Quantitative Proteomic Profiling of Small Molecule Treated Mesenchymal Stem Cells Using Chemical Probes

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Results
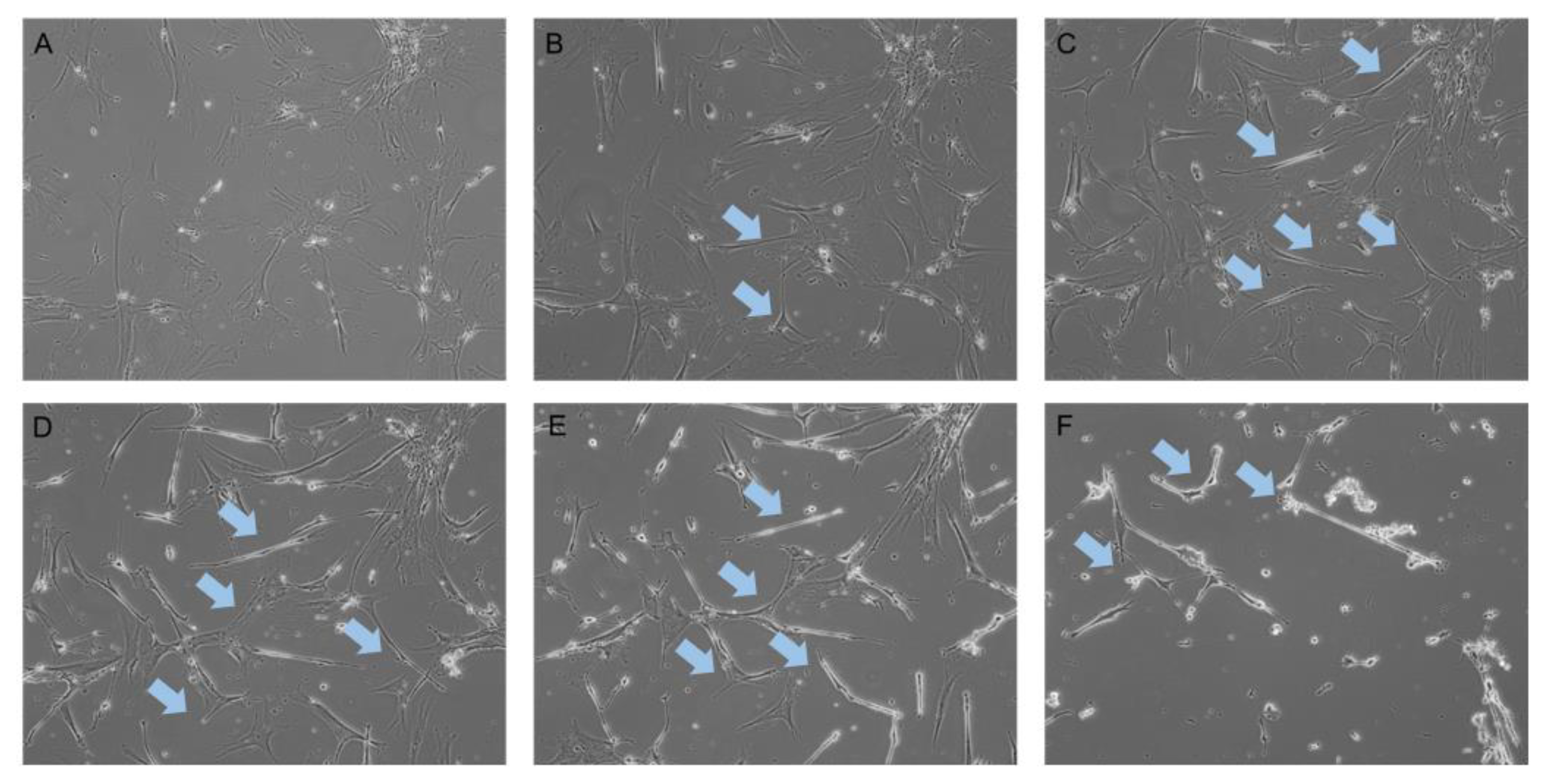
2.1. Neurogenic Differentiation of hADSCs
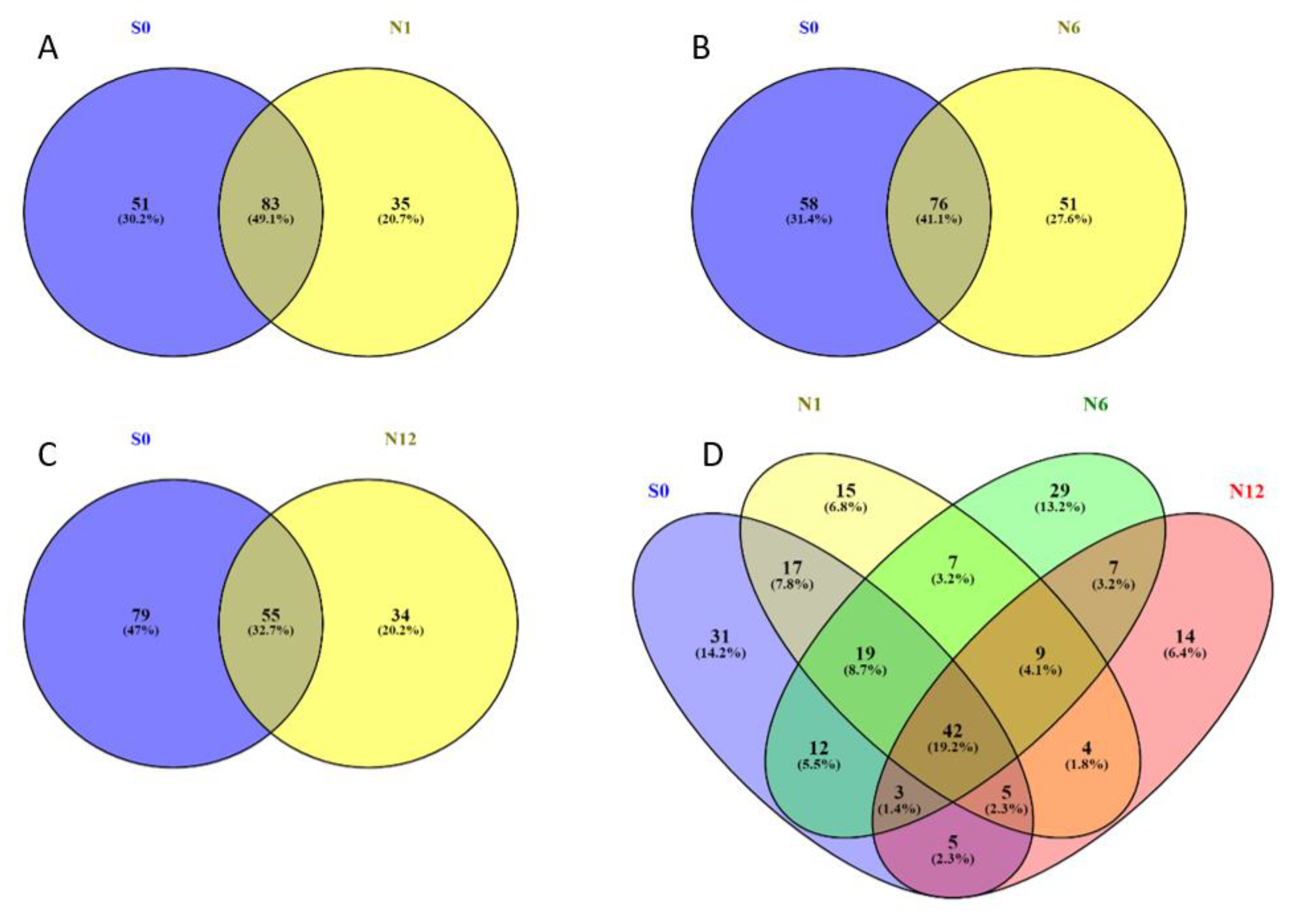
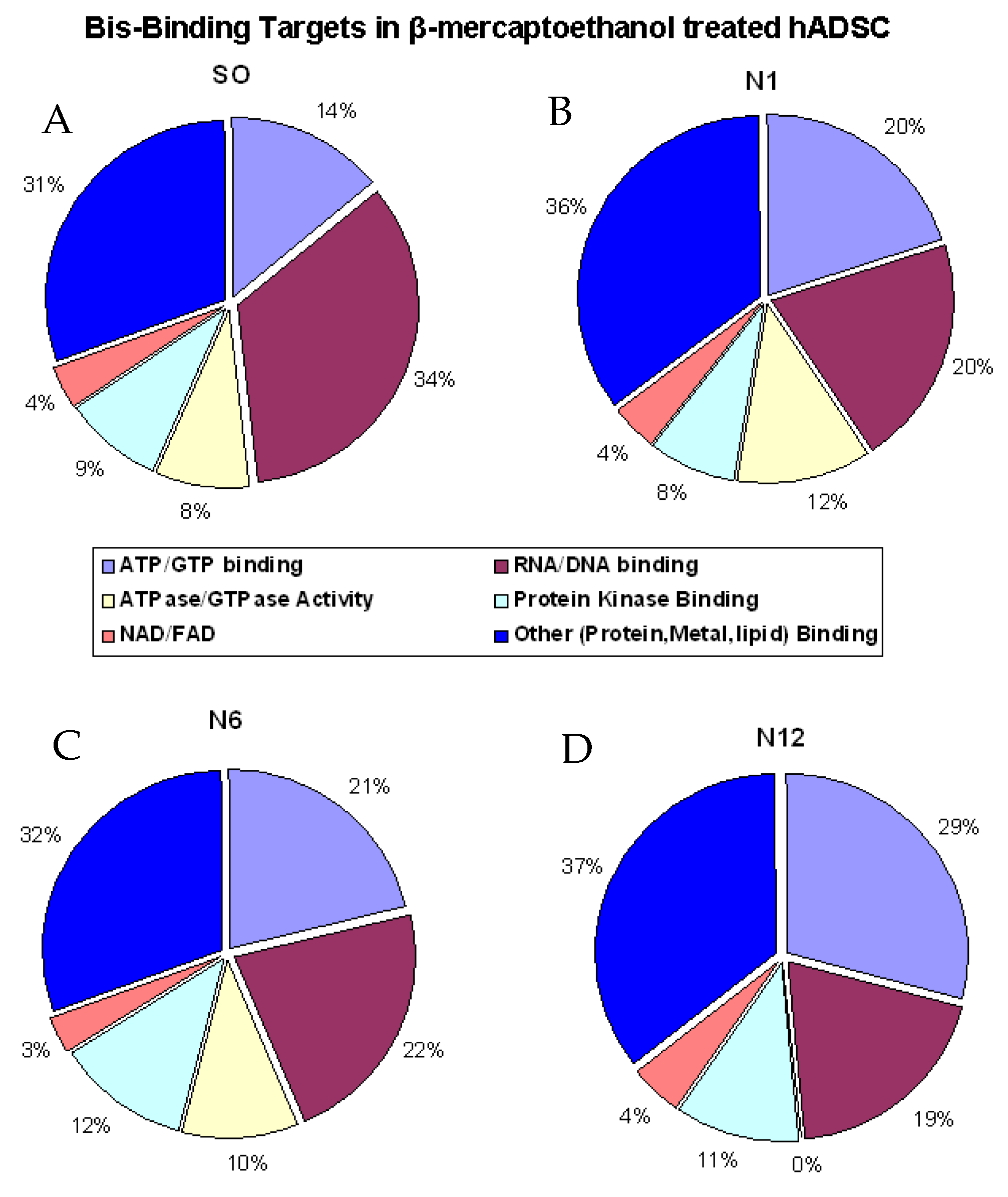
2.2. Bis-Probe Proteome Characterization
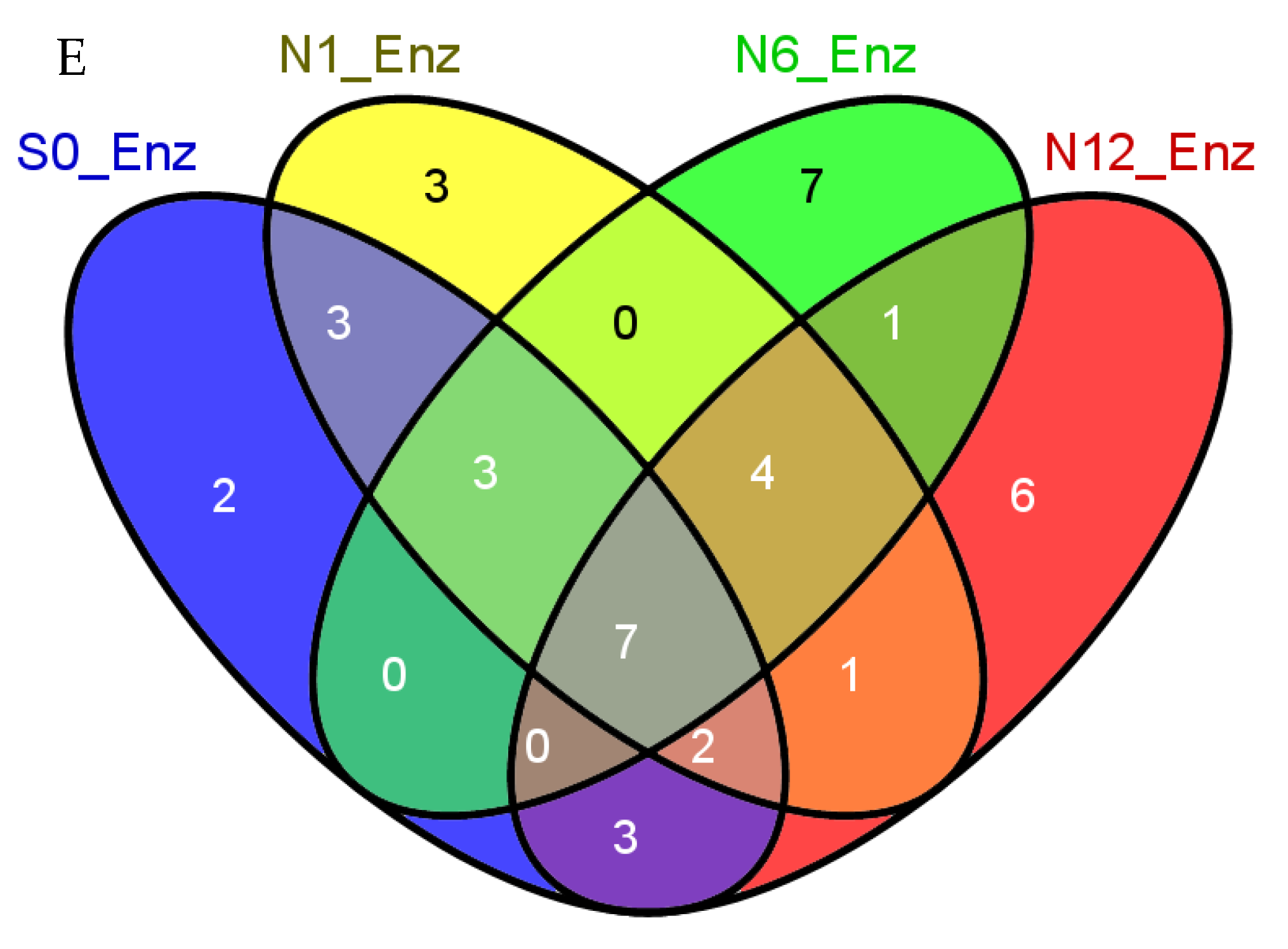
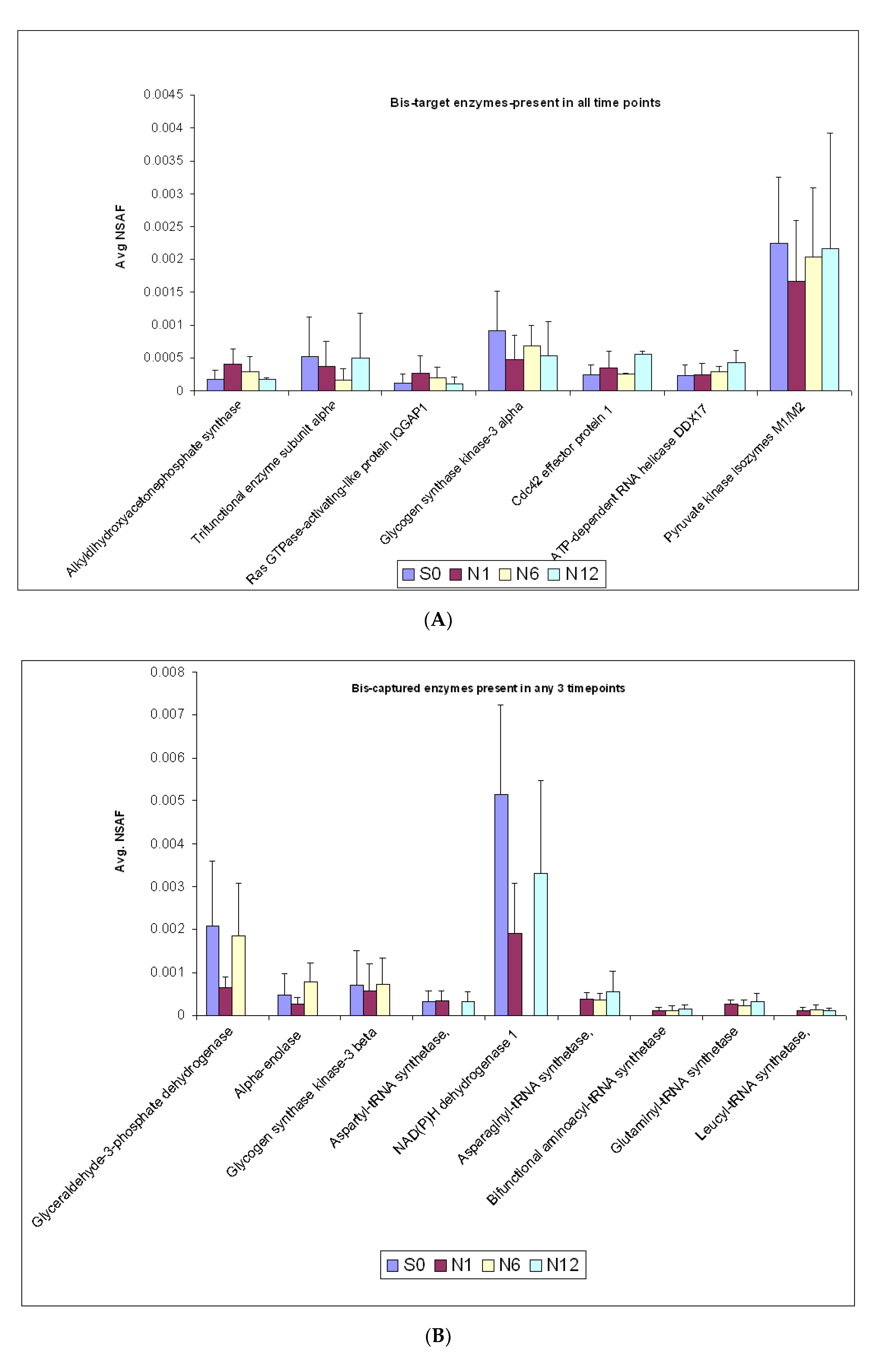
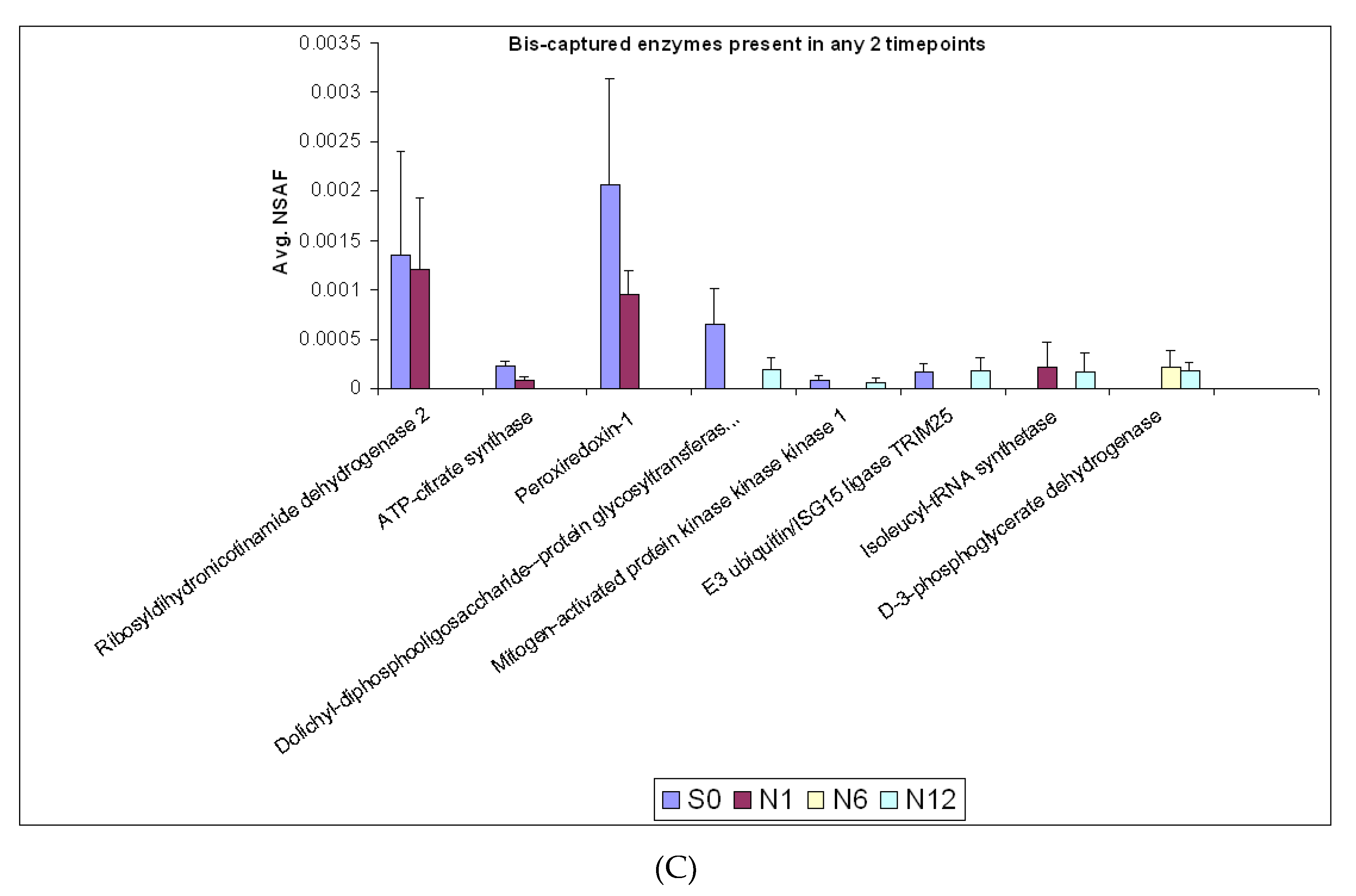
2.3. Enzymes Captured by the Bis-Probe
2.4. Identified Enzymes
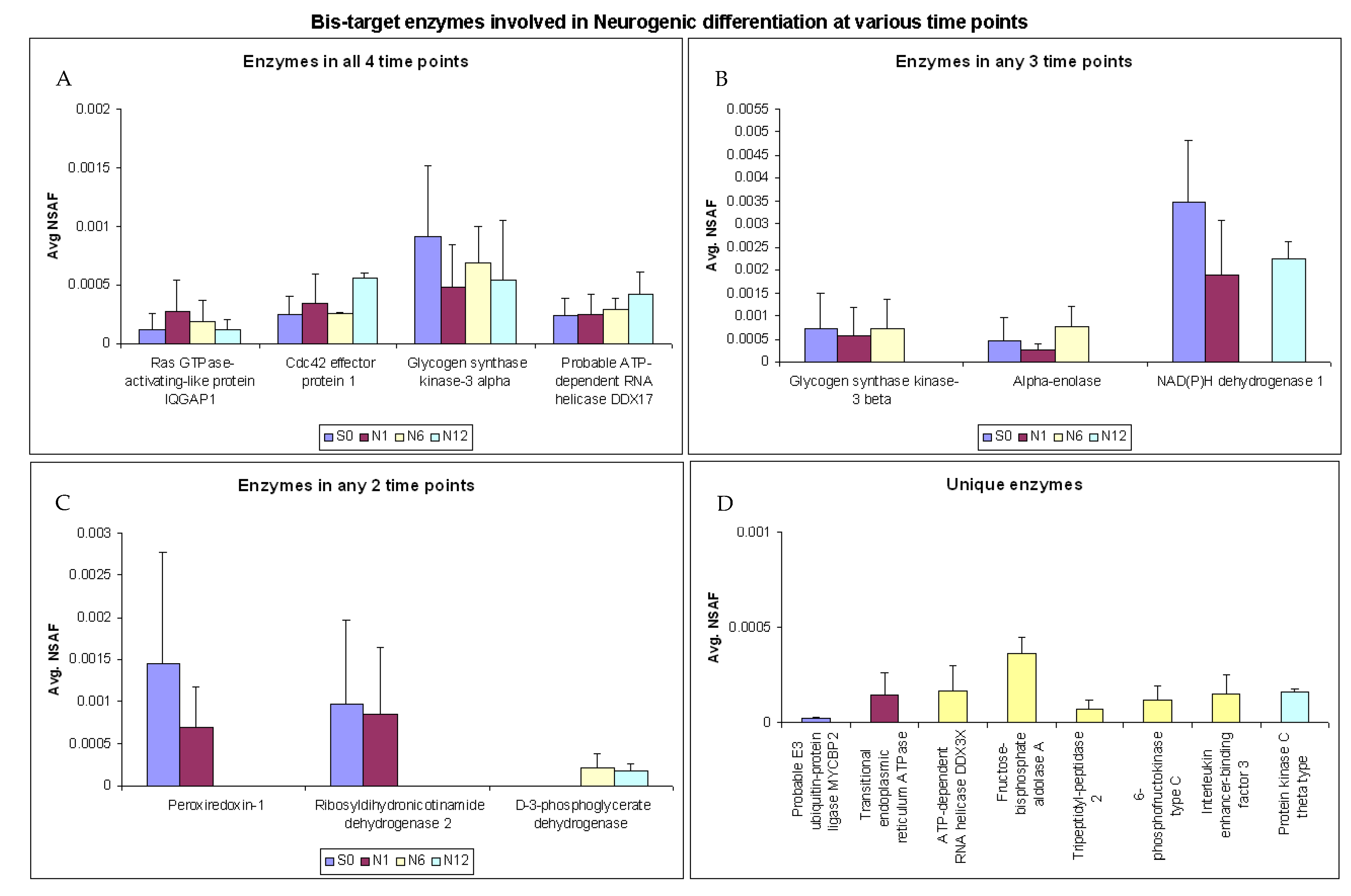
2.5. Enzymes Involved in Neurogenic Differentiation
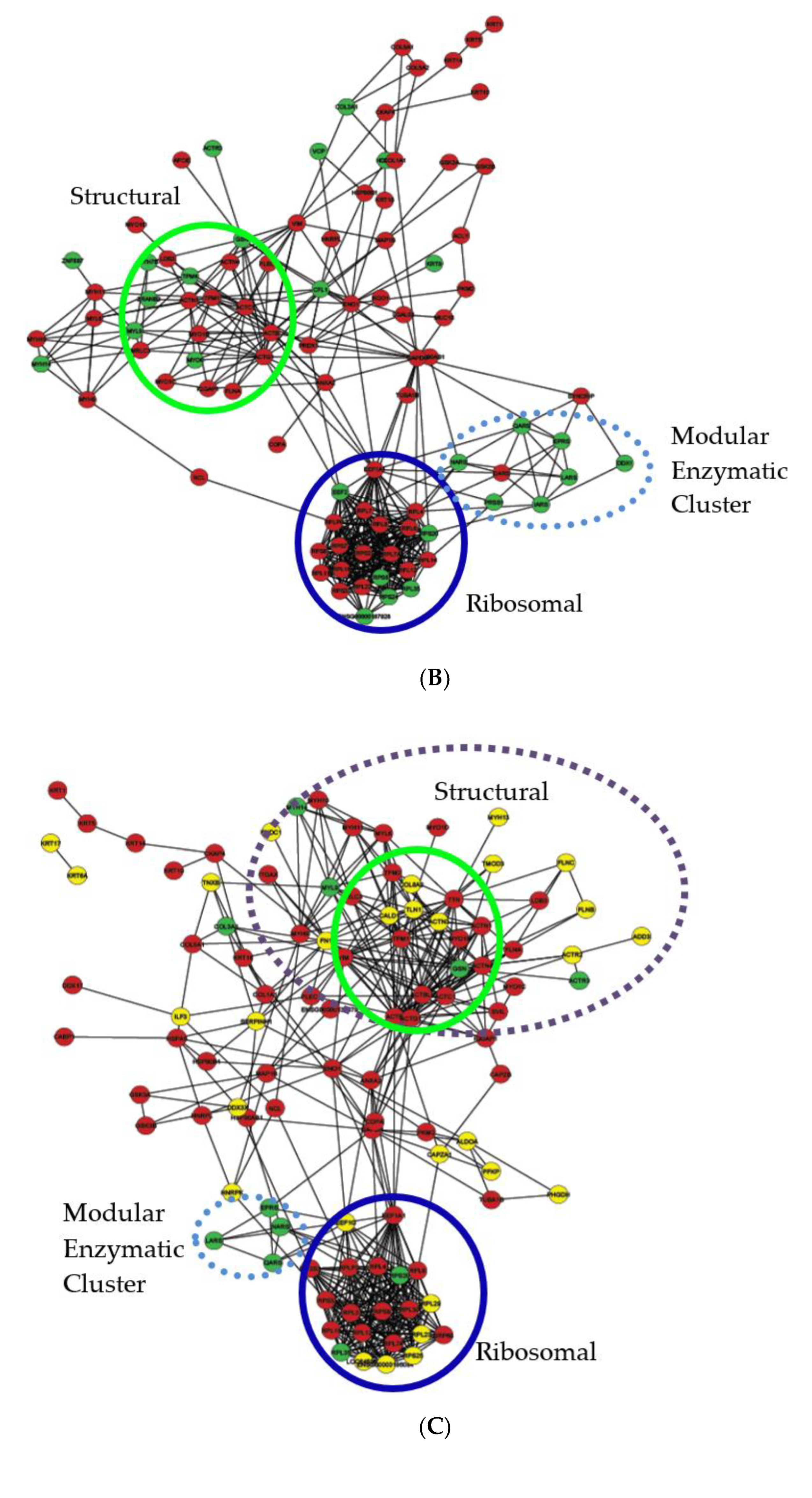
2.6. Protein–Protein Interaction Network Analysis of Bis-Enriched hADSC and Neural Differentiated Proteomes
3. Discussion
4. Methods
4.1. Cell Culture
4.2. Neurogenic Differentiation
4.3. Sample Preparation
4.4. Synthesis of Bis-probe, Affinity Pull down, In-gel Clean up and Digestion
4.5. Mass Spectrometry, Protein Identification and Data Analysis
4.6. Calculation of Protein Normalized Spectral Abundance Factor (NSAF)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BME | Beta-mercaptoethanol |
| DMEM | Delbuccos modified eagle medium |
| FBS | Fetal bovine serum |
| FDR | False discovery rate |
| hADSCs | Human adipose-derived stem cells |
| LTQ XL | Linear Ion trap mass spectrometer |
| MSC | Mesenchymal stem cells |
| NSAF | Normalized spectral abundance factors |
| PEO | Polyethelene oxide |
| PIM | Pre-induction media |
| SVF | Stromal vascular fraction |
References
- Ratanavaraporn, J.; Kanokpanont, S.; Tabata, Y.; Damrongsakkul, S. Growth and osteogenic differentiation of adipose-derived and bone marrow-derived stem cells on chitosan and chitooligosaccharide films. Carbohydr. Polym. 2009, 78, 873–878. [Google Scholar] [CrossRef]
- Conrad, C.; Huss, R. Adult stem cell lines in regenerative medicine and reconstructive surgery. J. Surg. Res. 2005, 124, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A.M.; Beck, S.C.; Murphy, J.M.; Barry, F.P.; Chichester, C.O.; Pittenger, M.F. Chondrogenic Differentiation of Cultured Human Mesenchymal Stem Cells from Marrow. Tissue Eng. 1998, 4, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Vallée, M.; Cote, J.-F.; Fradette, J. Adipose-tissue engineering: Taking advantage of the properties of human adipose-derived stem/stromal cells. Pathol. Biol. 2009, 57, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Woodbury, D.; Schwarz, E.J.; Prockop, D.J.; Black, I.B. Adult rat and human bone marrow stromal cells differentiate into neurons. J. Neurosci. Res. 2000, 61, 364–370. [Google Scholar] [CrossRef]
- Fujimura, J.; Ogawa, R.; Mizuno, H.; Fukunaga, Y.; Suzuki, H. Neural differentiation of adipose-derived stem cells isolated from GFP transgenic mice. Biochem. Biophys. Res. Commun. 2005, 333, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Zuk, P.A.; Min, Z.; Peter, A.; Daniel, A.U.; Jerry, I.H.; Hiroshi, M.; Zeni, C.A.; John, K.F.; et al. Human Adipose Tissue Is a Source of Multipotent Stem Cells. Mol. Biol. Cell 2002, 13, 4279–4295. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.; Milthorpe, B.K.; Herbert, B.R.; Padula, M.P. Proteomic Analysis of Human Adipose Derived Stem Cells during Small Molecule Chemical Stimulated Pre-neuronal Differentiation. Int. J. Stem Cells 2017, 10, 193–217. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Ramos, J.R.; Shijie, S.S.; Kamath, G.; Tanja, Z.; Alison, W.; Fernando, C.; Todd, S.; Chopp, M.; Sanberg, P.R. Expression of Neural Markers in Human Umbilical Cord Blood. Exp. Neurol. 2001, 171, 109–115. [Google Scholar] [CrossRef]
- Barnabé, G.F.; Schwindt, T.T.; Calcagnotto, M.E.; Motta, F.L.; De Oliveira, A.C.; Keim, L.M.N.; D’Almeida, V.; Mendez-Otero, R.; Mello, L.E. Chemically-Induced RAT Mesenchymal Stem Cells Adopt Molecular Properties of Neuronal-Like Cells but Do Not Have Basic Neuronal Functional Properties. PLoS ONE 2009, 4, e5222. [Google Scholar] [CrossRef] [Green Version]
- Radtke, C.; Schmitz, B.; Spies, M.; Kocsis, J.; Vogt, P. Peripheral glial cell differentiation from neurospheres derived from adipose mesenchymal stem cells. Int. J. Dev. Neurosci. 2009, 27, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Medrano, S.; Burns-Cusato, M.; Atienza, M.B.; Rahimi, D.; Scrable, H. Regenerative capacity of neural precursors in the adult mammalian brain is under the control of p53. Neurobiol. Aging 2009, 30, 483–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.-M.; Fu, Y.-M.; Shan, Z.; Shen, J.-L.; Zhang, X.-M.; Lei, L.; Jin, L.-H. MSCs guide neurite directional extension and promote oligodendrogenesis in NSCs. Biochem. Biophys. Res. Commun. 2009, 384, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Konishi, M.; Asaki, T.; Furukawa, N.; Ukai, K.; Mori, M.; Hirasawa, A.; Tsujimoto, G.; Ohta, M.; Itoh, N.; et al. Neudesin, an extracellular heme-binding protein, suppresses adipogenesis in 3T3-L1 cells via the MAPK cascade. Biochem. Biophys. Res. Commun. 2009, 381, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.P.F.; Zandonai, A.F.; Bonatto, D.; Machado, D.C.; Henriques, J.A.P. Differentiation of human adipose-derived adult stem cells into neuronal tissue: Does it work? Differentiation 2009, 77, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, L.; Li, Y.; Zhou, C.; Xiong, F.; Liu, Z.; Gu, R.; Hou, X.; Zhang, C. Myelin-forming ability of Schwann cell-like cells induced from rat adipose-derived stem cells in vitro. Brain Res. 2008, 1239, 49–55. [Google Scholar] [CrossRef]
- Gunhanlar, N.; Shpak, G.; Van Der Kroeg, M.; Gouty-Colomer, L.; Munshi, S.T.; Lendemeijer, B.; Ghazvini, M.; Dupont, C.; Hoogendijk, W.J.G.; Gribnau, J.; et al. A simplified protocol for differentiation of electrophysiologically mature neuronal networks from human induced pluripotent stem cells. Mol. Psychiatry 2017, 23, 1336–1344. [Google Scholar] [CrossRef] [Green Version]
- Al-Shammari, A.; Mohammad, M.; Al-Juboory, A.A.; Yaseen, N.Y. Characterization of neural stemness status through the neurogenesis process for bone marrow mesenchymal stem cells. Stem Cells Cloning 2016, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, A.; Yasuhara, R.; Akino, R.; Mishima, K.; Nasu, M.; Sekizawa, A. Engraftment potential of maternal adipose-derived stem cells for fetal transplantation. Heliyon 2020, 6, e03409. [Google Scholar] [CrossRef]
- Jackson-Holmes, E.L.; Schaefer, A.W.; McDevitt, T.C.; Lu, H. Microfluidic perfusion modulates growth and motor neuron differentiation of stem cell aggregates. Analysis 2020, 145, 4815–4826. [Google Scholar] [CrossRef]
- Dolai, S.; Xu, Q.; Liu, F.; Molloy, M.P. Quantitative chemical proteomics in small-scale culture of phorbol ester stimulated basal breast cancer cells. Proteomics 2011, 11, 2683–2692. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Kamada, T.; Kawai, S. Advanced graphics for visualization of shielding relations Computer Vision, Graph, Image Process. Vis. Comput. 1988, 43, 294–312. [Google Scholar]
- Eades, P. Springs Algorithms and symmetry. In Proceedings of the Congressus Numerantium, International Computing and Combinatorics Conference, New York, NY, USA, 1984; pp. 149–160. [Google Scholar]
- Swanson, K.D. Transcription Factor Phosphorylation by pp90 rsk2. Identificaiton of Fos kinase and NGF-B kineas as pp90 rsk2. J. Biol. Chem. 1999, 274, 3385–3395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, C.; Zhen, E.; Higgs, R.E.; Hale, J.E. An Immuno-Chemo-Proteomics Method for Drug Target Deconvolution. J. Proteome Res. 2008, 7, 3490–3497. [Google Scholar] [CrossRef] [PubMed]
- Brehmer, D.; Godl, K.; Zech, B.; Wissing, J.; Daub, H. Proteome-wide Identification of Cellular Targets Affected by Bisindolylmaleimide-type Protein Kinase C Inhibitors. Mol. Cell. Proteom. 2004, 3, 490–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, S.-E.; Schenone, M.; Margolin, A.A.; Li, X.; Do, K.; Doud, M.K.; Mani, D.R.; Kuai, L.; Wang, X.; Wood, J.L.; et al. Identifying the proteins to which small-molecule probes and drugs bind in cells. Proc. Natl. Acad. Sci. USA 2009, 106, 4617–4622. [Google Scholar] [CrossRef] [Green Version]
- Wissing, J.; Godl, K.; Brehmer, D.; Blencke, S.; Weber, M.; Habenberger, P.; Stein-Gerlach, M.; Missio, A.; Cotten, M.; Müller, S.; et al. Chemical Proteomic Analysis Reveals Alternative Modes of Action for Pyrido[2,3-d]pyrimidine Kinase Inhibitors. Mol. Cell. Proteom. 2004, 3, 1181–1193. [Google Scholar] [CrossRef] [Green Version]
- Wissing, J.; Jänsch, L.; Nimtz, M.; Dieterich, G.; Hornberger, R.; Kéri, G.; Wehland, J.; Daub, H. Proteomics Analysis of Protein Kinases by Target Class-selective Prefractionation and Tandem Mass Spectrometry. Mol. Cell. Proteom. 2006, 6, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.; Rameshwar, P.; Sadoshima, J. Distinct roles of glycogen synthase kinase (GSK)-3alpha and GSK-3beta in mediating cardiomyocyte differentiation in murine bone marrow-derived mesenchymal stem cells. J. Biol. Chem. 2009, 284, 36647–36658. [Google Scholar] [CrossRef] [Green Version]
- Daub, H.; Olsen, J.V.; Bairlein, M.; Gnad, F.; Oppermann, F.S.; Körner, R.; Greff, Z.; Kéri, G.; Stemmann, O.; Mann, M. Kinase-Selective Enrichment Enables Quantitative Phosphoproteomics of the Kinome across the Cell Cycle. Mol. Cell 2008, 31, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Lie, D.C.; Colamarino, S.A.; Song, H.-J.; Désiré, L.; Mira, H.; Consiglio, A.; Lein, E.S.; Jessberger, S.; Lansford, H.; Dearie, A.R.; et al. Wnt signalling regulates adult hippocampal neurogenesis. Nature 2005, 437, 1370–1375. [Google Scholar] [CrossRef] [PubMed]
- Ronowska, A.; Andrzej, S.; Hanna, B.; Sylwia, G.-H.; Joanna, K.L.; Aleksandra, D.; Marlena, Z.; Agnieszka, J.K. The Regulatory Effects of Acetyl-CoA Distribution in the Healthy and Diseased Brain. Front. Cell. Neurosci. 2018, 12, 169. [Google Scholar] [CrossRef] [Green Version]
- Maurer, M.H.; Brömme, J.O.; Feldmann, R.E.; Järve, A.; Sabouri, F.; Bürgers, H.F.; Schelshorn, D.W.; Krüger, C.; Schneider, A.; Kuschinsky, W. Glycogen Synthase Kinase 3β (GSK3β) Regulates Differentiation and Proliferation in Neural Stem Cells from the Rat Subventricular Zone. J. Proteome Res. 2007, 6, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Sparatore, B.; Patrone, M.; Passalacqua, M.; Pedrazzi, M.; Pontremoli, S.; Melloni, E. Neuronal Differentiation of PC12 Cells Involves Changes in Protein Kinase C-θ Distribution and Molecular Properties. Biochem. Biophys. Res. Commun. 2000, 275, 149–153. [Google Scholar] [CrossRef]
- Tsutsumi, H.; Tani, K.; Fujii, H.; Miwa, S. Expression of L- and M-type pyruvate kinase in human tissues. Genomes 1988, 2, 86–89. [Google Scholar] [CrossRef]
- Todd, A.G.; Robert, M.; Debra, J.S.; Samuel, M.; Howard, S.; Philip, J.Y. SMN, Gemin2 and Gemin3 Associate with [beta]-Actin mRNA in the Cytoplasm of Neuronal Cells In Vitro. J. Mol. Biol. 2010, 401, 681–689. [Google Scholar] [CrossRef]
- Zhu, M.; Yang, T.; Wei, S.; De Wan, A.T.; Morell, R.J.; Elfenbein, J.L.; Fisher, R.A.; Leal, S.M.; Smith, R.J.H.; Friderici, K.H. Mutations in the Gamma-Actin Gene (ACTG1) Are Associated with Dominant Progressive Deafness (DFNA20/26). Am. J. Hum. Genet. 2003, 73, 1082–1091. [Google Scholar] [CrossRef] [Green Version]
- Kakita, A.; Hayashi, S.; Moro, F.; Guerrini, R.; Ozawa, T.; Ono, K.; Kameyama, S.A.; Walsh, C.; Takahashi, H. Bilateral periventricular nodular heterotopia due to filamin 1 gene mutation: widespread glomeruloid microvascular anomaly and dysplastic cytoarchitecture in the cerebral cortex. Acta Neuropathol. 2002, 104, 649–657. [Google Scholar] [CrossRef]
- Schultz, A.; Jonsson, J.I.; Larsson, C. The regulatory domain of protein kinase Ctheta localises to the Golgi complex and induces apoptosis in neuroblastoma and Jurkat cells. Cell Death Differ. 2003, 10, 662–675. [Google Scholar] [CrossRef]
- Briggs, M.W.; Sacks, D.B. IQGAP proteins are integral components of cytoskeletal regulation. EMBO Rep. 2003, 4, 571–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swart-Mataraza, J.M.; Li, Z.; Sacks, D.B. IQGAP1 Is a Component of Cdc42 Signaling to the Cytoskeleton. J. Biol. Chem. 2002, 277, 24753–24763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; McNulty, D.E.; Marler, K.J.M.; Lim, L.; Hall, C.; Annan, R.S.; Sacks, D.B. IQGAP1 Promotes Neurite Outgrowth in a Phosphorylation-dependent Manner. J. Biol. Chem. 2005, 280, 13871–13878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, J.S.; Dotti, C.G. Breaking the neuronal sphere: regulation of the actin cytoskeleton in neuritogenesis. Nat. Rev. Neurosci. 2002, 3, 694–704. [Google Scholar] [CrossRef]
- Helfand, B.T.; Mendez, M.G.; Pugh, J.; Delsert, C.; Goldman, R.D. A Role for Intermediate Filaments in Determining and Maintaining the Shape of Nerve Cells V. Mol. Biol. Cell 2003, 14, 5069–5081. [Google Scholar] [CrossRef]
- Bashour, A.-M.; Fullerton, A.T.; Hart, M.J.; Bloom, G.S. IQGAP1, a Rac- and Cdc42-binding Protein, Directly Binds and Cross-links Microfilaments. J. Cell Biol. 1997, 137, 1555–1566. [Google Scholar] [CrossRef] [Green Version]
- Erickson, J.W.; Cerione, R.A.; Hart, M.J. Identification of an Actin Cytoskeletal Complex That Includes IQGAP and the Cdc42 GTPase. J. Biol. Chem. 1997, 272, 24443–24447. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Wu, T.; Lau, A.; Jiang, T.; Huang, Z.; Wang, X.-J.; Chen, W.; Wong, P.K.; Zhang, D.D. Nrf2 promotes neuronal cell differentiation. Free. Radic. Biol. Med. 2009, 47, 867–879. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Blesch, A.; Tuszynski, M.H. Induction of bone marrow stromal cells to neurons: Differentiation, transdifferentiation, or artifact? J. Neurosci. Res. 2004, 77, 174–191. [Google Scholar] [CrossRef]
- Yamasaki, M.; Yamada, K.; Furuya, S.; Mitoma, J.; Hirabayashi, Y.; Watanabe, M. 3-Phosphoglycerate Dehydrogenase, a Key Enzyme forl-Serine Biosynthesis, Is Preferentially Expressed in the Radial Glia/Astrocyte Lineage and Olfactory Ensheathing Glia in the Mouse Brain. J. Neurosci. 2001, 21, 7691–7704. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.; Sankaran, B.; Poole, L.B.; Karplus, P.A. Structural Changes Common to Catalysis in the Tpx Peroxiredoxin Subfamily. J. Mol. Biol. 2009, 393, 867–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, H.H.; Kyun, O.L.; Yong, H.C.; Bae, G.J.; Soo, K.P.; Jin, H.P.; Jung, R.L.; Seung, S.L. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell 2004, 117, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Sabharwal, P.; Rao, M.; Sockanathan, S. The Antioxidant Enzyme Prdx1 Controls Neuronal Differentiation by Thiol-Redox-Dependent Activation of GDE2. Cell 2009, 138, 1209–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beavis, R.C. Using the Global Proteome Machine for Protein Identification. New Emerg. Proteomic Technol. 2006, 328, 217–228. [Google Scholar] [CrossRef]
- Zybailov, B.L.; Florens, L.; Washburn, M.P. Quantitative shotgun proteomics using a protease with broad specificity and normalized spectral abundance factors. Mol. BioSyst. 2007, 3, 354–360. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Number of Proteins | Protein FDR | Peptide FDR |
|---|---|---|---|
| S0 | 133 | 0.11949686 | 0.0082224 |
| N1 | 118 | 0.17567568 | 0.0106044 |
| N6 | 127 | 0.15584416 | 0.0076948 |
| N12 | 89 | 0.11949686 | 0.0082224 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, J.; Dolai, S.; O’Rourke, M.B.; Liu, F.; Padula, M.P.; Molloy, M.P.; Milthorpe, B.K. Quantitative Proteomic Profiling of Small Molecule Treated Mesenchymal Stem Cells Using Chemical Probes. Int. J. Mol. Sci. 2021, 22, 160. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010160
Santos J, Dolai S, O’Rourke MB, Liu F, Padula MP, Molloy MP, Milthorpe BK. Quantitative Proteomic Profiling of Small Molecule Treated Mesenchymal Stem Cells Using Chemical Probes. International Journal of Molecular Sciences. 2021; 22(1):160. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010160
Chicago/Turabian StyleSantos, Jerran, Sibasish Dolai, Matthew B. O’Rourke, Fei Liu, Matthew P. Padula, Mark P. Molloy, and Bruce K. Milthorpe. 2021. "Quantitative Proteomic Profiling of Small Molecule Treated Mesenchymal Stem Cells Using Chemical Probes" International Journal of Molecular Sciences 22, no. 1: 160. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010160