New Insight into Mechanisms of Protein Adaptation to High Temperatures: A Comparative Molecular Dynamics Simulation Study of Thermophilic and Mesophilic Subtilisin-Like Serine Proteases



Abstract
:1. Introduction
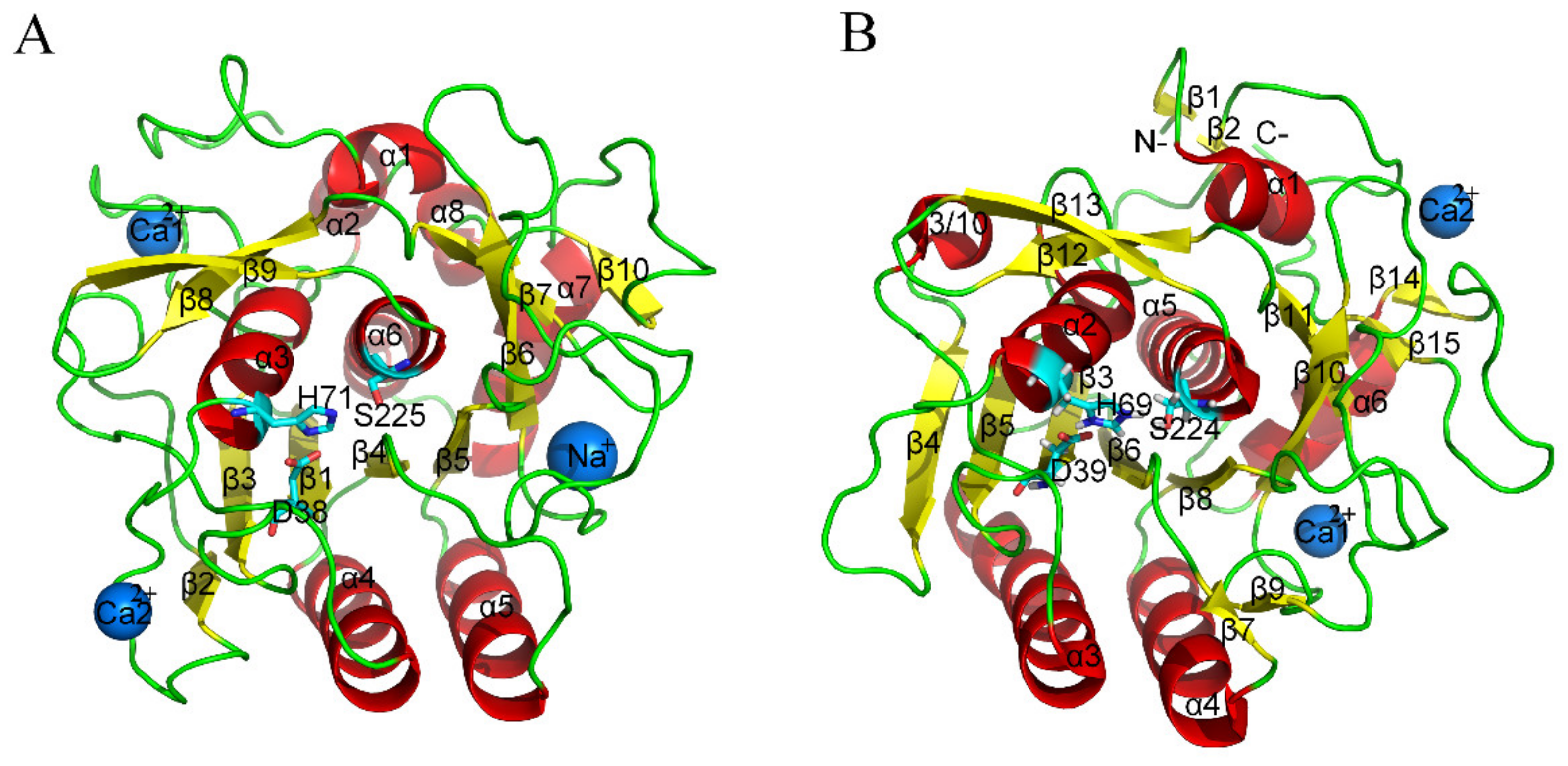
2. Results
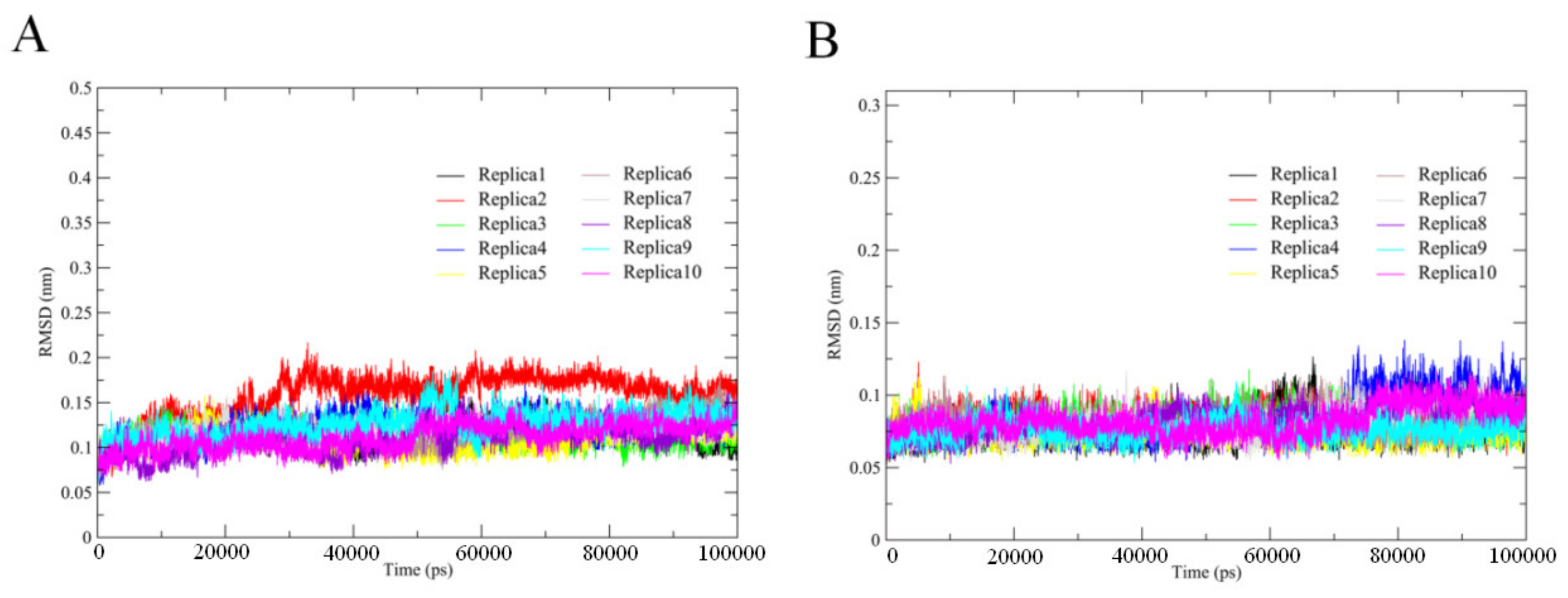
2.1. Trajectory Stability and Evaluation of Conformation Sampling
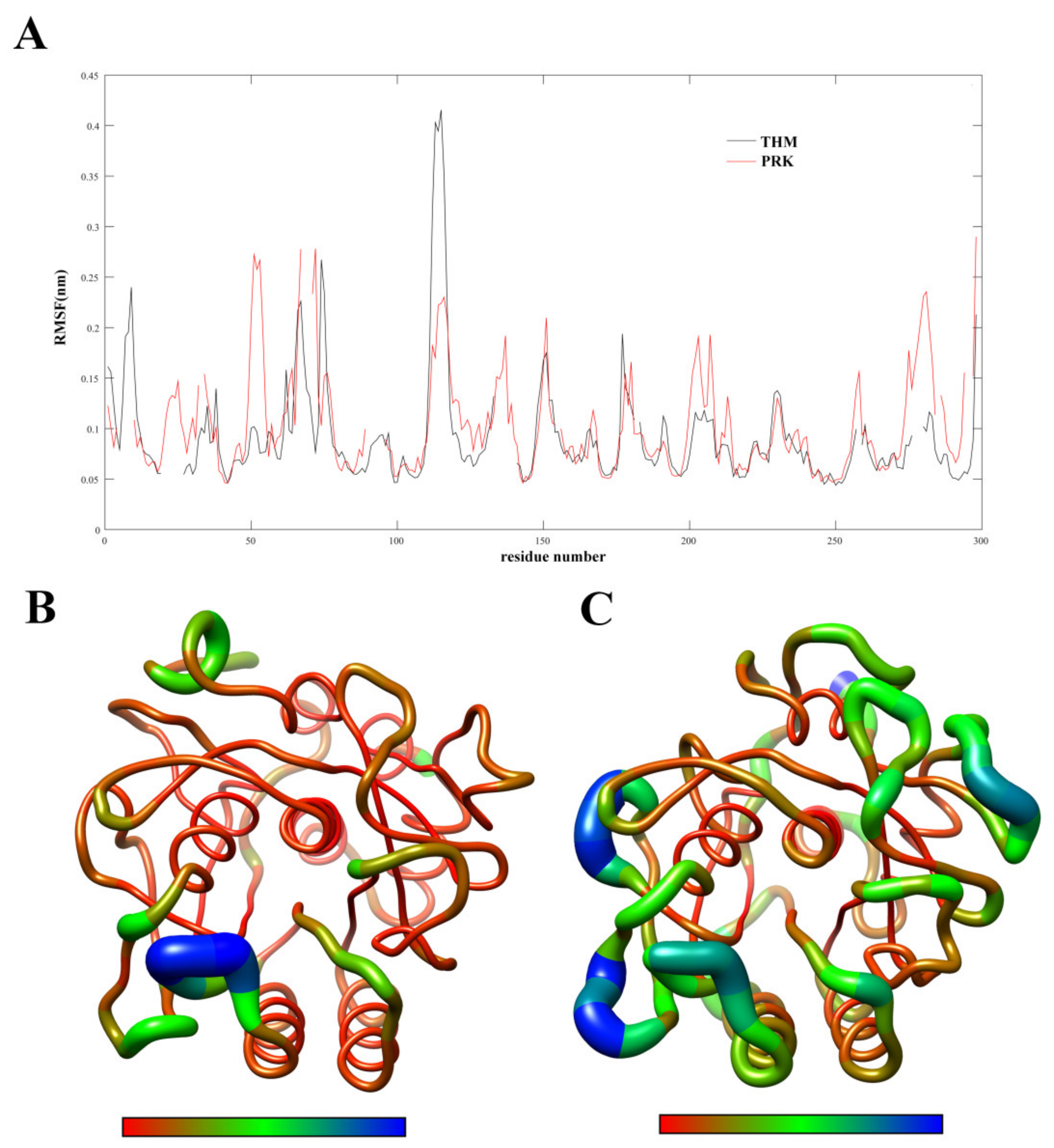
2.2. Flexibility Analysis
2.3. Geometrical and Structural Properties
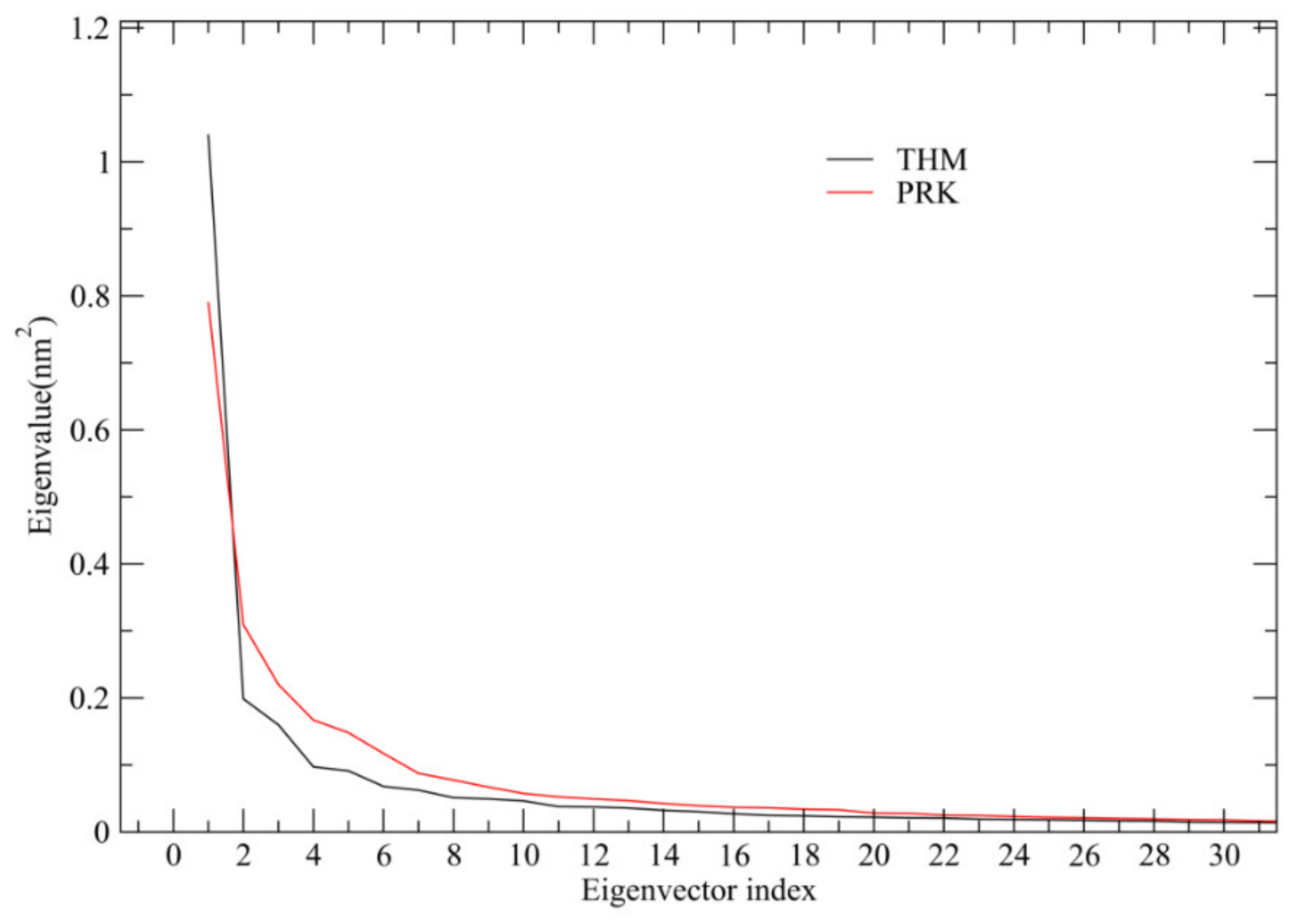
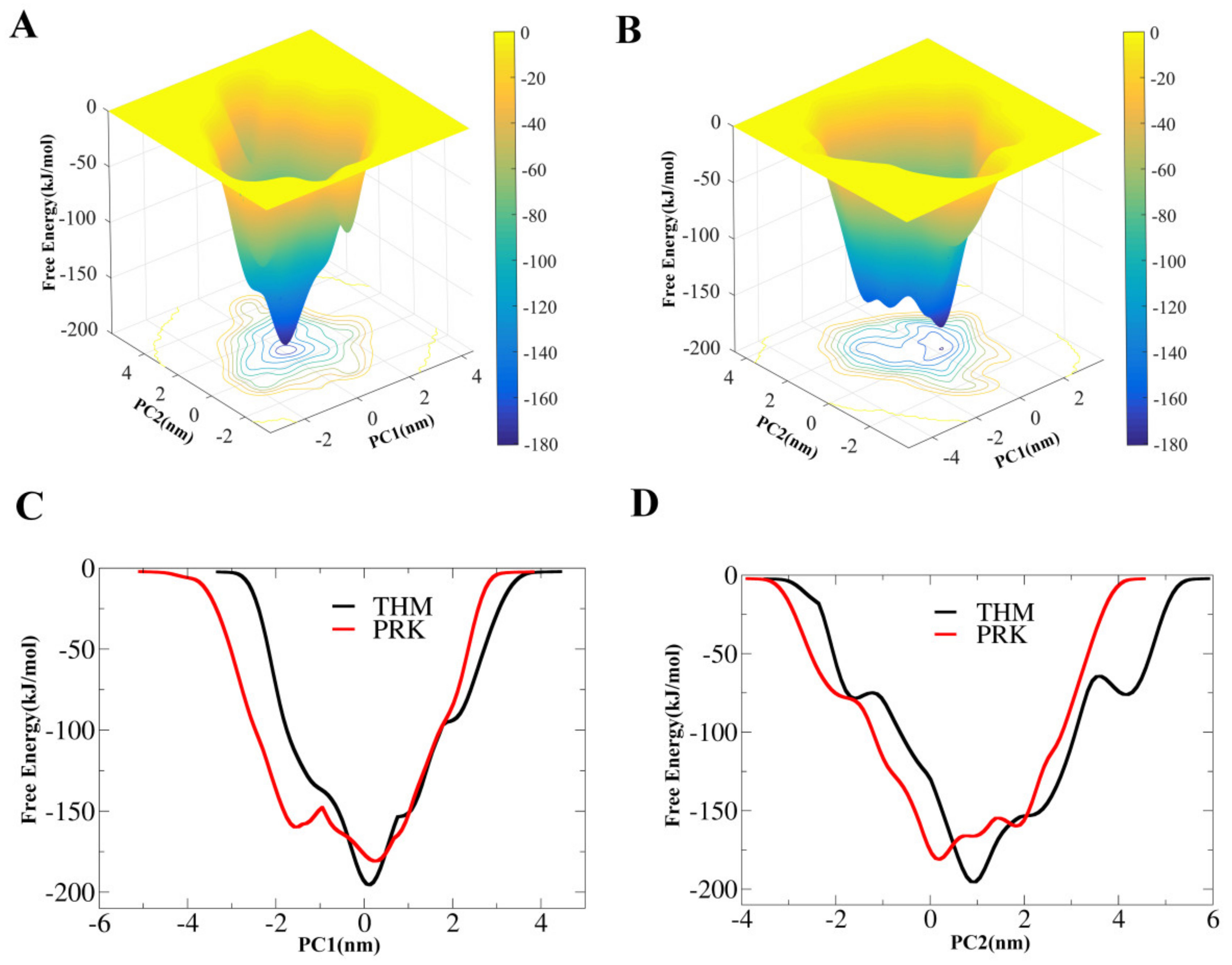
2.4. Essential Dynamics (ED) Analyses and Free-energy Landscapes
3. Discussion
4. Materials and Methods
4.1. Preparation of the Simulated System
4.2. MD Simulations
4.3. Structural and Geometrical Properties
4.4. Essential Dynamics (ED)
4.5. Free-Energy Calculations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Babu, P.; Chandel, A.K.; Singh, O.V. Survival mechanisms of extremophiles. In Extremophiles and Their Applications in Medical Processes; Springer International Publishing: Cham, Switzerland, 2015. [Google Scholar]
- Sang, P.; Du, X.; Yang, L.-Q.; Meng, Z.-H.; Liu, S.-Q. Molecular motions and free-energy landscape of serine proteinase K in relation to its cold-adaptation: A comparative molecular dynamics simulation study and the underlying mechanisms. RSC Adv. 2017, 7, 28580–28590. [Google Scholar] [CrossRef] [Green Version]
- Tilton, R.F., Jr.; Dewan, J.C.; Petsko, G.A. Effects of temperature on protein structure and dynamics: X-ray crystallographic studies of the protein ribonuclease-A at nine different temperatures from 98 to 320K. Biochemistry 1992, 31, 2469–2481. [Google Scholar] [CrossRef] [PubMed]
- Sang, P.; Yang, Q.; Du, X.; Yang, N.; Yang, L.-Q.; Ji, X.-L.; Fu, Y.-X.; Meng, Z.-H.; Liu, S.-Q. Effect of the solvent temperatures on dynamics of serine protease proteinase K. Int. J. Mol. Sci. 2016, 17, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonhienne, T.; Gerday, C.; Feller, G. Psychrophilic enzymes: Revisiting the thermodynamic parameters of activation may explain local flexibility. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2000, 1543, 1–10. [Google Scholar] [CrossRef]
- Papaleo, E.; Tiberti, M.; Invernizzi, G.; Pasi, M.; Ranzani, V. Molecular determinants of enzyme cold adaptation: Comparative structural and computational studies of cold-and warm-adapted enzymes. Curr. Protein Pept. Sci. 2011, 12, 657–683. [Google Scholar] [CrossRef]
- Sterner, R.; Liebl, W. Thermophilic adaptation of proteins. Crit. Rev. Biochem. Mol. Biol. 2001, 36, 39–106. [Google Scholar] [CrossRef]
- Xia, Y.-L.; Sun, J.-H.; Ai, S.-M.; Li, Y.; Du, X.; Sang, P.; Yang, L.-Q.; Fu, Y.-X.; Liu, S.-Q. Insights into the role of electrostatics in temperature adaptation: A comparative study of psychrophilic, mesophilic, and thermophilic subtilisin-like serine proteases. RSC Adv. 2018, 8, 29698–29713. [Google Scholar] [CrossRef] [Green Version]
- Tiberti, M.; Papaleo, E. Dynamic properties of extremophilic subtilisin-like serine-proteases. J. Struct. Biol. 2011, 174, 69–83. [Google Scholar] [CrossRef]
- Kumar, S.; Nussinov, R. How do thermophilic proteins deal with heat? Cell. Mol. Life Sci. CMLS 2001, 58, 1216–1233. [Google Scholar] [CrossRef]
- Vieille, C.; Zeikus, G.J. Hyperthermophilic enzymes: Sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev. 2001, 65, 1–43. [Google Scholar] [CrossRef] [Green Version]
- Pica, A.; Graziano, G. Shedding light on the extra thermal stability of thermophilic proteins. Biopolymers 2016, 105, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Mattos, C. Protein–water interactions in a dynamic world. Trends Biochem. Sci. 2002, 27, 203–208. [Google Scholar] [CrossRef]
- Li, H.; Xie, Y.; Liu, C.; Liu, S. Physicochemical bases for protein folding, dynamics, and protein-ligand binding. Sci. China Life Sci. 2014, 57, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A. Dominant forces in protein folding. Biochemistry 1990, 29, 7133–7155. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Sang, P.; Xia, Y.-L.; Li, Y.; Liang, J.; Ai, S.-M.; Ji, X.-L.; Fu, Y.-X.; Liu, S.-Q. Comparative thermal unfolding study of psychrophilic and mesophilic subtilisin-like serine proteases by molecular dynamics simulations. J. Biomol. Struct. Dyn. 2017, 35, 1500–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitkup, D.; Ringe, D.; Petsko, G.A.; Karplus, M. Solvent mobility and the protein’glass’ transition. Nat. Struct. Mol. Biol. 2000, 7, 34. [Google Scholar]
- Finkelstein, A.V.; Ptitsyn, O. Protein Physics: A Course of Lectures; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Chaplin, M.F. Water’s hydrogen bond strength. arXiv 2007, arXiv:preprint cond-mat/0706.1355. [Google Scholar]
- Yang, L.-Q.; Ji, X.-L.; Liu, S.-Q. The free energy landscape of protein folding and dynamics: A global view. J. Biomol. Struct. Dyn. 2013, 31, 982–992. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.-Q.; Sang, P.; Tao, Y.; Fu, Y.-X.; Zhang, K.-Q.; Xie, Y.-H.; Liu, S.-Q. Protein dynamics and motions in relation to their functions: Several case studies and the underlying mechanisms. J. Biomol. Struct. Dyn. 2014, 32, 372–393. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, S.; Marx, J.-C.; Gerday, C.; Feller, G. Activity-stability relationships in extremophilic enzymes. J. Biol. Chem. 2003, 278, 7891–7896. [Google Scholar] [CrossRef] [Green Version]
- Hedstrom, L. Serine protease mechanism and specificity. Chem. Rev. 2002, 102, 4501–4524. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, Version 1.8.; Schrodinger, LLC: New York, NY, USA, 2015.
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pace, C.N. Contribution of the hydrophobic effect to globular protein stability. J. Mol. Biol. 1992, 226, 29–35. [Google Scholar] [CrossRef]
- Reed, C.J.; Lewis, H.; Trejo, E.; Winston, V.; Evilia, C. Protein adaptations in archaeal extremophiles. Archaea 2013, 2013, 373275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaenicke, R. Protein stability and molecular adaptation to extreme conditions. In EJB Reviews 1991; Springer: Berlin, Germany, 1991; pp. 291–304. [Google Scholar]
- Quezada, A.G.; Díaz-Salazar, A.J.; Cabrera, N.; Pérez-Montfort, R.; Piñeiro, Á.; Costas, M. Interplay between protein thermal flexibility and kinetic stability. Structure 2017, 25, 167–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamerzell, T.J.; Middaugh, C.R. The complex inter-relationships between protein flexibility and stability. J. Pharm. Sci. 2008, 97, 3494–3517. [Google Scholar] [CrossRef] [PubMed]
- Karshikoff, A.; Nilsson, L.; Ladenstein, R. Rigidity versus flexibility: The dilemma of understanding protein thermal stability. FEBS J. 2015, 282, 3899–3917. [Google Scholar] [CrossRef]
- Xie, B.-B.; Bian, F.; Chen, X.-L.; He, H.-L.; Guo, J.; Gao, X.; Zeng, Y.-X.; Chen, B.; Zhou, B.-C.; Zhang, Y.-Z. Cold adaptation of zinc metalloproteases in the thermolysin family from deep sea and arctic sea ice bacteria revealed by catalytic and structural properties and molecular dynamics new insights into relationship between conformational flexibility and hydrogen bonding. J. Biol. Chem. 2009, 284, 9257–9269. [Google Scholar]
- Teplyakov, A.V.; Kuranova, I.P.; Harutyunyan, E.H.; Vainshtein, B.K.; Fro, C.; Ho, W.E.; Wilson, K.S. Crystal structure of thermitase at 1.4 A resolution. J. Mol. Biol. 1990, 214, 261–279. [Google Scholar] [CrossRef]
- Betzel, C.; Gourinath, S.; Kumar, P.; Kaur, P.; Perbandt, M.; Eschenburg, S.; Singh, T.P. Structure of a serine protease proteinase K from Tritirachium album limber at 0.98 Å resolution. Biochemistry 2001, 40, 3080–3088. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.; Postma, J.; van Gunsteren, W.F.; DiNola, A.; Haak, J. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Lins, L.; Thomas, A.; Brasseur, R. Analysis of accessible surface of residues in proteins. Protein Sci. 2003, 12, 1406–1417. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.; Berendsen, H.J. Essential dynamics of proteins. Proteins Struct. Funct. Bioinform. 1993, 17, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-tempered metadynamics: A smoothly converging and tunable free-energy method. Phys. Rev. Lett. 2008, 100, 020603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laio, A.; Gervasio, F.L. Metadynamics: A method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys. 2008, 71, 126601. [Google Scholar] [CrossRef]
- Sicard, F.; Senet, P. Reconstructing the free-energy landscape of Met-enkephalin using dihedral principal component analysis and well-tempered metadynamics. J. Chem. Phys. 2013, 138, 06B610_1. [Google Scholar] [CrossRef] [Green Version]
- Spiwok, V.; Lipovová, P.; Králová, B. Metadynamics in essential coordinates: Free energy simulation of conformational changes. J. Phys. Chem. B 2007, 111, 3073–3076. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trajectory | THM | PRK | ||
|---|---|---|---|---|
| eig.1 | eig.2 | eig.1 | eig.2 | |
| 1 | 0.7491 | 0.3862 | 0.4935 | 0.3572 |
| 2 | 0.2527 | 0.6482 | 0.1579 | 0.5803 |
| 3 | 0.5723 | 0.3765 | 0.2015 | 0.1097 |
| 4 | 0.4325 | 0.2513 | 0.8605 | 0.2531 |
| 5 | 0.1708 | 0.2517 | 0.3701 | 0.2017 |
| 6 | 0.7365 | 0.6804 | 0.4971 | 0.0671 |
| 7 | 0.0376 | 0.4086 | 0.2036 | 0.2471 |
| 8 | 0.3692 | 0.8974 | 0.1912 | 0.4076 |
| 9 | 0.6702 | 0.3975 | 0.6081 | 0.2992 |
| 10 | 0.6091 | 0.4839 | 0.3582 | 0.5703 |
| joined | 0.0019 | 0.0257 | 0.0309 | 0.0093 |
| Protein | NNC a | SASA b (Å2) | NSB c | Rg d (Å) | NHB e | |
|---|---|---|---|---|---|---|
| Stat f | Dyna g | |||||
| THM | 135,765 (1139) | 10,334 (191) | 14 (0.08) | 16.5 (0.05) | 208 (7.7) | 1592 |
| PRK | 133,764 (982) | 11,007 (222) | 10 (0.07) | 16.7 (0.05) | 202 (7.6) | 1952 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sang, P.; Liu, S.-Q.; Yang, L.-Q. New Insight into Mechanisms of Protein Adaptation to High Temperatures: A Comparative Molecular Dynamics Simulation Study of Thermophilic and Mesophilic Subtilisin-Like Serine Proteases. Int. J. Mol. Sci. 2020, 21, 3128. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093128
Sang P, Liu S-Q, Yang L-Q. New Insight into Mechanisms of Protein Adaptation to High Temperatures: A Comparative Molecular Dynamics Simulation Study of Thermophilic and Mesophilic Subtilisin-Like Serine Proteases. International Journal of Molecular Sciences. 2020; 21(9):3128. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093128
Chicago/Turabian StyleSang, Peng, Shu-Qun Liu, and Li-Quan Yang. 2020. "New Insight into Mechanisms of Protein Adaptation to High Temperatures: A Comparative Molecular Dynamics Simulation Study of Thermophilic and Mesophilic Subtilisin-Like Serine Proteases" International Journal of Molecular Sciences 21, no. 9: 3128. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093128