Deubiquitinase MYSM1 in the Hematopoietic System and beyond: A Current Review

 , , and
, , and 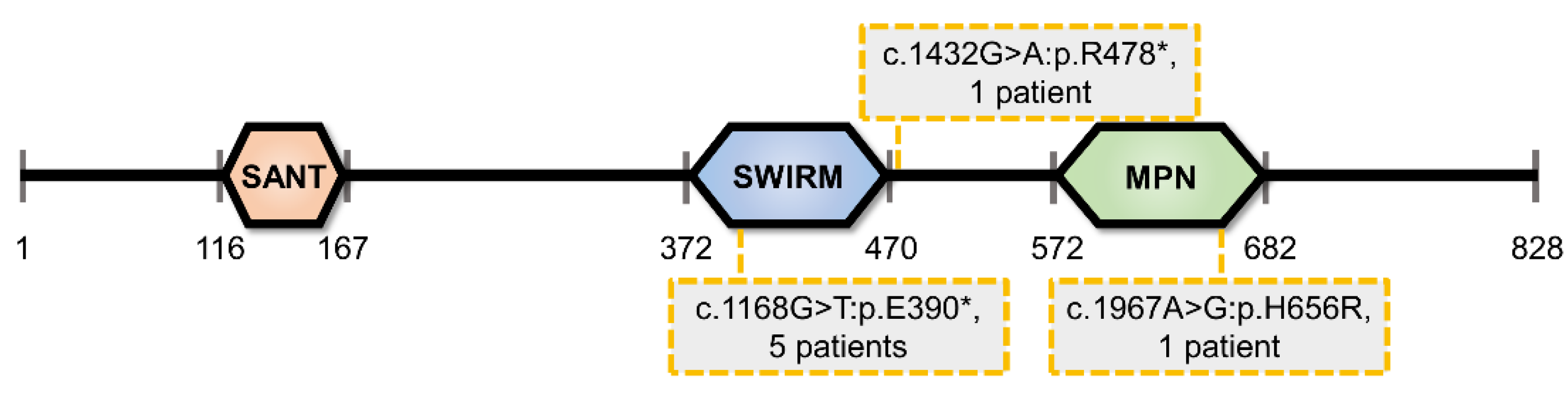{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Overview of MYSM1 Protein Structure and Catalytic Activity
1.2. MYSM1-Deficiency in Human and Mouse: Mechanistic Insights and Biomedical Significance
2. Molecular Functions of MYSM1 Protein
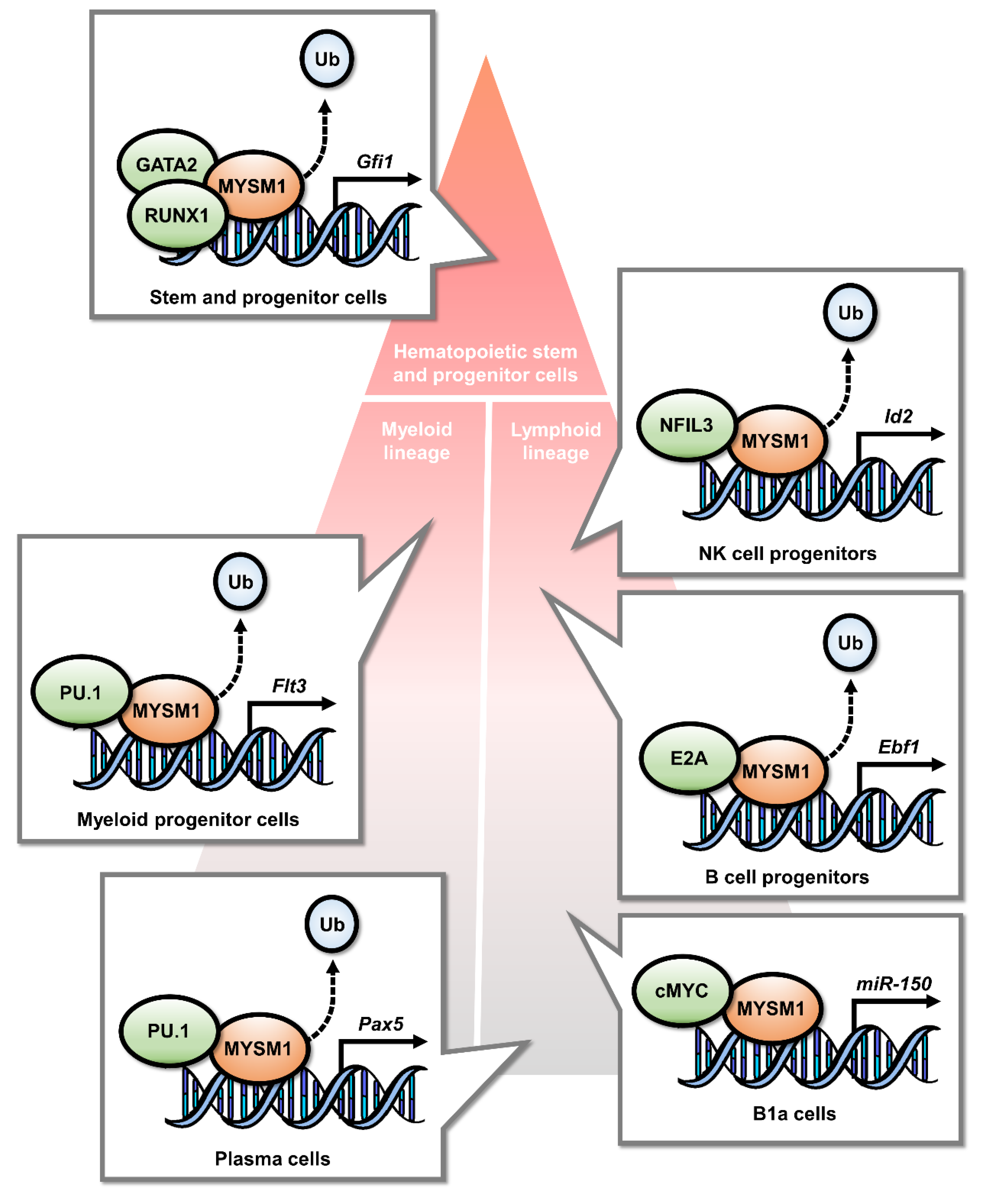
2.1. MYSM1 Is a Transcriptional Regulator in Hematopoietic Lineage Specification
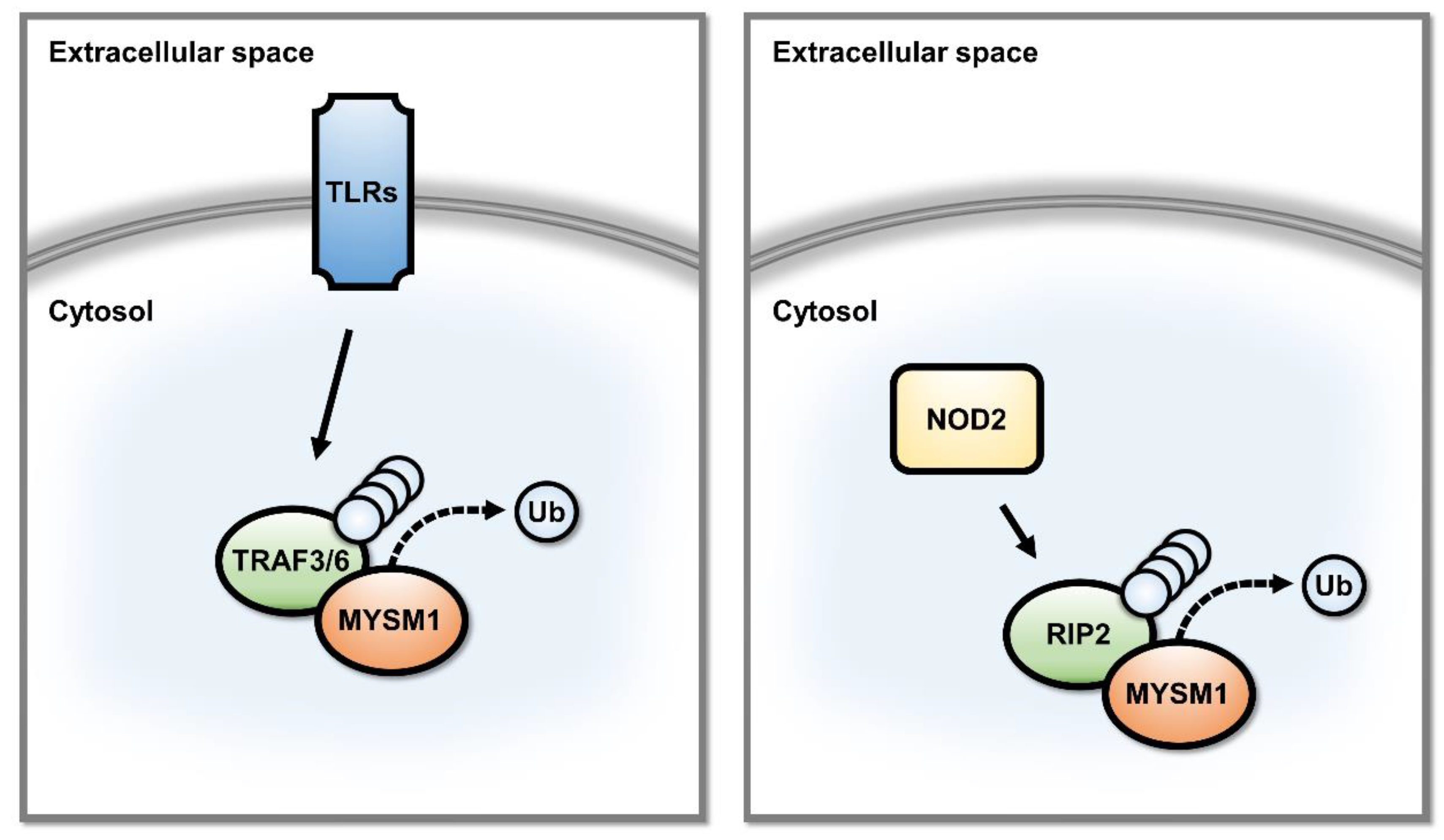
2.2. MYSM1 Is a Regulator of Signal Transduction Pathways in Innate Immunity
2.3. Putative Roles of MYSM1 in DNA Repair
2.4. MYSM1 and the p53 Stress Response Pathway
3. MYSM1 Regulated Checkpoints in Mammalian Physiology
3.1. Essential and Cell Intrinsic Functions of MYSM1 in Hematopoietic Stem Cells
3.2. MYSM1 in B Cell Development and Humoral Immune Response
3.3. MYSM1 in T Cell and NK Cell Development
3.4. MYSM1 in Myeloid Lineage Cell Development and Innate Immune Response
3.5. MYSM1 Functions beyond the Hematopoietic System
3.6. Possible Roles of MYSM1 in Cancer
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ogata, K.; Morikawa, S.; Nakamura, H.; Sekikawa, A.; Inoue, T.; Kanai, H.; Sarai, A.; Ishii, S.; Nishimura, Y. Solution structure of a specific DNA complex of the Myb DNA-binding domain with cooperative recognition helices. Cell 1994, 79, 639–648. [Google Scholar] [CrossRef]
- Yoneyama, M.; Tochio, N.; Umehara, T.; Koshiba, S.; Inoue, M.; Yabuki, T.; Aoki, M.; Seki, E.; Matsuda, T.; Watanabe, S.; et al. Structural and functional differences of SWIRM domain subtypes. J. Mol. Biol. 2007, 369, 222–238. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; Nilsson, J.A.; Gekara, N.O. Deubiquitinase MYSM1 Regulates Innate Immunity through Inactivation of TRAF3 and TRAF6 Complexes. Immunity 2015, 43, 647–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, C.; Zhang, Q.; Li, S.; Zeng, L.; Walsh, M.J.; Zhou, M.M. Structure and chromosomal DNA binding of the SWIRM domain. Nat. Struct. Mol. Biol. 2005, 12, 1078–1085. [Google Scholar] [CrossRef]
- Aravind, L.; Iyer, L.M. The SWIRM domain: A conserved module found in chromosomal proteins points to novel chromatin-modifying activities. Genome Biol. 2002, 3, RESEARCH0039. [Google Scholar] [CrossRef] [PubMed]
- Birol, M.; Echalier, A. Structure and Function of MPN (Mpr1/Pad1 N-terminal) Domain-Containing Proteins. Curr. Protein Pept. Sc. 2014, 15, 504–517. [Google Scholar] [CrossRef]
- Zhu, P.; Zhou, W.; Wang, J.; Puc, J.; Ohgi, K.A.; Erdjument-Bromage, H.; Tempst, P.; Glass, C.K.; Rosenfeld, M.G. A histone H2A deubiquitinase complex coordinating histone acetylation and H1 dissociation in transcriptional regulation. Mol. Cell 2007, 27, 609–621. [Google Scholar] [CrossRef] [Green Version]
- Panda, S.; Gekara, N.O. The deubiquitinase MYSM1 dampens NOD2-mediated inflammation and tissue damage by inactivating the RIP2 complex. Nat. Commun. 2018, 9, 4654. [Google Scholar] [CrossRef] [Green Version]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [Green Version]
- Steffen, P.A.; Ringrose, L. What are memories made of? How Polycomb and Trithorax proteins mediate epigenetic memory. Nat. Rev. Mol. Cell Biol. 2014, 15, 340–356. [Google Scholar] [CrossRef]
- Vidal, M.; Starowicz, K. Polycomb complexes PRC1 and their function in hematopoiesis. Exp. Hematol. 2017, 48, 12–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef]
- Pavri, R.; Zhu, B.; Li, G.; Trojer, P.; Mandal, S.; Shilatifard, A.; Reinberg, D. Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell 2006, 125, 703–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekharan, M.B.; Huang, F.; Sun, Z.W. Histone H2B ubiquitination and beyond: Regulation of nucleosome stability, chromatin dynamics and the trans-histone H3 methylation. Epigenetics 2010, 5, 460–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belle, J.I.; Nijnik, A. H2A-DUBbing the mammalian epigenome: Expanding frontiers for histone H2A deubiquitinating enzymes in cell biology and physiology. Int. J. Biochem. Cell Biol. 2014, 50, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Durocher, D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell 2013, 49, 795–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Zhang, H.; Zhang, H.; Wang, Z.; Zhou, H.; Zhang, Z. A Cul4 E3 ubiquitin ligase regulates histone hand-off during nucleosome assembly. Cell 2013, 155, 817–829. [Google Scholar] [CrossRef] [Green Version]
- Sarcinella, E.; Zuzarte, P.C.; Lau, P.N.; Draker, R.; Cheung, P. Monoubiquitylation of H2A.Z distinguishes its association with euchromatin or facultative heterochromatin. Mol. Cell Biol. 2007, 27, 6457–6468. [Google Scholar] [CrossRef] [Green Version]
- Nijnik, A.; Clare, S.; Hale, C.; Raisen, C.; McIntyre, R.E.; Yusa, K.; Everitt, A.R.; Mottram, L.; Podrini, C.; Lucas, M.; et al. The critical role of histone H2A-deubiquitinase Mysm1 in hematopoiesis and lymphocyte differentiation. Blood 2012, 119, 1370–1379. [Google Scholar] [CrossRef]
- Jiang, X.X.; Nguyen, Q.; Chou, Y.; Wang, T.; Nandakumar, V.; Yates, P.; Jones, L.; Wang, L.; Won, H.; Lee, H.R.; et al. Control of B cell development by the histone H2A deubiquitinase MYSM1. Immunity 2011, 35, 883–896. [Google Scholar] [CrossRef] [Green Version]
- Skarnes, W.; Rosen, B.; West, A.; Koutsourakis, M.; Bushell, W.; Iyer, V.; Cox, T.; Jackson, D.; Severin, J.; Biggs, P.; et al. A conditional knockout resource for genome-wide analysis of mouse gene function. Nature 2011, 474, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.K.; Gerdin, A.K.; Karp, N.A.; Ryder, E.; Buljan, M.; Bussell, J.N.; Salisbury, J.; Clare, S.; Ingham, N.J.; Podrini, C.; et al. Genome-wide Generation and Systematic Phenotyping of Knockout Mice Reveals New Roles for Many Genes. Cell 2013, 154, 452–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickinson, M.E.; Flenniken, A.M.; Ji, X.; Teboul, L.; Wong, M.D.; White, J.K.; Meehan, T.F.; Weninger, W.J.; Westerberg, H.; Adissu, H.; et al. High-throughput discovery of novel developmental phenotypes. Nature 2016, 537, 508–514. [Google Scholar] [CrossRef] [PubMed]
- International Mouse Phenotyping Consortium. Available online: https://www.mousephenotype.org/data/genes/MGI:2444584 (accessed on 28 March 2020).
- Alsultan, A.; Shamseldin, H.E.; Osman, M.E.; Aljabri, M.; Alkuraya, F.S. MYSM1 is mutated in a family with transient transfusion-dependent anemia, mild thrombocytopenia, and low NK- and B-cell counts. Blood 2013, 122, 3844–3845. [Google Scholar] [CrossRef]
- Le Guen, T.; Touzot, F.; Andre-Schmutz, I.; Lagresle-Peyrou, C.; France, B.; Kermasson, L.; Lambert, N.; Picard, C.; Nitschke, P.; Carpentier, W.; et al. An in vivo genetic reversion highlights the crucial role of Myb-Like, SWIRM, and MPN domains 1 (MYSM1) in human hematopoiesis and lymphocyte differentiation. J. Allergy Clin. Immunol. 2015, 136, 1619–1626 e1615. [Google Scholar] [CrossRef]
- Bahrami, E.; Witzel, M.; Racek, T.; Puchalka, J.; Hollizeck, S.; Greif-Kohistani, N.; Kotlarz, D.; Horny, H.P.; Feederle, R.; Schmidt, H.; et al. Myb-like, SWIRM, and MPN domains 1 (MYSM1) deficiency: Genotoxic stress-associated bone marrow failure and developmental aberrations. J. Allergy Clin. Immunol. 2017, 140, 1112–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanda, A.; Al-Abboh, H.; Zahra, A.; Al-Sabah, H.; Gupta, A.; Adekile, A.D. Neutrophilic Panniculitis in a child with MYSM1 deficiency. Pediatr. Dermatol. 2019, 36, 258–259. [Google Scholar] [CrossRef] [PubMed]
- Ulirsch, J.C.; Verboon, J.M.; Kazerounian, S.; Guo, M.H.; Yuan, D.; Ludwig, L.S.; Handsaker, R.E.; Abdulhay, N.J.; Fiorini, C.; Genovese, G.; et al. The Genetic Landscape of Diamond-Blackfan Anemia. Am. J. Hum. Genet. 2018, 103, 930–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.X.; Chou, Y.; Jones, L.; Wang, T.; Sanchez, S.; Huang, X.F.; Zhang, L.; Wang, C.; Chen, S.Y. Epigenetic Regulation of Antibody Responses by the Histone H2A Deubiquitinase MYSM1. Sci. Rep. 2015, 5, 13755. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.X.; Liu, Y.; Li, H.; Gao, Y.; Mu, R.; Guo, J.; Zhang, J.; Yang, Y.M.; Xiao, F.; Liu, B.; et al. MYSM1/miR-150/FLT3 inhibits B1a cell proliferation. Oncotarget 2016, 7, 68086–68096. [Google Scholar] [CrossRef] [Green Version]
- Nandakumar, V.; Chou, Y.; Zang, L.; Huang, X.F.; Chen, S.Y. Epigenetic control of natural killer cell maturation by histone H2A deubiquitinase, MYSM1. Proc. Natl. Acad. Sci. USA 2013, 110, E3927–E3936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, H.; Nandakumar, V.; Yates, P.; Sanchez, S.; Jones, L.; Huang, X.F.; Chen, S.Y. Epigenetic control of dendritic cell development and fate determination of common myeloid progenitor by Mysm1. Blood 2014, 124, 2647–2656. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Nandakumar, V.; Jiang, X.X.; Jones, L.; Yang, A.G.; Huang, X.F.; Chen, S.Y. The control of hematopoietic stem cell maintenance, self-renewal, and differentiation by Mysm1-mediated epigenetic regulation. Blood 2013, 122, 2812–2822. [Google Scholar] [CrossRef]
- Wang, Y.H.; Huang, X.H.; Yang, Y.M.; He, Y.; Dong, X.H.; Yang, H.X.; Zhang, L.; Wang, Y.; Zhou, J.; Wang, C.; et al. Mysm1 epigenetically regulates the immunomodulatory function of adipose-derived stem cells in part by targeting miR-150. J. Cell Mol. Med. 2019, 23, 3737–3746. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Jiang, X.; Chen, J. The role of miR-150 in normal and malignant hematopoiesis. Oncogene 2014, 33, 3887–3893. [Google Scholar] [CrossRef] [PubMed]
- Jacq, X.; Kemp, M.; Martin, N.M.B.; Jackson, S.P. Deubiquitylating Enzymes and DNA Damage Response Pathways. Cell Biochem. Biophys. 2013, 67, 25–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Nishi, R.; Wijnhoven, P.; le Sage, C.; Tjeertes, J.; Galanty, Y.; Forment, J.V.; Clague, M.J.; Urbe, S.; Jackson, S.P. Systematic characterization of deubiquitylating enzymes for roles in maintaining genome integrity. Nat. Cell Biol. 2014, 16, 1016–1026. [Google Scholar] [CrossRef] [Green Version]
- Wilms, C.; Kroeger, C.M.; Hainzl, A.V.; Banik, I.; Bruno, C.; Krikki, I.; Farsam, V.; Wlaschek, M.; Gatzka, M.V. MYSM1/2A-DUB is an epigenetic regulator in human melanoma and contributes to tumor cell growth. Oncotarget 2017, 8, 67287–67299. [Google Scholar] [CrossRef] [Green Version]
- Belle, J.I.; Petrov, J.C.; Langlais, D.; Robert, F.; Cencic, R.; Shen, S.; Pelletier, J.; Gros, P.; Nijnik, A. Repression of p53-target gene Bbc3/PUMA by MYSM1 is essential for the survival of hematopoietic multipotent progenitors and contributes to stem cell maintenance. Cell Death Differ. 2016, 23, 759–775. [Google Scholar] [CrossRef]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belle, J.I.; Langlais, D.; Petrov, J.C.; Pardo, M.; Jones, R.G.; Gros, P.; Nijnik, A. p53 mediates loss of hematopoietic stem cell function and lymphopenia in Mysm1 deficiency. Blood 2015, 125, 2344–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatzka, M.; Tasdogan, A.; Hainzl, A.; Allies, G.; Maity, P.; Wilms, C.; Wlaschek, M.; Scharffetter-Kochanek, K. Interplay of H2A deubiquitinase 2A-DUB/Mysm1 and the p19/p53 axis in hematopoiesis, early T-cell development and tissue differentiation. Cell Death Differ. 2015, 22, 1451–1462. [Google Scholar] [CrossRef] [PubMed]
- Forster, M.; Belle, J.I.; Petrov, J.C.; Ryder, E.J.; Clare, S.; Nijnik, A. Deubiquitinase MYSM1 Is Essential for Normal Fetal Liver Hematopoiesis and for the Maintenance of Hematopoietic Stem Cells in Adult Bone Marrow. Stem Cells Dev. 2015, 24, 1865–1877. [Google Scholar] [CrossRef]
- Wilms, C.; Krikki, I.; Hainzl, A.; Kilo, S.; Alupei, M.; Makrantonaki, E.; Wagner, M.; Kroeger, C.M.; Brinker, T.J.; Gatzka, M. 2A-DUB/Mysm1 Regulates Epidermal Development in Part by Suppressing p53-Mediated Programs. Int. J. Mol. Sci. 2018, 19, 687. [Google Scholar] [CrossRef] [Green Version]
- Haffner-Luntzer, M.; Kovtun, A.; Fischer, V.; Prystaz, K.; Hainzl, A.; Kroeger, C.M.; Krikki, I.; Brinker, T.J.; Ignatius, A.; Gatzka, M. Loss of p53 compensates osteopenia in murine Mysm1 deficiency. FASEB J. 2018, 32, 1957–1968. [Google Scholar] [CrossRef] [Green Version]
- Forster, M.; Boora, R.K.; Petrov, J.C.; Fodil, N.; Albanese, I.; Kim, J.; Gros, P.; Nijnik, A. A role for the histone H2A deubiquitinase MYSM1 in maintenance of CD8(+) T cells. Immunology 2017, 151, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Forster, M.; Farrington, K.; Petrov, J.C.; Belle, J.I.; Mindt, B.C.; Witalis, M.; Duerr, C.U.; Fritz, J.H.; Nijnik, A. MYSM1-dependent checkpoints in B cell lineage differentiation and B cell-mediated immune response. J. Leukoc. Biol. 2017, 101, 643–654. [Google Scholar] [CrossRef]
- Petrov, J.C.; Nijnik, A. Mysm1 expression in the bone marrow niche is not essential for hematopoietic maintenance. Exp. Hematol. 2017, 47, 76–82 e73. [Google Scholar] [CrossRef]
- Huo, Y.; Li, B.Y.; Lin, Z.F.; Wang, W.; Jiang, X.X.; Chen, X.; Xi, W.J.; Yang, A.G.; Chen, S.Y.; Wang, T. MYSM1 Is Essential for Maintaining Hematopoietic Stem Cell (HSC) Quiescence and Survival. Med. Sci. Monit. 2018, 24, 2541–2549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandanpour, C.; Phelan, J.D.; Vassen, L.; Schutte, J.; Chen, R.; Horman, S.R.; Gaudreau, M.C.; Krongold, J.; Zhu, J.; Paul, W.E.; et al. Growth factor independence 1 antagonizes a p53-induced DNA damage response pathway in lymphoblastic leukemia. Cancer Cell 2013, 23, 200–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadnais, C.; Chen, R.; Fraszczak, J.; Hamard, P.J.; Manfredi, J.J.; Moroy, T. A novel regulatory circuit between p53 and GFI1 controls induction of apoptosis in T cells. Sci. Rep. 2019, 9, 6304. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.F.; Nandakumar, V.; Tumurkhuu, G.; Wang, T.; Jiang, X.; Hong, B.; Jones, L.; Won, H.; Yoshii, H.; Ozato, K.; et al. Mysm1 is required for interferon regulatory factor expression in maintaining HSC quiescence and thymocyte development. Cell Death Dis. 2016, 7, e2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Huang, X.H.; Dong, X.H.; Wang, Y.H.; Yang, H.X.; Wang, Y.; He, Y.; Liu, S.; Zhou, J.; Wang, C.; et al. Deubiquitinase Mysm1 regulates macrophage survival and polarization. Mol. Biol. Rep. 2018, 45, 2393–2401. [Google Scholar] [CrossRef] [PubMed]
- Liakath-Ali, K.; Vancollie, V.E.; Heath, E.; Smedley, D.P.; Estabel, J.; Sunter, D.; Ditommaso, T.; White, J.K.; Ramirez-Solis, R.; Smyth, I.; et al. Novel skin phenotypes revealed by a genome-wide mouse reverse genetic screen. Nat. Commun. 2014, 5, 3540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiTommaso, T.; Jones, L.K.; Cottle, D.L.; Program, W.M.G.; Gerdin, A.K.; Vancollie, V.E.; Watt, F.M.; Ramirez-Solis, R.; Bradley, A.; Steel, K.P.; et al. Identification of genes important for cutaneous function revealed by a large scale reverse genetic screen in the mouse. PLoS Genet. 2014, 10, e1004705. [Google Scholar] [CrossRef]
- Martins, V.C.; Busch, K.; Juraeva, D.; Blum, C.; Ludwig, C.; Rasche, V.; Lasitschka, F.; Mastitsky, S.E.; Brors, B.; Hielscher, T.; et al. Cell competition is a tumour suppressor mechanism in the thymus. Nature 2014, 509, 465–470. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Liu, H.; Liu, Y.; Cui, B. Expression of MYSM1 is associated with tumor progression in colorectal cancer. PLoS ONE 2017, 12, e0177235. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiore, A.; Liang, Y.; Lin, Y.H.; Tung, J.; Wang, H.; Langlais, D.; Nijnik, A. Deubiquitinase MYSM1 in the Hematopoietic System and beyond: A Current Review. Int. J. Mol. Sci. 2020, 21, 3007. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21083007
Fiore A, Liang Y, Lin YH, Tung J, Wang H, Langlais D, Nijnik A. Deubiquitinase MYSM1 in the Hematopoietic System and beyond: A Current Review. International Journal of Molecular Sciences. 2020; 21(8):3007. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21083007
Chicago/Turabian StyleFiore, Amanda, Yue Liang, Yun Hsiao Lin, Jacky Tung, HanChen Wang, David Langlais, and Anastasia Nijnik. 2020. "Deubiquitinase MYSM1 in the Hematopoietic System and beyond: A Current Review" International Journal of Molecular Sciences 21, no. 8: 3007. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21083007