhen, D.; Werhahn, K.J.; David, M.; Werner, C.; Engelhard, K. Impaired cerebrovascular autoregulation in patients with severe sepsis and sepsis-associated delirium. Crit. Care 2012, 16, R181. [Google Scholar] [CrossRef] [Green Version]
- Duncan, J.; Younes, S.T.; Hildebrandt, E.; Ryan, M.J.; Granger, J.P.; Drummond, H.A. Tumor necrosis factor-alpha impairs cerebral blood flow in pregnant rats: Role of vascular beta-epithelial Na(+) channel. Am. J. Physiol. Heart Circ. Physiol. 2020. [Google Scholar] [CrossRef]
- Cheng, G.; Wei, L.; Xiurong, W.; Xiangzhen, L.; Shiguang, Z.; Songbin, F. Il-17 stimulates migration of carotid artery vascular smooth muscle cells in an mmp-9 dependent manner via p38 mapk and erk1/2-dependent nf-kappab and ap-1 activation. Cell. Mol. Neurobiol. 2009, 29, 1161–1168. [Google Scholar] [CrossRef]
- De Oliveira, P.S.; Cardoso, P.R.; Lima, E.V.; Pereira, M.C.; Duarte, A.L.; Pitta Ida, R.; Rego, M.J.; Pitta, M.G. Il-17a, il-22, il-6, and il-21 serum levels in plaque-type psoriasis in brazilian patients. Mediat. Inflamm. 2015, 2015, 819149. [Google Scholar] [CrossRef] [Green Version]
- Cornelius, D.C.; Hogg, J.P.; Scott, J.; Wallace, K.; Herse, F.; Moseley, J.; Wallukat, G.; Dechend, R.; LaMarca, B. Administration of interleukin-17 soluble receptor c suppresses th17 cells, oxidative stress, and hypertension in response to placental ischemia during pregnancy. Hypertension 2013, 62, 1068–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Sommerville, L.J.; Cuneo, A.; Kelemen, S.E.; Autieri, M.V. Expression and suppressive effects of interleukin-19 on vascular smooth muscle cell pathophysiology and development of intimal hyperplasia. Am. J. Pathol. 2008, 173, 901–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazighi, M.; Pelle, A.; Gonzalez, W.; el Mtairag, M.; Philippe, M.; Henin, D.; Michel, J.B.; Feldman, L.J. Il-10 inhibits vascular smooth muscle cell activation in vitro and in vivo. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H866–H871. [Google Scholar] [CrossRef] [PubMed]
- Cuneo, A.A.; Herrick, D.; Autieri, M.V. Il-19 reduces vsmc activation by regulation of mRNA regulatory factor hur and reduction of mRNA stability. J. Mol. Cell. Cardiol. 2010, 49, 647–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, C.; Wu, L.; Li, X. Il-17 family: Cytokines, receptors and signaling. Cytokine 2013, 64, 477–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noubade, R.; Krementsov, D.N.; Del Rio, R.; Thornton, T.; Nagaleekar, V.; Saligrama, N.; Spitzack, A.; Spach, K.; Sabio, G.; Davis, R.J.; et al. Activation of p38 mapk in cd4 t cells controls il-17 production and autoimmune Encephalomyelitis. Blood 2011, 118, 3290–3300. [Google Scholar] [CrossRef] [PubMed]
- Li, J.K.; Nie, L.; Zhao, Y.P.; Zhang, Y.Q.; Wang, X.; Wang, S.S.; Liu, Y.; Zhao, H.; Cheng, L. IL-17 mediates inflammatory reactions via p38/c-Fos and JNK/c-Jun activation in an Ap-1-dependent manner in human nucleus pulposus cells. J. Transl. Med. 2016, 14, 77. [Google Scholar]
- Iyoda, M.; Shibata, T.; Kawaguchi, M.; Hizawa, N.; Yamaoka, T.; Kokubu, F.; Akizawa, T. IL-17a and IL-17f stimulate chemokines via mapk pathways (erk1/2 and p38 but not JNK) in mouse cultured mesangial cells: Synergy with TNF-alpha and IL-1beta. Am. J. Physiol. Renal Physiol. 2010, 298, F779–F787. [Google Scholar] [CrossRef]
- Applegarth, A. On structures. J. Am. Psychoanal Assoc. 1989, 37, 1097–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, S.; Li, J.; Wang, J.H.; Wu, Q.; Yang, P.; Hsu, H.C.; Smythies, L.E.; Mountz, J.D. IL-17 activates the canonical NF-kappab signaling pathway in autoimmune b cells of bxd2 mice to upregulate the expression of regulators of g-protein signaling 16. J. Immunol. 2010, 184, 2289–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, F.; Hu, Z.; Goswami, J.; Gaffen, S.L. Identification of common transcriptional regulatory elements in interleukin-17 target genes. J. Biol. Chem. 2006, 281, 24138–24148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonder, S.U.; Saret, S.; Tang, W.; Sturdevant, D.E.; Porcella, S.F.; Siebenlist, U. IL-17-induced NF-kappab activation via CIKS/Act1: Physiologic significance and signaling mechanisms. J. Biol. Chem. 2011, 286, 12881–12890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fey, D.; Croucher, D.R.; Kolch, W.; Kholodenko, B.N. Crosstalk and signaling switches in mitogen-activated protein kinase cascades. Front. Physiol. 2012, 3, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrowski, E.; Bender, B.; Huppert, J.; White, R.; Luhmann, H.J.; Kuhlmann, C.R. Pro-inflammatory effects of interleukin-17A on vascular smooth muscle cells involve NAD(P)H- oxidase derived reactive oxygen species. J. Vasc. Res. 2011, 48, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.I.; Kotsias, B.A. Expression of the epithelial sodium channel sensitive to amiloride (Enac) in normal and preeclamptic human placenta. Placenta 2013, 34, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.J.; Gilbert, E.L.; Glover, P.H.; George, E.M.; Masterson, C.W.; McLemore, G.R., Jr.; LaMarca, B.; Granger, J.P.; Drummond, H.A. Placental ischemia impairs middle cerebral artery myogenic responses in the pregnant rat. Hypertension 2011, 58, 1126–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; He, G.; Yang, Y.; Liu, Y.; Diao, R.; Sheng, K.; Liu, X.; Xu, W. Reduced expression of Enac in placenta tissues of patients with severe preeclampsia is related to compromised trophoblastic cell migration and invasion during pregnancy. PLoS ONE 2013, 8, e72153. [Google Scholar] [CrossRef] [Green Version]
- Warrington, J.P.; Fan, F.; Murphy, S.R.; Roman, R.J.; Drummond, H.A.; Granger, J.P.; Ryan, M.J. Placental ischemia in pregnant rats impairs cerebral blood flow autoregulation and increases blood-brain barrier permeability. Physiol. Rep. 2014, 2, e12134. [Google Scholar] [CrossRef]
- Kalantar, F.; Rajaei, S.; Heidari, A.B.; Mansouri, R.; Rashidi, N.; Izad, M.H.; Mirahmadian, M. Serum levels of tumor necrosis factor-alpha, interleukin-15 and interleukin-10 in patients with pre-eclampsia in comparison with normotensive pregnant women. Iran. J. Nurs. Midwifery Res. 2013, 18, 463–466. [Google Scholar]
- LaMarca, B.; Speed, J.; Fournier, L.; Babcock, S.A.; Berry, H.; Cockrell, K.; Granger, J.P. Hypertension in response to chronic reductions in uterine perfusion in pregnant rats: Effect of tumor necrosis factor-alpha blockade. Hypertension 2008, 52, 1161–1167. [Google Scholar] [CrossRef] [Green Version]
- Molvarec, A.; Czegle, I.; Szijarto, J.; Rigo, J., Jr. Increased circulating interleukin-17 levels in preeclampsia. J. Reprod. Immunol. 2015, 112, 53–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelius, D.C.; Amaral, L.M.; Harmon, A.; Wallace, K.; Thomas, A.J.; Campbell, N.; Scott, J.; Herse, F.; Haase, N.; Moseley, J.; et al. An increased population of regulatory T cells improves the pathophysiology of placental ischemia in a rat model of preeclampsia. Am. J. Physiol. Regul. Integr Comp. Physiol. 2015, 309, R884–R891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarty, A.; Chakrabarti, S.D. The neurology of eclampsia: Some observations. Neurol. India 2002, 50, 128–135. [Google Scholar] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duncan, J.W.; Granger, J.P.; Ryan, M.J.; Drummond, H.A. Interleukin-17 Reduces βENaC via MAPK Signaling in Vascular Smooth Muscle Cells. Int. J. Mol. Sci. 2020, 21, 2953. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082953
Duncan JW, Granger JP, Ryan MJ, Drummond HA. Interleukin-17 Reduces βENaC via MAPK Signaling in Vascular Smooth Muscle Cells. International Journal of Molecular Sciences. 2020; 21(8):2953. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082953
Chicago/Turabian StyleDuncan, Jeremy W., Joey P. Granger, Michael J. Ryan, and Heather A. Drummond. 2020. "Interleukin-17 Reduces βENaC via MAPK Signaling in Vascular Smooth Muscle Cells" International Journal of Molecular Sciences 21, no. 8: 2953. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082953