Therapeutic Potential of the Molecular Chaperone and Matrix Metalloproteinase Inhibitor Clusterin for Dry Eye

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Ocular Surface Disease
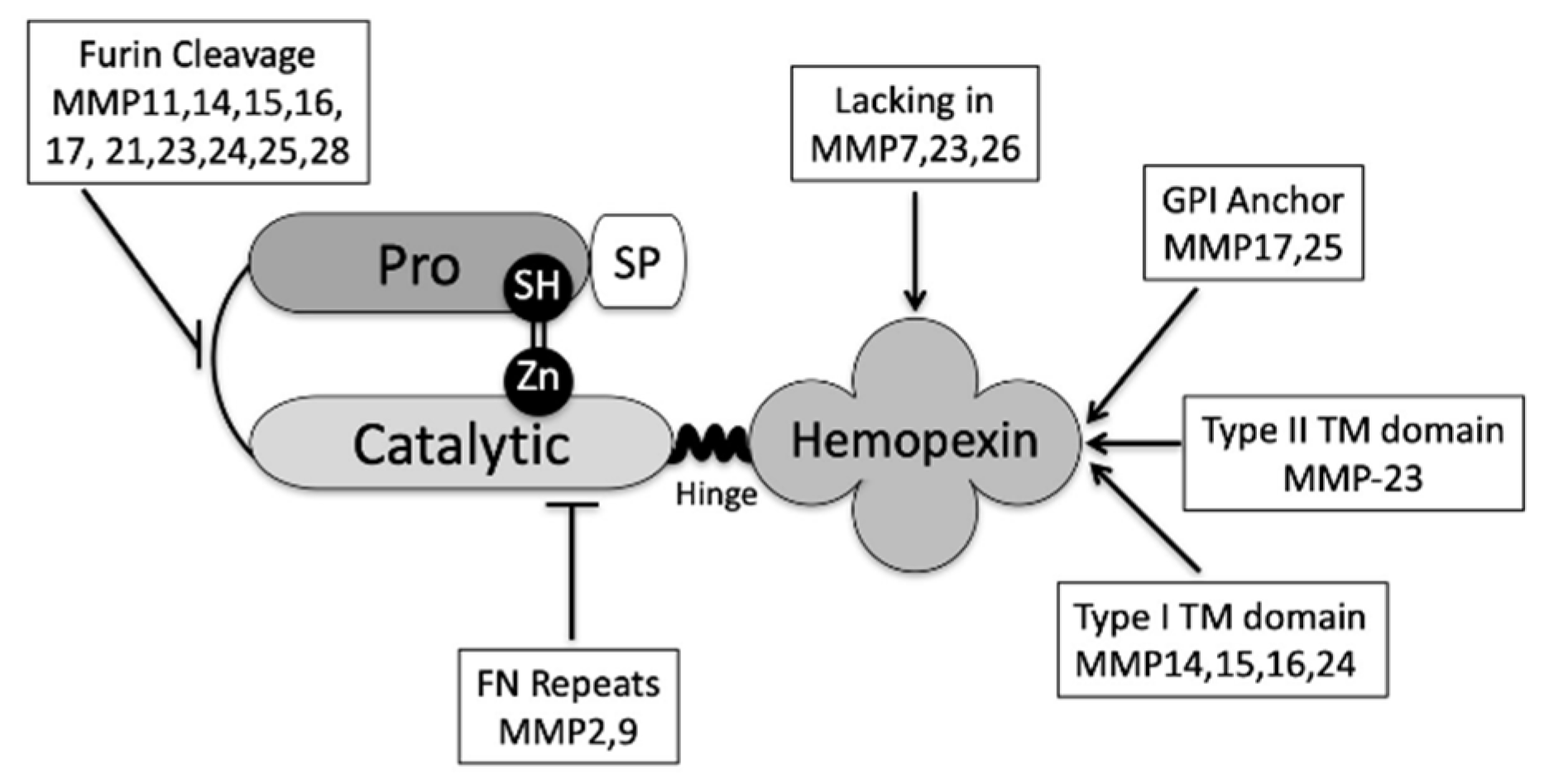
2. MMP9 Is Causal in Ocular Surface Disease
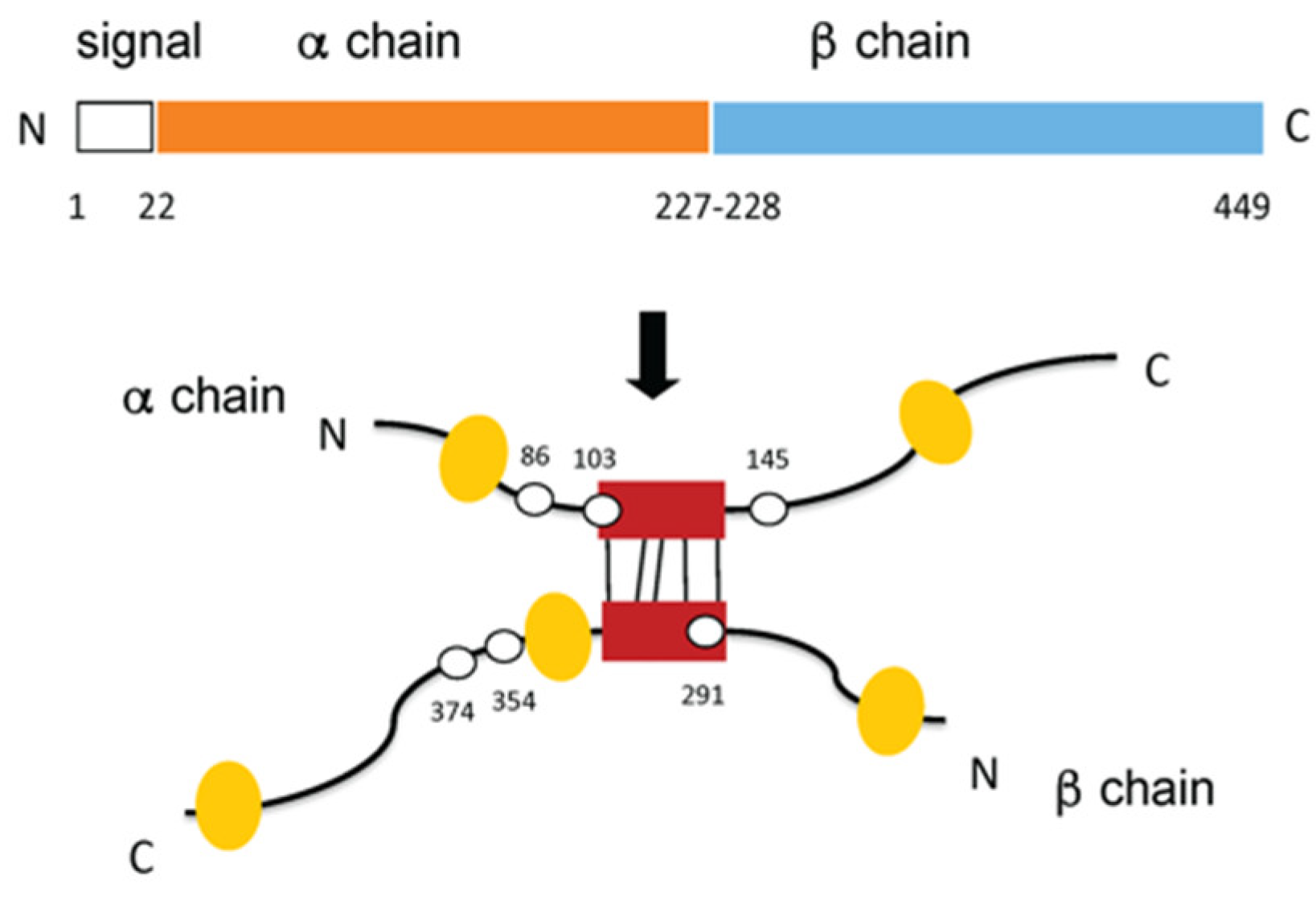
3. The Molecular Chaperone Clusterin Is a MMP9 Inhibitor
4. Clusterin Protects the Ocular Surface in Mouse Dry Eye
5. Clusterin in Human Tears Is Reduced in Dry Eye
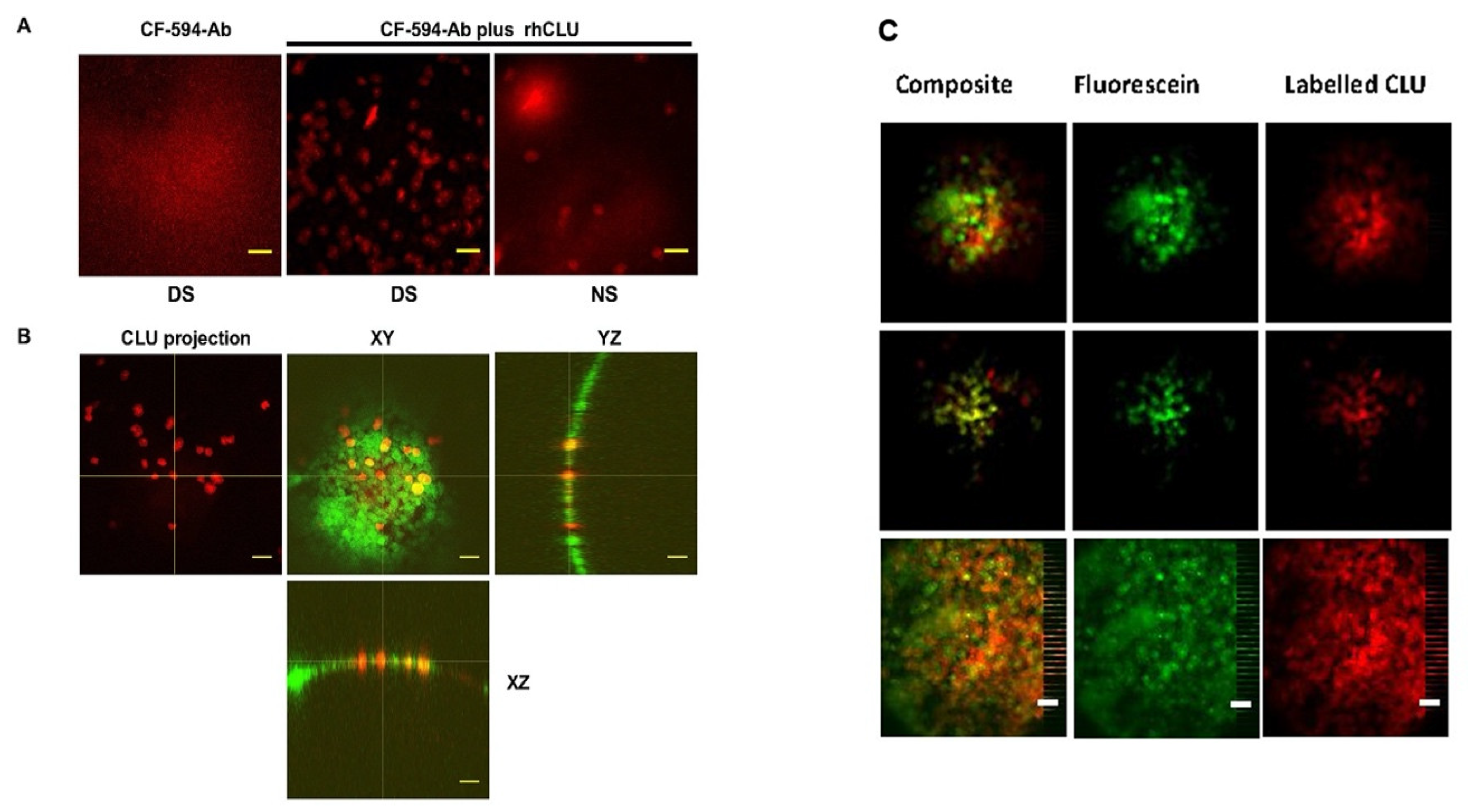
6. Clusterin Binds Selectively to the Damaged Ocular Surface to Protect and Seal
7. Clusterin Dampens the Autoimmune Response
8. Therapeutic Potential of Clusterin
9. Conclusions
10. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AST | autologous serum tears |
| DEWS II | Dry Eye Workshop |
| ECM | extracellular matrix |
| ER | endoplasmic reticulum |
| FDA | Food and Drug Administration |
| GMP | Good Manufacturing Practices |
| MMP | matrix metalloproteinase |
| MGD | Meibomian gland disease |
| TIMP | tissue inhibitors of metalloproteinases |
References
- Gipson, I.K. The Ocular Surface: The Challenge to Enable and Protect Vision: The Friedenwald lecture. Investig. Opthalmol. Vis. Sci. 2007, 48, 4391–4398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argüeso, P.; Guzman-Aranguez, A.; Mantelli, F.; Cao, Z.; Ricciuto, J.; Panjwani, N. Association of Cell Surface Mucins with Galectin-3 Contributes to the Ocular Surface Epithelial Barrier. J. Biol. Chem. 2009, 284, 23037–23045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blalock, T.D.; Spurr-Michaud, S.J.; Tisdale, A.S.; Heimer, S.R.; Gilmore, M.S.; Ramesh, V.; Gipson, I.K. Functions of MUC16 in Corneal Epithelial Cells. Investig. Opthalmol. Vis. Sci. 2007, 48, 4509–4518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gipson, I.K.; Spurr-Michaud, S.; Tisdale, A.; Menon, B.B. Comparison of the Transmembrane Mucins MUC1 and MUC16 in Epithelial Barrier Function. PLoS ONE 2014, 9, e100393. [Google Scholar] [CrossRef]
- Ren, H.; Wilson, G. Apoptosis in the corneal epithelium. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1017–1025. [Google Scholar]
- Hanna, C.; O’Brien, J.E. Cell Production and Migration in the Epithelial Layer of the Cornea. Arch. Ophthalmol. 1960, 64, 536–539. [Google Scholar] [CrossRef]
- Hanna, C.; Bicknell, D.S.; O’Brien, J.E. Cell Turnover in the Adult Human Eye. Arch. Ophthalmol. 1961, 65, 695–698. [Google Scholar] [CrossRef]
- Lee, J.; Koh, J.H.; Kwok, S.K.; Park, S.H. The EULAR Sjogren’s Syndrome Patient-Reported Index is an independent determinant of health-related utility values of Korean patients with primary Sjogren’s syndrome. Clin. Exp. Rheumatol. 2016, 34, 663–667. [Google Scholar]
- Saboo, U.S.; Amparo, F.; Abud, T.B.; Schaumberg, D.A.; Dana, R. Vision-Related Quality of Life in Patients with Ocular Graft-versus-Host Disease. Ophthalmology 2015, 122, 1669–1674. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.-C.; Chai, X.; Inamoto, Y.; Pidala, J.; Martin, P.J.; Flowers, M.E.; Shen, T.T.; Lee, S.J.; Jagasia, M. Impact of Ocular Chronic Graft-versus-Host Disease on Quality of Life. Biol. Blood Marrow Transplant. 2015, 21, 1687–1691. [Google Scholar] [CrossRef] [Green Version]
- TFOS. Report of the International Dry Eye Workshop (DEWS). Ocul. Surf. 2007, 5, 65–204. [Google Scholar]
- Craig, J.P.; Nichols, K.K.; Akpek, E.K.; Caffery, B.; Dua, H.S.; Joo, C.-K.; Liu, Z.; Nelson, J.D.; Nichols, J.J.; Tsubota, K.; et al. TFOS DEWS II Definition and Classification Report. Ocul. Surf. 2017, 15, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Abelson, M.B.; Ingerman, A. The Dye-Namics of Dry-Eye Diagnosis. Rev. Ophthalmol. 2005. Available online: https://www.reviewofophthalmology.com/article/the-dye-namics-of-dry-eye-diagnosis (accessed on 10 November 2020).
- Le, Q.; Ge, L.; Li, M.; Wu, L.; Xu, J.; Hong, J.; Gong, L. Comparison on the vision-related quality of life between outpatients and general population with dry eye syndrome. Acta Ophthalmol. 2014, 92, e124–e132. [Google Scholar] [CrossRef]
- Uchino, M.; Uchino, Y.; Dogru, M.; Kawashima, M.; Yokoi, N.; Komuro, A.; Sonomura, Y.; Kato, H.; Kinoshita, S.; Schaumberg, D.A.; et al. Dry Eye Disease and Work Productivity Loss in Visual Display Users: The Osaka Study. Am. J. Ophthalmol. 2014, 157, 294–300. [Google Scholar] [CrossRef]
- Lemp, M.A. Report of the National Eye Institute/Industry workshop on Clinical Trials in Dry Eyes. CLAO J. 1995, 21, 221–232. [Google Scholar]
- Özcura, F.; Aydin, S.; Helvaci, M.R. Ocular Surface Disease Index for the Diagnosis of Dry Eye Syndrome. Ocul. Immunol. Inflamm. 2007, 15, 389–393. [Google Scholar] [CrossRef]
- Amparo, F.; Schaumberg, D.A.; Dana, R. Comparison of Two Questionnaires for Dry Eye Symptom Assessment: The Ocular Surface Disease Index and the Symptom Assessment in Dry Eye. Ophthalmology 2015, 122, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Barton, K.; Monroy, D.C.; Nava, A.; Pflugfelder, S.C. Inflammatory Cytokines in the Tears of Patients with Ocular Rosacea. Ophthalmology 1997, 104, 1868–1874. [Google Scholar] [CrossRef]
- Yan, C.; Boyd, D.D. Regulation of matrix metalloproteinase gene expression. J. Cell Physiol. 2007, 211, 19–26. [Google Scholar] [CrossRef]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cieplak, P.; Strongin, A.Y. Matrix metalloproteinases - From the cleavage data to the prediction tools and beyond. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1952–1963. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.H. Matrix metalloproteinases: Effectors of development and normal physiology. Genes Dev. 2000, 14, 2123–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brew, K.; Dinakarpandian, D.; Nagase, H. Tissue inhibitors of metalloproteinases: Evolution, structure and function. Biochim. Biophys. Acta BBA Protein Struct. Mol. Enzymol. 2000, 1477, 267–283. [Google Scholar] [CrossRef]
- Bode, W.; Maskos, K. Structural Basis of the Matrix Metalloproteinases and Their Physiological Inhibitors, the Tissue Inhibitors of Metalloproteinases. Biol. Chem. 2003, 384. [Google Scholar] [CrossRef]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef]
- Massova, I.; Kotra, L.P.; Fridman, R.; Mobashery, S. Matrix metalloproteinases: Structures, evolution, and diversification. FASEB J. 1998, 12, 1075–1095. [Google Scholar] [CrossRef] [Green Version]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [CrossRef] [Green Version]
- Woessner, J.F. Matrix Metalloproteinases; Parks, W.C.M., Ed.; Academic Press Inc.: San Diego, CA, USA, 1998; pp. 1–14. [Google Scholar]
- Ra, H.-J.; Parks, W.C. Control of matrix metalloproteinase catalytic activity. Matrix Biol. 2007, 26, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Gill, S.E.; Kassim, S.Y.; Birkland, T.P.; Parks, W.C. Mouse Models of MMP and TIMP Function. Methods Mol. Biol. 2010, 622, 31–52. [Google Scholar] [CrossRef]
- Chen, P.; Parks, W.C. Role of matrix metalloproteinases in epithelial migration. J. Cell. Biochem. 2009, 108, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Fini, M.E.; Girard, M.T. The pattern of metalloproteinase expression by corneal fibroblasts is altered by passage in cell culture. J. Cell Sci. 1990, 97 Pt 2, 373–383. [Google Scholar]
- Fini, M.E.; Girard, M.T. Expression of collagenolytic/gelatinolytic metalloproteinases by normal cornea. Investig. Ophthalmol. Vis. Sci. 1990, 31, 1779–1788. [Google Scholar]
- Girard, M.T.; Matsubara, M.; Fini, M.E. Transforming growth factor-beta and interleukin-1 modulate metalloproteinase expression by corneal stromal cells. Investig. Ophthalmol. Vis. Sci. 1991, 32, 2441–2454. [Google Scholar]
- Matsubara, M.; Girard, M.T.; Kublin, C.L.; Cintron, C.; Fini, M. Differential roles for two gelatinolytic enzymes of the matrix metalloproteinase family in the remodelling cornea. Dev. Biol. 1991, 147, 425–439. [Google Scholar] [CrossRef]
- Matsubara, M.; Zieske, J.D.; Fini, M.E. Mechanism of basement membrane dissolution preceding corneal ulceration. Investig. Ophthalmol. Vis. Sci. 1991, 32, 3221–3237. [Google Scholar]
- Fini, M.E.; Parks, W.C.; Rinehart, W.B.; Girard, M.T.; Matsubara, M.; Cook, J.R.; West-Mays, J.A.; Sadow, P.M.; Burgeson, R.E.; Jeffrey, J.J.; et al. Role of matrix metalloproteinases in failure to re-epithelialize after corneal injury. Am. J. Pathol. 1996, 149, 1287–1302. [Google Scholar]
- Afonso, A.A.; Sobrin, L.; Monroy, D.C.; Selzer, M.; Lokeshwar, B.; Pflugfelder, S.C. Tear fluid gelatinase B activity correlates with IL-1alpha concentration and fluorescein clearance in ocular rosacea. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2506–2512. [Google Scholar]
- Sobrin, L.; Liu, Z.; Monroy, D.C.; Solomon, A.; Selzer, M.G.; Lokeshwar, B.L.; Pflugfelder, S.C. Regulation of MMP-9 activity in human tear fluid and corneal epithelial culture supernatant. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1703–1709. [Google Scholar]
- Solomon, A.; Dursun, D.; Liu, Z.; Xie, Y.; Macri, A.; Pflugfelder, S.C. Pro- and anti-inflammatory forms of interleukin-1 in the tear fluid and conjunctiva of patients with dry-eye disease. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2283–2292. [Google Scholar]
- Pflugfelder, S.C.; Solomon, A.; Dursun, D.; Li, D.-Q. Dry eye and Delayed Tear Clearance: “A Call to Arms”. Adv. Exp. Med. Biol. 2002, 506, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Chotikavanich, S.; De Paiva, C.S.; Li, D.Q.; Chen, J.J.; Bian, F.; Farley, W.J.; Pflugfelder, S.C. Production and Activity of Matrix Metalloproteinase-9 on the Ocular Surface Increase in Dysfunctional Tear Syndrome. Investig. Opthalmol. Vis. Sci. 2009, 50, 3203–3209. [Google Scholar] [CrossRef] [PubMed]
- Dursun, D.; Wang, M.; Monroy, D.; Li, D.Q.; Lokeshwar, B.L.; Stern, M.E. A mouse model of keratoconjunctivitis sicca. Investig. Ophthalmol. Vis. Sci. 2002, 43, 632–638. [Google Scholar]
- Luo, L.; Li, D.-Q.; Doshi, A.; Farley, W.; Corrales, R.M.; Pflugfelder, S. Experimental Dry Eye Stimulates Production of Inflammatory Cytokines and MMP-9 and Activates MAPK Signaling Pathways on the Ocular Surface. Investig. Opthalmol. Vis. Sci. 2004, 45, 4293–4301. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.H.; Shipley, J.M.; Bergers, G.; Berger, J.E.; Helms, J.A.; Hanahan, D.; Shapiro, S.D.; Senior, R.M.; Werb, Z. MMP-9/Gelatinase B Is a Key Regulator of Growth Plate Angiogenesis and Apoptosis of Hypertrophic Chondrocytes. Cell 1998, 93, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Chintala, S.K.; Zhang, X.; Austin, J.S.; Fini, M.E. Deficiency in Matrix Metalloproteinase Gelatinase B (MMP-9) Protects against Retinal Ganglion Cell Death after Optic Nerve Ligation. J. Biol. Chem. 2002, 277, 47461–47468. [Google Scholar] [CrossRef] [Green Version]
- Pflugfelder, S.C.; Farley, W.; Luo, L.; Chen, L.Z.; De Paiva, C.S.; Olmos, L.C.; Li, D.-Q.; Fini, M.E. Matrix Metalloproteinase-9 Knockout Confers Resistance to Corneal Epithelial Barrier Disruption in Experimental Dry Eye. Am. J. Pathol. 2005, 166, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.; Ledee, D.R.; Gordon, G.M.; Itakura, T.; Patel, N.; Martin, A.; Fini, M.E. Interaction of Clusterin and Matrix Metalloproteinase-9 and Its Implication for Epithelial Homeostasis and Inflammation. Am. J. Pathol. 2012, 180, 2028–2039. [Google Scholar] [CrossRef] [Green Version]
- Aronow, B.J.; Lund, S.D.; Brown, T.L.; Harmony, J.A.; Witte, D.P. Apolipoprotein J expression at fluid-tissue interfaces: Potential role in barrier cytoprotection. Proc. Natl. Acad. Sci. USA 1993, 90, 725–729. [Google Scholar] [CrossRef] [Green Version]
- Trougakos, I.P.; Gonos, E.S. Clusterin/Apolipoprotein J in human aging and cancer. Int. J. Biochem. Cell Biol. 2002, 34, 1430–1448. [Google Scholar] [CrossRef]
- Yerbury, J.J.; Poon, S.; Meehan, S.; Thompson, B.; Kumita, J.R.; Dobson, C.M.; Wilson, M.R. The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 2007, 21, 2312–2322. [Google Scholar] [CrossRef] [PubMed]
- Yerbury, J.J.; Stewart, E.M.; Wyatt, A.R.; Wilson, M.R. Quality control of protein folding in extracellular space. EMBO Rep. 2005, 6, 1131–1136. [Google Scholar] [CrossRef] [Green Version]
- Blaschuk, O.; Burdzy, K.; Fritz, I.B. Purification and characterization of a cell-aggregating factor (clusterin), the major glycoprotein in ram rete testis fluid. J. Biol. Chem. 1983, 258, 7714–7720. [Google Scholar] [PubMed]
- Fritz, I.B.; Burdzy, K.; Setchell, B.; Blaschuk, O. Ram rete testis fluid contains a protein (clusterin) which influences cell-cell interactions in vitro. Biol. Reprod. 1983, 28, 1173–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fini, M.E.; Bauskar, A.; Jeong, S.; Wilson, M.R. Clusterin in the eye: An old dog with new tricks at the ocular surface. Exp. Eye Res. 2016, 147, 57–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphreys, D.T.; Carver, J.A.; Easterbrook-Smith, S.B.; Wilson, M.R. Clusterin Has Chaperone-like Activity Similar to That of Small Heat Shock Proteins. J. Biol. Chem. 1999, 274, 6875–6881. [Google Scholar] [CrossRef] [Green Version]
- Pardue, M.L. The heat shock response in biology and human disease: A meeting review. Genes Dev. 1988, 2, 783–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabbs, R.A.; Wyatt, A.R.; Yerbury, J.J.; Ecroyd, H.; Wilson, M.R. Extracellular Chaperones. Top. Curr. Chem. 2011, 328, 241–268. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.R.; Yerbury, J.J.; Ecroyd, H.; Wilson, M.R. Extracellular Chaperones and Proteostasis. Annu. Rev. Biochem. 2013, 82, 295–322. [Google Scholar] [CrossRef] [Green Version]
- Poon, S.; Rybchyn, M.S.; Easterbrook-Smith, S.B.; Carver, J.A.; Pankhurst, G.J.; Wilson, M.R. Mildly Acidic pH Activates the Extracellular Molecular Chaperone Clusterin. J. Biol. Chem. 2002, 277, 39532–39540. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, M.E.; Girton, R.; Finkel, D.; Chmielewski, D.; Barrie, A., 3rd; Witte, D.P.; Zhu, G.; Bissler, J.J.; Harmony, J.A.K.; Aronow, B.J. Apolipoprotein J/clusterin prevents a progressive glomerulopathy of aging. Mol. Cell. Biol. 2002, 22, 1893–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, R.W.; Aronow, B.; Harmony, J.A.; Griswold, M.D. Heat Shock-Initiated Apoptosis Is Accelerated and Removal of Damaged Cells Is Delayed in the Testis of Clusterin/ApoJ Knock-Out Mice1. Biol. Reprod. 2002, 66, 1042–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, L.; Zhu, G.; Mistry, M.; Ley-Ebert, C.; Stuart, W.D.; Florio, C.J.; Groen, P.A.; Witt, S.A.; Kimball, T.R.; Witte, D.P.; et al. Apolipoprotein J/clusterin limits the severity of murine autoimmune myocarditis. J. Clin. Investig. 2000, 106, 1105–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehrli, P.; Charnay, Y.; Vallet, P.; Zhu, G.; Harmony, J.; Aronow, B.; Tschopp, J.; Bouras, C.; Viard-Leveugle, I.; French, L.E.; et al. Inhibition of post-ischemic brain injury by clusterin overexpression. Nat. Med. 2001, 7, 977–978. [Google Scholar] [CrossRef] [PubMed]
- Kounnas, M.Z.; Loukinova, E.B.; Stefansson, S.; Harmony, J.A.K.; Brewer, B.H.; Strickland, D.K.; Argraves, W.S. Identification of Glycoprotein 330 as an Endocytic Receptor for Apolipoprotein J/Clusterin. J. Biol. Chem. 1995, 270, 13070–13075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leeb, C.; Eresheim, C.; Nimpf, J. Clusterin Is a Ligand for Apolipoprotein E Receptor 2 (ApoER2) and Very Low Density Lipoprotein Receptor (VLDLR) and Signals via the Reelin-signaling Pathway. J. Biol. Chem. 2014, 289, 4161–4172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, E.M.; Dangla-Valls, A.; Lovestone, S.; Ribe, E.M.; Buckley, N.J. Clusterin in Alzheimer’s Disease: Mechanisms, Genetics, and Lessons From Other Pathologies. Front. Neurosci. 2019, 13, 164. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, A.; Itoh, Y.; Koshikawa, N.; Akizawa, T.; Yana, I.; Seiki, M. Clusterin, an Abundant Serum Factor, Is a Possible Negative Regulator of MT6-MMP/MMP-25 Produced by Neutrophils. J. Biol. Chem. 2003, 278, 36350–36357. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, A.R.; Constantinescu, P.; Ecroyd, H.; Dobson, C.M.; Wilson, M.R.; Kumita, J.R.; Yerbury, J.J. Protease-activated alpha-2-macroglobulin can inhibit amyloid formation via two distinct mechanisms. FEBS Lett. 2013, 587, 398–403. [Google Scholar] [CrossRef] [Green Version]
- Baker, A.H.; Edwards, D.R.; Murphy, G. Metalloproteinase inhibitors: Biological actions and therapeutic opportunities. J. Cell Sci. 2002, 115 Pt 19, 3719–3727. [Google Scholar] [CrossRef] [Green Version]
- Nishida, K.; Adachi, W.; Shimizu-Matsumoto, A.; Kinoshita, S.; Mizuno, K.; Matsubara, K.; Okubo, K. A gene expression profile of human corneal epithelium and the isolation of human keratin 12 cDNA. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1800–1809. [Google Scholar]
- Nishida, K.; Kawasaki, S.; Adachi, W.; Kinoshita, S. Apolipoprotein J expression in human ocular surface epithelium. Investig. Ophthalmol. Vis. Sci. 1996, 37, 2285–2292. [Google Scholar]
- Li, N.; Wang, N.; Zheng, J.; Liu, X.M.; Lever, O.W.; Erickson, P.M.; Li, L. Characterization of Human Tear Proteome Using Multiple Proteomic Analysis Techniques. J. Proteome Res. 2005, 4, 2052–2061. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhao, S.Z.; Koh, S.K.; Chen, L.; Vaz, C.; Tanavde, V.; Li, X.R.; Beuerman, R.W. In-depth analysis of the human tear proteome. J. Proteom. 2012, 75, 3877–3885. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Nishida, K.; Dota, A.; Kinoshita, S. Changes in conjunctival clusterin expression in severe ocular surface disease. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1702–1707. [Google Scholar]
- Nishida, K.; Kawasaki, S.; Kinoshita, S. Clusterin may be essential for maintaining ocular surface epithelium as a non-keratinizing epithelium. Adv. Exp. Med. Biol. 1998, 438, 629–635. [Google Scholar] [CrossRef]
- Bauskar, A.; Mack, W.J.; Mauris, J.; Argüeso, P.; Heur, M.; Nagel, B.A.; Kolar, G.R.; Gleave, M.E.; Nakamura, T.; Kinoshita, S.; et al. Clusterin Seals the Ocular Surface Barrier in Mouse Dry Eye. PLoS ONE 2015, 10, e0138958. [Google Scholar] [CrossRef] [Green Version]
- Yu, V.; Bhattacharya, D.; Webster, A.; Bauskar, A.; Flowers, C.; Heur, M.; Chintala, S.K.; Itakura, T.; Wilson, M.R.; Barr, J.T.; et al. Clusterin from human clinical tear samples: Positive correlation between tear concentration and Schirmer strip test results. Ocul. Surf. 2018, 16, 478–486. [Google Scholar] [CrossRef]
- Zhou, L.; Beuerman, R.W. Tear analysis in ocular surface diseases. Prog. Retin. Eye Res. 2012, 31, 527–550. [Google Scholar] [CrossRef]
- Mokhtarzadeh, M.; Casey, R.; Glasgow, B.J. Fluorescein Punctate Staining Traced to Superficial Corneal Epithelial Cells by Impression Cytology and Confocal Microscopy. Investig. Opthalmol. Vis. Sci. 2011, 52, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- Bron, A.J.; Argüeso, P.; Irkec, M.; Bright, F. Clinical staining of the ocular surface: Mechanisms and interpretations. Prog. Retin. Eye Res. 2015, 44, 36–61. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, A.R.; Yerbury, J.J.; Wilson, M.R. Structural Characterization of Clusterin-Chaperone Client Protein Complexes. J. Biol. Chem. 2009, 284, 21920–21927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segrest, J.P.; De Loof, H.; Dohlman, J.G.; Brouillette, C.G.; Ananthara-Maiah, G.M. Amphipathic helix motif: Classes and properties. Proteins 1990, 8, 103–117. [Google Scholar] [CrossRef]
- Reddy, S.T.; Anantharamaiah, G.M.; Navab, M.; Hama, S.; Hough, G.; Grijalva, V.; Garber, D.W.; Datta, G.; Fogelman, A.M. Oral amphipathic peptides as therapeutic agents. Expert Opin. Investig. Drugs 2006, 15, 13–21. [Google Scholar] [CrossRef]
- Cunin, P.; Beauvillain, C.; Miot, C.; Augusto, J.-F.; Preisser, L.; Blanchard, S.; Pignon, P.; Scotet, M.; Garo, E.; Fremaux, I.; et al. Clusterin facilitates apoptotic cell clearance and prevents apoptotic cell-induced autoimmune responses. Cell Death Dis. 2016, 7, e2215. [Google Scholar] [CrossRef] [Green Version]
- Baudouin, C. A new approach for better comprehension of diseases of the ocular surface. J. Fr. Ophtalmol. 2007, 30, 239–246. [Google Scholar] [CrossRef]
- Pflugfelder, S.C.; de Paiva, C.S. The Pathophysiology of Dry Eye Disease: What We Know and Future Directions for Research. Ophthalmology 2017, 124, S4–S13. [Google Scholar] [CrossRef]
- Van Lenten, B.J.; Navab, M.; Anantharamaiah, G.; Buga, G.M.; Reddy, S.T.; Fogelman, A.M. Multiple indications for anti-inflammatory apolipoprotein mimetic peptides. Curr. Opin. Investig. Drugs 2008, 9, 1157–1162. [Google Scholar]
- Navab, M.; Anantharamaiah, G.; Reddy, S.T.; Van Lenten, B.J.; Wagner, A.C.; Hama, S.; Hough, G.; Bachini, E.; Garber, D.W.; Mishra, V.K.; et al. An Oral ApoJ Peptide Renders HDL Antiinflammatory in Mice and Monkeys and Dramatically Reduces Atherosclerosis in Apolipoprotein E–Null Mice. Arter. Thromb. Vasc. Biol. 2005, 25, 1932–1937. [Google Scholar] [CrossRef] [Green Version]
- Heller, A.R.; Fiedler, F.; Braun, P.; Stehr, S.N.; Bödeker, H.; Koch, T. Clusterin Protects the Lung from Leukocyte-Induced Injury. Shock 2003, 20, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Newkirk, M.M.; Apostolakos, P.; Neville, C.; Fortin, P.R. Systemic lupus erythematosus, a disease associated with low levels of clusterin/apoJ, an antiinflammatory protein. J. Rheumatol. 1999, 26, 597–603. [Google Scholar] [PubMed]
- Fini, M.E.; Jeong, S.; Gong, H.; Martinez-Carrasco, R.; Laver, N.M.V.; Hijikata, M.; Keicho, N.; Argüeso, P. Membrane-associated mucins of the ocular surface: New genes, new protein functions and new biological roles in human and mouse. Prog. Retin. Eye Res. 2019, 75, 100777. [Google Scholar] [CrossRef]
- Fini, M.E.; Schwartz, S.G.; Gao, X.; Jeong, S.; Patel, N.; Itakura, T.; Price, M.O.; Price, F.W.; Varma, R.; Stamer, W.D. Steroid-induced ocular hypertension/glaucoma: Focus on pharmacogenomics and implications for precision medicine. Prog. Retin. Eye Res. 2017, 56, 58–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, V.L.; Pflugfelder, S.; Zhang, S.; Shojaei, A.; Haque, R. Lifitegrast, a Novel Integrin Antagonist for Treatment of Dry Eye Disease. Ocul. Surf. 2016, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Noble, B.A.; Loh, R.S.K.; MacLennan, S.; Pesudovs, K.; Reynolds, A.; Bridges, L.R.; Burr, J.; Stewart, O.; Quereshi, S. Comparison of autologous serum eye drops with conventional therapy in a randomised controlled crossover trial for ocular surface disease. Br. J. Ophthalmol. 2004, 88, 647–652. [Google Scholar] [CrossRef]
- Urzua, C.A.; Vasquez, D.H.; Huidobro, A.; Hernandez, H.; Alfaro, J. Randomized Double-Blind Clinical Trial of Autologous Serum Versus Artificial Tears in Dry Eye Syndrome. Curr. Eye Res. 2012, 37, 684–688. [Google Scholar] [CrossRef] [PubMed]
- Celebi, H.S.A.R.C.; Ulusoy, C.; Mirza, G.E. The efficacy of autologous serum eye drops for severe dry eye syndrome: A randomized double-blind crossover study. Graefes Arch. Clin. Exp. Ophthalmol. 2014, 252, 619–626. [Google Scholar] [CrossRef]
- Pan, Q.; Angelina, A.; Zambrano, A.; Marrone, M.; Stark, W.J.; Heflin, T.; Tang, L.; Akpek, E.K. Autologous serum eye drops for dry eye. Cochrane Database Syst. Rev. 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Dogru, M.; Tsubota, K. Pharmacotherapy of dry eye. Expert Opin. Pharmacother. 2011, 12, 325–334. [Google Scholar] [CrossRef]
- Tsubota, K.; Goto, E.; Fujita, H.; Ono, M.; Inoue, H.; Saito, I.; Shimmura, S. Treatment of dry eye by autologous serum application in Sjogren’s syndrome. Br. J. Ophthalmol. 1999, 83, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Okamoto, S.; Mori, T.; Yamada, M.; Mashima, Y.; Watanabe, R.; Kuwana, M.; Tsubota, K.; Ikeda, Y.; Oguchi, Y. Autologous serum eye drops for the treatment of severe dry eye in patients with chronic graft-versus-host disease. Bone Marrow Transplant. 2003, 31, 579–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, C.-C.; Lin, J.-M.; Chen, W.-L.; Tsai, Y.-Y. Allogeneic Serum Eye Drops for the Treatment of Severe Dry Eye in Patients with Chronic Graft-Versus-Host Disease. Cornea 2007, 26, 861–863. [Google Scholar] [CrossRef] [PubMed]
- Na, K.-S.; Kim, M.S. Allogeneic serum eye drops for the treatment of dry eye patients with chronic graft-versus-host disease. J. Ocul. Pharmacol. Ther. 2012, 28, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.; Chung, S.-H.; Jeon, S.; Kwok, S.-K.; Park, S.-H.; Kim, M.-S. Comparison of Clinical Efficacies of Autologous Serum Eye Drops in Patients with Primary and Secondary Sjögren Syndrome. Cornea 2014, 33, 663–667. [Google Scholar] [CrossRef]
- Harloff, S.; Hartwig, D.; Kasper, K.; Wedel, T.; Müller, M.; Geerling, G. Epitheliotrophe Kapazität von Serum-Augentropfen gesunder versus immunsupprimierter Patienten mit rheumatoider Arthritis. Klin. Mon. Augenheilkd. 2008, 225, 200–206. [Google Scholar] [CrossRef]
- Hussain, M.; Shtein, R.M.; Sugar, A.; Soong, H.K.; Woodward, M.A.; DeLoss, K.; Mian, S.I. Long-term Use of Autologous Serum 50% Eye Drops for the Treatment of Dry Eye Disease. Cornea 2014, 33, 1245–1251. [Google Scholar] [CrossRef]
- Poon, A.C.; Geerling, G.; Dart, J.K.G.; Fraenkel, G.E.; Daniels, J.T. Autologous serum eyedrops for dry eyes and epithelial defects: Clinical and in vitro toxicity studies. Br. J. Ophthalmol. 2001, 85, 1188–1197. [Google Scholar] [CrossRef] [Green Version]
- McDonnell, P.J.; Schanzlin, D.J.; Rao, N.A. Immunoglobulin Deposition in the Cornea after Application of Autologous Serum. Arch. Ophthalmol. 1988, 106, 1423–1425. [Google Scholar] [CrossRef]
- Ali, T.K.; Gibbons, A.; Cartes, C.; Zarei-Ghanavati, S.; Gomaa, M.; Gonzalez, I.; Gonzalez, A.E.; Ozturk, H.E.; Betancurt, C.; Perez, V.L. Use of Autologous Serum Tears for the Treatment of Ocular Surface Disease From Patients With Systemic Autoimmune Diseases. Am. J. Ophthalmol. 2018, 189, 65–70. [Google Scholar] [CrossRef]
- Rodney, J.; Chien, J.; Ho, R.J.Y. Trends in Translational Medicine and Drug Targeting and Delivery: New Insights on an Old Concept—Targeted Drug Delivery with Antibody–Drug Conjugates for Cancers. J. Pharm. Sci. 2014, 103, 71–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Satapathy, S.; Dabbs, R.A.; Wilson, M.R. Rapid high-yield expression and purification of fully post-translationally modified recombinant clusterin and mutants. Sci. Rep. 2020, 10, 14243. [Google Scholar] [CrossRef] [PubMed]
- Azharuddin, M.; Khandelwal, J.; Datta, H.; Dasgupta, A.K.; Raja, S.O. Dry Eye: A Protein Conformational Disease. Investig. Opthalmol. Vis. Sci. 2015, 56, 1423–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalia, S.K.; Kalia, L.V.; McLean, P.J. Molecular chaperones as rational drug targets for Parkinson’s disease therapeutics. CNS Neurol. Disord. Drug Targets 2010, 9, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Saidi, L.-J.; Wahlster, L. Molecular chaperones and protein folding as therapeutic targets in Parkinson’s disease and other synucleinopathies. Acta Neuropathol. Commun. 2013, 1, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Wiggs, J.L.; Kang, J.H.; Fan, B.; Levkovitch-Verbin, H.; Pasquale, L.R. A Role for Clusterin in Exfoliation Syndrome and Exfoliation Glaucoma? J. Glaucoma 2018, 27, S61–S66. [Google Scholar] [CrossRef]
- Wilson, M.R.; Zoubeidi, A. Clusterin as a therapeutic target. Expert Opin. Ther. Targets 2017, 21, 201–213. [Google Scholar] [CrossRef]
- Vargas, A.; Kim, H.S.; Baral, E.; Yu, W.-Q.; Craft, C.M.; Lee, E.-J. Protective effect of clusterin on rod photoreceptor in rat model of retinitis pigmentosa. PLoS ONE 2017, 12, e0182389. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fini, M.E.; Jeong, S.; Wilson, M.R. Therapeutic Potential of the Molecular Chaperone and Matrix Metalloproteinase Inhibitor Clusterin for Dry Eye. Int. J. Mol. Sci. 2021, 22, 116. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010116
Fini ME, Jeong S, Wilson MR. Therapeutic Potential of the Molecular Chaperone and Matrix Metalloproteinase Inhibitor Clusterin for Dry Eye. International Journal of Molecular Sciences. 2021; 22(1):116. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010116
Chicago/Turabian StyleFini, M. Elizabeth, Shinwu Jeong, and Mark R. Wilson. 2021. "Therapeutic Potential of the Molecular Chaperone and Matrix Metalloproteinase Inhibitor Clusterin for Dry Eye" International Journal of Molecular Sciences 22, no. 1: 116. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010116