Amino Acid Transporters Are a Vital Focal Point in the Control of mTORC1 Signaling and Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
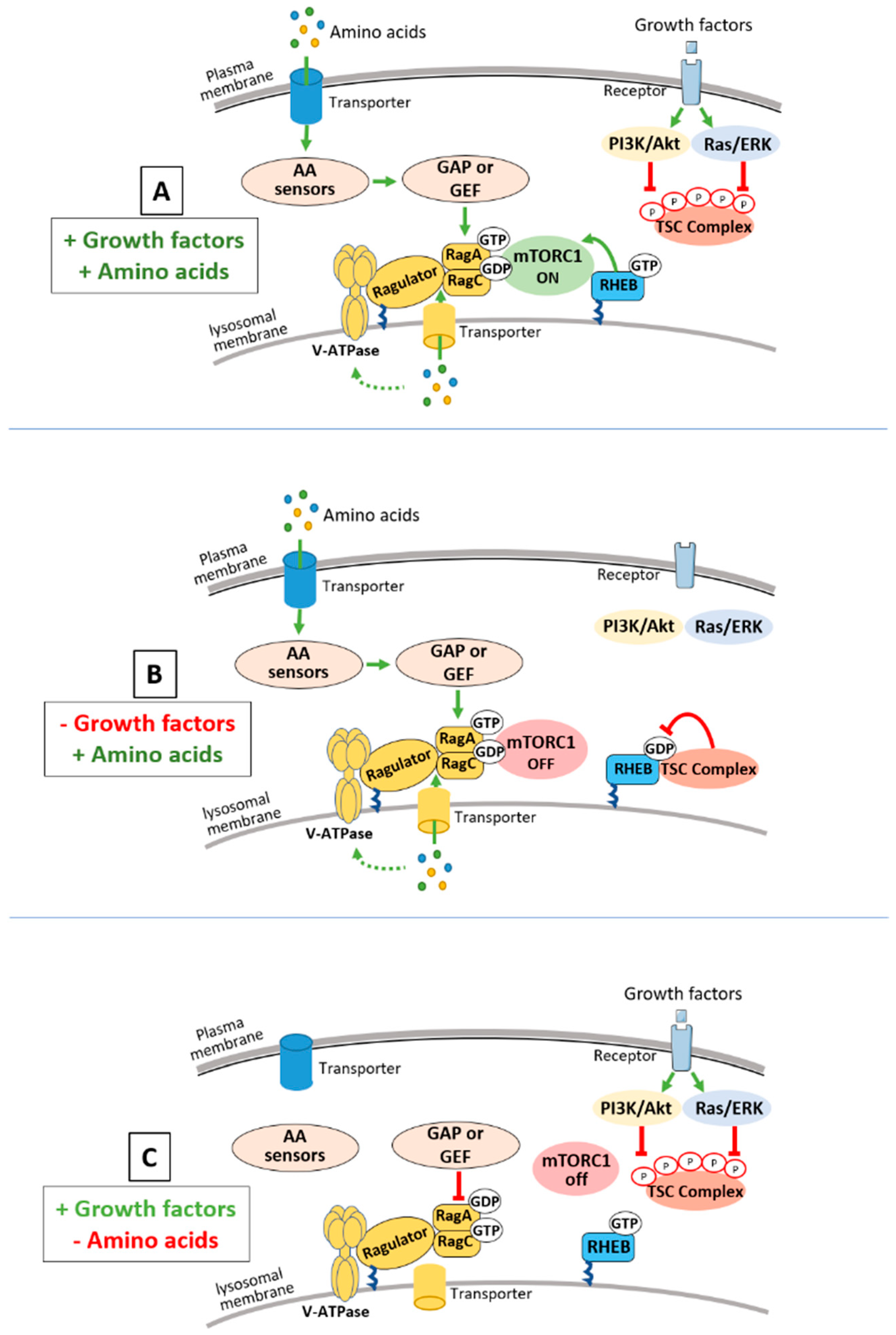
:1. Introduction: mTORC1 and Nutrient Sensing
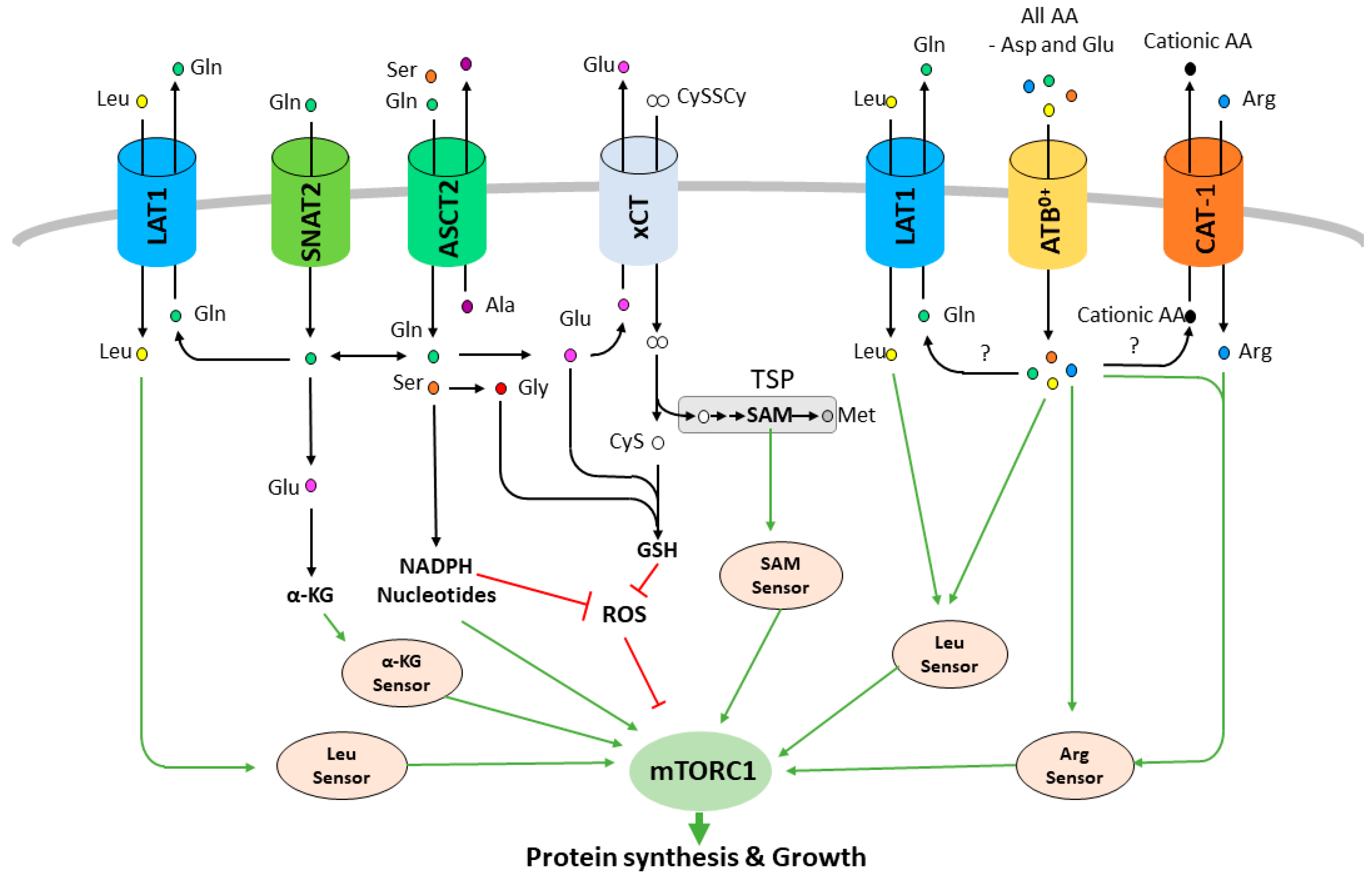
2. Plasma Membrane AA Transporters Regulate mTORC1 Activity and Are Promising Therapeutic Targets
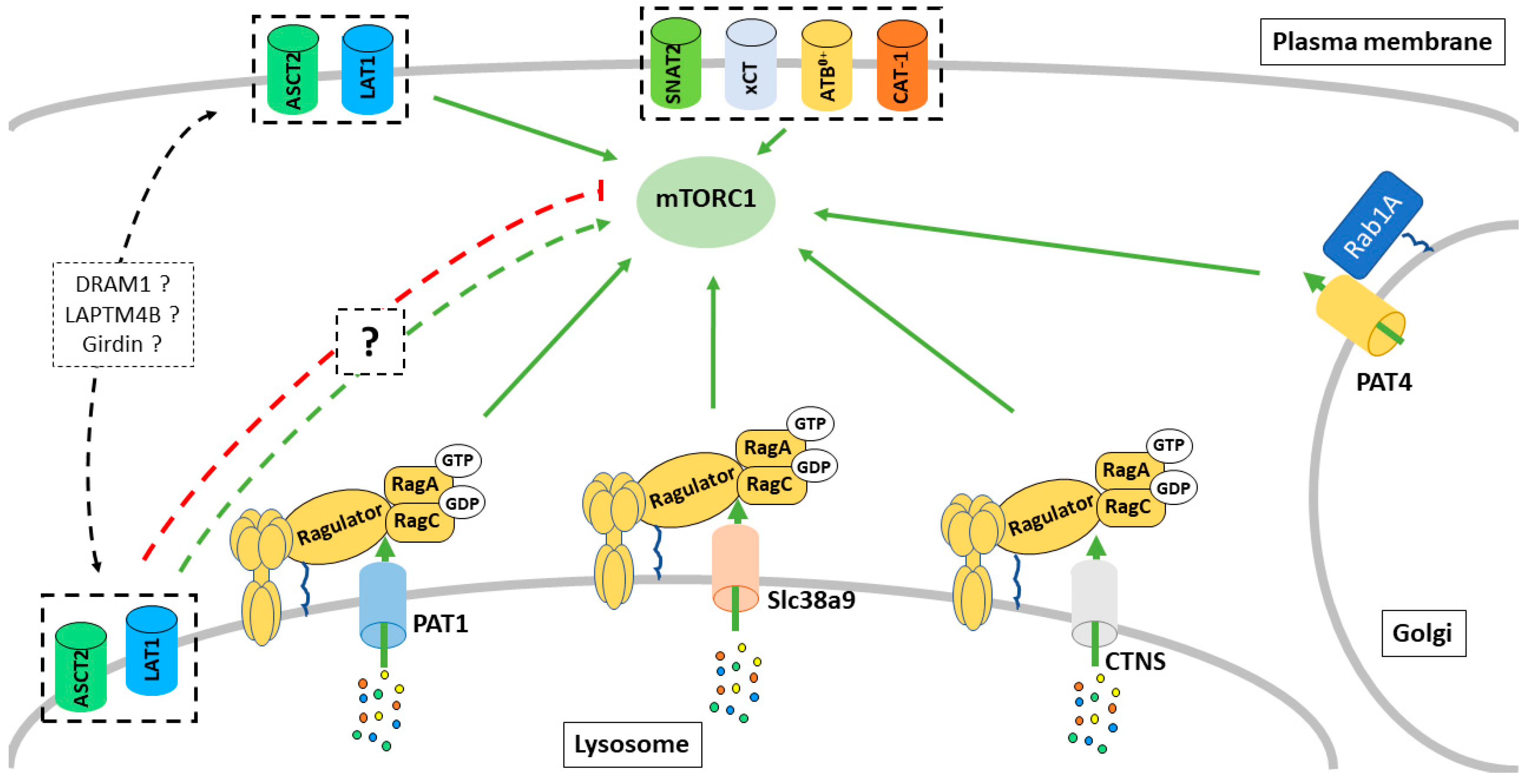
3. Intracellular Transporters Regulate mTORC1 Activity
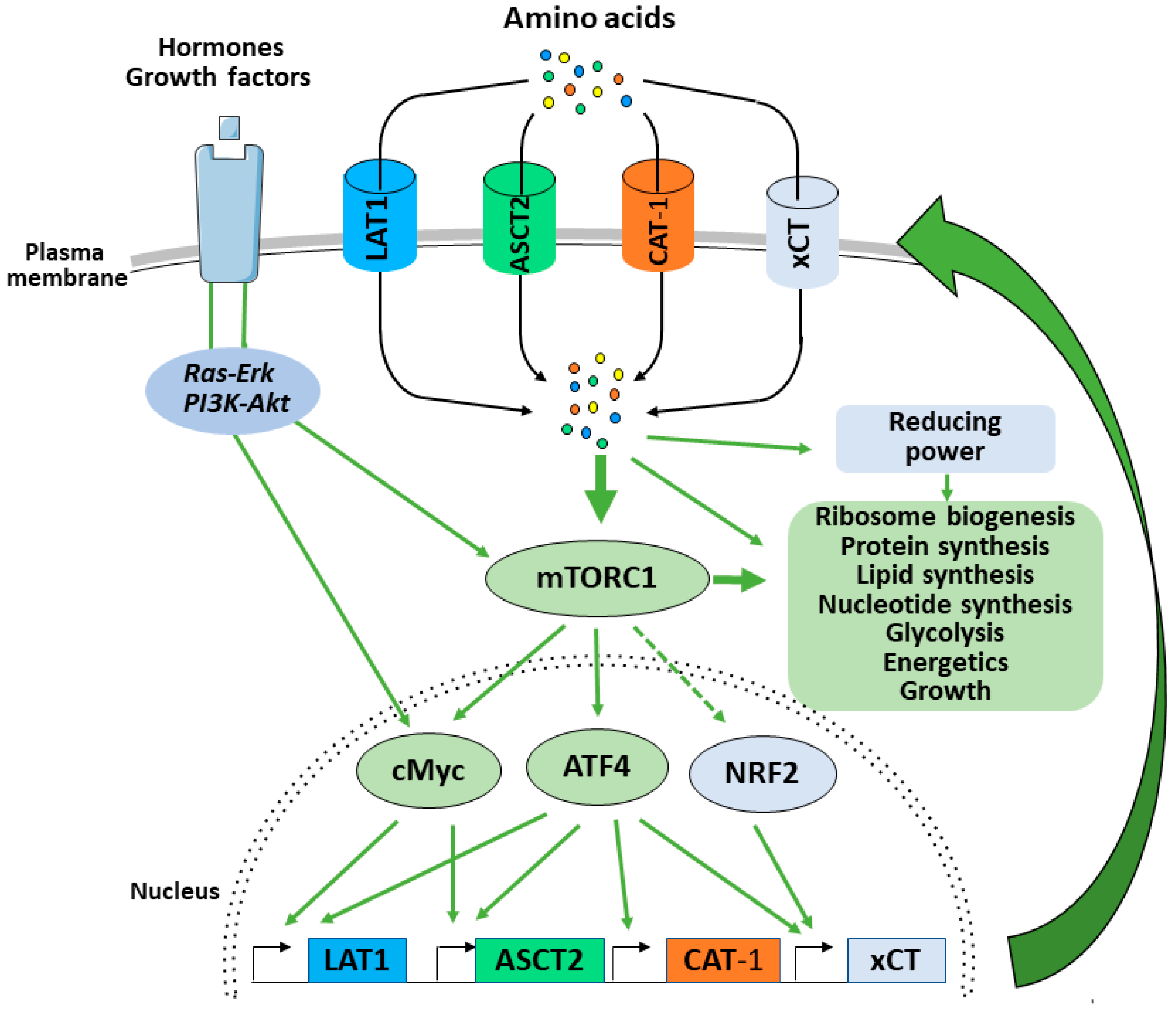
4. mTORC1 Balances Supply and Demand by Promoting Amino Acid Uptake
5. Concluding Remarks and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Manning, B.D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol. 2017, 45, 72–82. [Google Scholar] [CrossRef] [Green Version]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Dibble, C.C.; Manning, B.D. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 2013, 15, 555–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfson, R.L.; Sabatini, D.M. The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metab. 2017, 26, 301–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Orozco, J.M.; Saxton, R.A.; Condon, K.J.; Liu, G.Y.; Krawczyk, P.A.; Scaria, S.M.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 2017, 358, 813–818. [Google Scholar] [CrossRef] [Green Version]
- Duran, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.D.; Zhang, Y.J.; Wei, Y.H.; Cho, J.H.; Morris, L.E.; Wang, H.Y.; Zheng, X.F.S. Rab1A Is an mTORC1 Activator and a Colorectal Oncogene. Cancer Cell 2016, 30, 181–182. [Google Scholar] [CrossRef] [Green Version]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.L. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 2015, 347, 194–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okosun, J.; Wolfson, R.L.; Wang, J.; Araf, S.; Wilkins, L.; Castellano, B.M.; Escudero-Ibarz, L.; Al Seraihi, A.F.; Richter, J.; Bernhart, S.H.; et al. Recurrent mTORC1-activating RRAGC mutations in follicular lymphoma. Nat. Genet. 2016, 48, 183–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsun, Z.Y.; Bar-Peled, L.; Chantranupong, L.; Zoncu, R.; Wang, T.; Kim, C.; Spooner, E.; Sabatini, D.M. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol. Cell 2013, 52, 495–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, E.; Sato, M.; Yang, H.; Miyagawa, F.; Harasaki, M.; Tomita, K.; Matsuoka, S.; Noma, A.; Iwai, K.; Minato, N. 4F2 (CD98) heavy chain is associated covalently with an amino acid transporter and controls intracellular trafficking and membrane topology of 4F2 heterodimer. J. Biol. Chem. 1999, 274, 3009–3016. [Google Scholar] [CrossRef] [Green Version]
- Scalise, M.; Console, L.; Rovella, F.; Galluccio, M.; Pochini, L.; Indiveri, C. Membrane Transporters for Amino Acids as Players of Cancer Metabolic Rewiring. Cells 2020, 9, 28. [Google Scholar] [CrossRef]
- Broer, S. Amino Acid Transporters as Targets for Cancer Therapy: Why, Where, When, and How. Int. J. Mol. Sci. 2020, 21, 6156. [Google Scholar] [CrossRef]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino Acid transporters in cancer and their relevance to “glutamine addiction”: Novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef] [Green Version]
- Kanai, Y.; Segawa, H.; Miyamoto, K.; Uchino, H.; Takeda, E.; Endou, H. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). J. Biol. Chem. 1998, 273, 23629–23632. [Google Scholar] [CrossRef] [Green Version]
- Cormerais, Y.; Giuliano, S.; LeFloch, R.; Front, B.; Durivault, J.; Tambutte, E.; Massard, P.A.; de la Ballina, L.R.; Endou, H.; Wempe, M.F.; et al. Genetic Disruption of the Multifunctional CD98/LAT1 Complex Demonstrates the Key Role of Essential Amino Acid Transport in the Control of mTORC1 and Tumor Growth. Cancer Res. 2016, 76, 4481–4492. [Google Scholar] [CrossRef] [Green Version]
- Hafliger, P.; Charles, R.P. The L-Type Amino Acid Transporter LAT1-An Emerging Target in Cancer. Int. J. Mol. Sci. 2019, 20, 2428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elorza, A.; Soro-Arnaiz, I.; Melendez-Rodriguez, F.; Rodriguez-Vaello, V.; Marsboom, G.; de Carcer, G.; Acosta-Iborra, B.; Albacete-Albacete, L.; Ordonez, A.; Serrano-Oviedo, L.; et al. HIF2alpha acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol. Cell 2012, 48, 681–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, K.; Jutabha, P.; Endou, H.; Anzai, N. c-Myc is crucial for the expression of LAT1 in MIA Paca-2 human pancreatic cancer cells. Oncol. Rep. 2012, 28, 862–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timosenko, E.; Ghadbane, H.; Silk, J.D.; Shepherd, D.; Gileadi, U.; Howson, L.J.; Laynes, R.; Zhao, Q.; Strausberg, R.L.; Olsen, L.R.; et al. Nutritional Stress Induced by Tryptophan-Degrading Enzymes Results in ATF4-Dependent Reprogramming of the Amino Acid Transporter Profile in Tumor Cells. Cancer Res. 2016, 76, 6193–6204. [Google Scholar] [CrossRef] [Green Version]
- Poncet, N.; Halley, P.A.; Lipina, C.; Gierlinski, M.; Dady, A.; Singer, G.A.; Febrer, M.; Shi, Y.B.; Yamaguchi, T.P.; Taylor, P.M.; et al. Wnt regulates amino acid transporter Slc7a5 and so constrains the integrated stress response in mouse embryos. EMBO Rep. 2020, 21, e48469. [Google Scholar] [CrossRef]
- Hansen, C.G.; Ng, Y.L.; Lam, W.L.; Plouffe, S.W.; Guan, K.L. The Hippo pathway effectors YAP and TAZ promote cell growth by modulating amino acid signaling to mTORC1. Cell Res. 2015, 25, 1299–1313. [Google Scholar] [CrossRef]
- Rosilio, C.; Nebout, M.; Imbert, V.; Griessinger, E.; Neffati, Z.; Benadiba, J.; Hagenbeek, T.; Spits, H.; Reverso, J.; Ambrosetti, D.; et al. L-type amino-acid transporter 1 (LAT1): A therapeutic target supporting growth and survival of T-cell lymphoblastic lymphoma/T-cell acute lymphoblastic leukemia. Leukemia 2015, 29, 1253–1266. [Google Scholar] [CrossRef]
- Choi, D.W.; Kim, D.K.; Kanai, Y.; Wempe, M.F.; Endou, H.; Kim, J.K. JPH203, a selective L-type amino acid transporter 1 inhibitor, induces mitochondria-dependent apoptosis in Saos2 human osteosarcoma cells. Korean J. Physiol. Pharmacol. 2017, 21, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Yothaisong, S.; Dokduang, H.; Anzai, N.; Hayashi, K.; Namwat, N.; Yongvanit, P.; Sangkhamanon, S.; Jutabha, P.; Endou, H.; Loilome, W. Inhibition of l-type amino acid transporter 1 activity as a new therapeutic target for cholangiocarcinoma treatment. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, K.; Jutabha, P.; Maeda, S.; Supak, Y.; Ouchi, M.; Endou, H.; Fujita, T.; Chida, M.; Anzai, N. LAT1 acts as a crucial transporter of amino acids in human thymic carcinoma cells. J. Pharmacol. Sci. 2016, 132, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Okano, N.; Naruge, D.; Kawai, K.; Kobayashi, T.; Nagashima, F.; Endou, H.; Furuse, J. First-in-human phase I study of JPH203, an L-type amino acid transporter 1 inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, A.M.; Holmberg, J.C.; Feliciano, D.M. The amino acid transporter Slc7a5 regulates the mTOR pathway and is required for granule cell development. Hum. Mol. Genet. 2020, 29, 3003–3013. [Google Scholar] [CrossRef] [PubMed]
- Tarlungeanu, D.C.; Deliu, E.; Dotter, C.P.; Kara, M.; Janiesch, P.C.; Scalise, M.; Galluccio, M.; Tesulov, M.; Morelli, E.; Sonmez, F.M.; et al. Impaired Amino Acid Transport at the Blood Brain Barrier Is a Cause of Autism Spectrum Disorder. Cell 2016, 167, 1481–1494.e1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloetta, D.; Thomanetz, V.; Baranek, C.; Lustenberger, R.M.; Lin, S.; Oliveri, F.; Atanasoski, S.; Ruegg, M.A. Inactivation of mTORC1 in the developing brain causes microcephaly and affects gliogenesis. J. NeuroSci. 2013, 33, 7799–7810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poncet, N.; Mitchell, F.E.; Ibrahim, A.F.; McGuire, V.A.; English, G.; Arthur, J.S.; Shi, Y.B.; Taylor, P.M. The catalytic subunit of the system L1 amino acid transporter (slc7a5) facilitates nutrient signalling in mouse skeletal muscle. PLoS ONE 2014, 9, e89547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, K.; Jutabha, P.; Endou, H.; Sagara, H.; Anzai, N. LAT1 is a critical transporter of essential amino acids for immune reactions in activated human T cells. J. Immunol. 2013, 191, 4080–4085. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, L.V.; Rolf, J.; Emslie, E.; Shi, Y.B.; Taylor, P.M.; Cantrell, D.A. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 2013, 14, 500–508. [Google Scholar] [CrossRef] [Green Version]
- Yoon, B.R.; Oh, Y.J.; Kang, S.W.; Lee, E.B.; Lee, W.W. Role of SLC7A5 in Metabolic Reprogramming of Human Monocyte/Macrophage Immune Responses. Front. Immunol. 2018, 9, 53. [Google Scholar] [CrossRef] [Green Version]
- Torigoe, M.; Maeshima, K.; Ozaki, T.; Omura, Y.; Gotoh, K.; Tanaka, Y.; Ishii, K.; Shibata, H. l-Leucine influx through Slc7a5 regulates inflammatory responses of human B cells via mammalian target of rapamycin complex 1 signaling. Mod. Rheumatol. 2019, 29, 885–891. [Google Scholar] [CrossRef]
- Loftus, R.M.; Assmann, N.; Kedia-Mehta, N.; O’Brien, K.L.; Garcia, A.; Gillespie, C.; Hukelmann, J.L.; Oefner, P.J.; Lamond, A.I.; Gardiner, C.M.; et al. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat. Commun. 2018, 9, 2341. [Google Scholar] [CrossRef]
- Cormerais, Y.; Vucetic, M.; Pouyssegur, J. Targeting amino acids transporters (SLCs) to starve cancer cells to death. BioChem. Biophys. Res. Commun. 2019, 520, 691–693. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, B.C.; Bode, B.P. Amino acid transporters ASCT2 and LAT1 in cancer: Partners in crime? Semin. Cancer Biol. 2005, 15, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, R.; Shuai, Y.; Huang, Y.; Jin, R.; Wang, X.; Luo, J. ASCT2 (SLC1A5)-dependent glutamine uptake is involved in the progression of head and neck squamous cell carcinoma. Br. J. Cancer 2020, 122, 82–93. [Google Scholar] [CrossRef] [PubMed]
- van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Hardie, R.A.; Hoy, A.J.; van Geldermalsen, M.; Gao, D.; Fazli, L.; Sadowski, M.C.; Balaban, S.; Schreuder, M.; Nagarajah, R.; et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 2015, 236, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Ni, F.; Yu, W.M.; Li, Z.; Graham, D.K.; Jin, L.; Kang, S.; Rossi, M.R.; Li, S.; Broxmeyer, H.E.; Qu, C.K. Critical role of ASCT2-mediated amino acid metabolism in promoting leukaemia development and progression. Nat. Metab. 2019, 1, 390–403. [Google Scholar] [CrossRef]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Broer, A.; Gauthier-Coles, G.; Rahimi, F.; van Geldermalsen, M.; Dorsch, D.; Wegener, A.; Holst, J.; Broer, S. Ablation of the ASCT2 (SLC1A5) gene encoding a neutral amino acid transporter reveals transporter plasticity and redundancy in cancer cells. J. Biol. Chem. 2019, 294, 4012–4026. [Google Scholar] [CrossRef] [Green Version]
- Cormerais, Y.; Massard, P.A.; Vucetic, M.; Giuliano, S.; Tambutte, E.; Durivault, J.; Vial, V.; Endou, H.; Wempe, M.F.; Parks, S.K.; et al. The glutamine transporter ASCT2 (SLC1A5) promotes tumor growth independently of the amino acid transporter LAT1 (SLC7A5). J. Biol. Chem. 2018, 293, 2877–2887. [Google Scholar] [CrossRef] [Green Version]
- Hoxhaj, G.; Hughes-Hallett, J.; Timson, R.C.; Ilagan, E.; Yuan, M.; Asara, J.M.; Ben-Sahra, I.; Manning, B.D. The mTORC1 Signaling Network Senses Changes in Cellular Purine Nucleotide Levels. Cell Rep. 2017, 21, 1331–1346. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Labuschagne, C.F.; van den Broek, N.J.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vucetic, M.; Cormerais, Y.; Parks, S.K.; Pouyssegur, J. The Central Role of Amino Acids in Cancer Redox Homeostasis: Vulnerability Points of the Cancer Redox Code. Front. Oncol. 2017, 7, 319. [Google Scholar] [CrossRef] [PubMed]
- Parrott, L.A.; Templeton, D.J. Osmotic stress inhibits p70/85 S6 kinase through activation of a protein phosphatase. J. Biol. Chem. 1999, 274, 24731–24736. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Li, J.; Ke, Q.; Leonard, S.S.; Jiang, B.H.; Zhong, X.S.; Costa, M.; Castranova, V.; Shi, X. Ultraviolet-induced phosphorylation of p70(S6K) at Thr(389) and Thr(421)/Ser(424) involves hydrogen peroxide and mammalian target of rapamycin but not Akt and atypical protein kinase C. Cancer Res. 2002, 62, 5689–5697. [Google Scholar] [PubMed]
- Sarbassov, D.D.; Sabatini, D.M. Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. J. Biol. Chem. 2005, 280, 39505–39509. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, S.; Hong, S.; Suzuki, T.; Nada, S.; Mannan, A.M.; Wang, J.; Okada, M.; Guan, K.L.; Inoki, K. Redox regulates mammalian target of rapamycin complex 1 (mTORC1) activity by modulating the TSC1/TSC2-Rheb GTPase pathway. J. Biol. Chem. 2011, 286, 32651–32660. [Google Scholar] [CrossRef] [Green Version]
- Alexander, A.; Cai, S.L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.L.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 2010, 107, 4153–4158. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kim, J.; Alexander, A.; Cai, S.; Tripathi, D.N.; Dere, R.; Tee, A.R.; Tait-Mulder, J.; Di Nardo, A.; Han, J.M.; et al. A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat. Cell Biol. 2013, 15, 1186–1196. [Google Scholar] [CrossRef]
- Bannai, S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J. Biol. Chem. 1986, 261, 2256–2263. [Google Scholar]
- Banerjee, R.; Zou, C.G. Redox regulation and reaction mechanism of human cystathionine-beta-synthase: A PLP-dependent hemesensor protein. Arch. BioChem. Biophys. 2005, 433, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Shiiya, A.; Kimata, M.; Maebara, K.; Tamba, M.; Sakakura, Y.; Makino, N.; Sugiyama, F.; Yagami, K.; Moriguchi, T.; et al. Redox imbalance in cystine/glutamate transporter-deficient mice. J. Biol. Chem. 2005, 280, 37423–37429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, H.; Sato, H.; Kuriyama-Matsumura, K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 2002, 277, 44765–44771. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Nomura, S.; Maebara, K.; Sato, K.; Tamba, M.; Bannai, S. Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. BioChem. Biophys. Res. Commun. 2004, 325, 109–116. [Google Scholar] [CrossRef]
- Zong, Z.H.; Du, Z.X.; Li, N.; Li, C.; Zhang, Q.; Liu, B.Q.; Guan, Y.; Wang, H.Q. Implication of Nrf2 and ATF4 in differential induction of CHOP by proteasome inhibition in thyroid cancer cells. Biochim. Biophys. Acta 2012, 1823, 1395–1404. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.K.M.; Delaidelli, A.; Minaker, S.W.; Zhang, H.F.; Colovic, M.; Yang, H.; Negri, G.L.; von Karstedt, S.; Lockwood, W.W.; Schaffer, P.; et al. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc. Natl. Acad. Sci. USA 2019, 116, 9433–9442. [Google Scholar] [CrossRef] [Green Version]
- Verschoor, M.L.; Singh, G. Ets-1 regulates intracellular glutathione levels: Key target for resistant ovarian cancer. Mol. Cancer 2013, 12, 138. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daher, B.; Vucetic, M.; Pouyssegur, J. Cysteine Depletion, a Key Action to Challenge Cancer Cells to Ferroptotic Cell Death. Front. Oncol. 2020, 10, 723. [Google Scholar] [CrossRef] [PubMed]
- Daher, B.; Parks, S.K.; Durivault, J.; Cormerais, Y.; Baidarjad, H.; Tambutte, E.; Pouyssegur, J.; Vucetic, M. Genetic Ablation of the Cystine Transporter xCT in PDAC Cells Inhibits mTORC1, Growth, Survival, and Tumor Formation via Nutrient and Oxidative Stresses. Cancer Res. 2019, 79, 3877–3890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Shertzer, H.G.; Schneider, S.N.; Nebert, D.W.; Dalton, T.P. Glutamate cysteine ligase catalysis: Dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J. Biol. Chem. 2005, 280, 33766–33774. [Google Scholar] [CrossRef] [Green Version]
- Shin, C.S.; Mishra, P.; Watrous, J.D.; Carelli, V.; D’Aurelio, M.; Jain, M.; Chan, D.C. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat. Commun. 2017, 8, 15074. [Google Scholar] [CrossRef] [Green Version]
- Koppula, P.; Zhang, Y.; Shi, J.; Li, W.; Gan, B. The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J. Biol. Chem. 2017, 292, 14240–14249. [Google Scholar] [CrossRef] [Green Version]
- Spitz, D.R.; Sim, J.E.; Ridnour, L.A.; Galoforo, S.S.; Lee, Y.J. Glucose deprivation-induced oxidative stress in human tumor cells. A fundamental defect in metabolism? Ann. N. Y. Acad. Sci. 2000, 899, 349–362. [Google Scholar] [CrossRef]
- Arensman, M.D.; Yang, X.S.; Leahy, D.M.; Toral-Barza, L.; Mileski, M.; Rosfjord, E.C.; Wang, F.; Deng, S.; Myers, J.S.; Abraham, R.T.; et al. Cystine-glutamate antiporter xCT deficiency suppresses tumor growth while preserving antitumor immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 9533–9542. [Google Scholar] [CrossRef] [Green Version]
- Riess, C.; Shokraie, F.; Classen, C.F.; Kreikemeyer, B.; Fiedler, T.; Junghanss, C.; Maletzki, C. Arginine-Depleting Enzymes—An Increasingly Recognized Treatment Strategy for Therapy-Refractory Malignancies. Cell Physiol. BioChem. 2018, 51, 854–870. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, W.; Wang, J.; Yang, C.; Mao, H.; Fu, X.; Wu, Y.; Cai, J.; Han, J.; Xu, Z.; et al. Overexpression of arginine transporter CAT-1 is associated with accumulation of L-arginine and cell growth in human colorectal cancer tissue. PLoS ONE 2013, 8, e73866. [Google Scholar] [CrossRef]
- Abdelmagid, S.A.; Rickard, J.A.; McDonald, W.J.; Thomas, L.N.; Too, C.K. CAT-1-mediated arginine uptake and regulation of nitric oxide synthases for the survival of human breast cancer cell lines. J. Cell BioChem. 2011, 112, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Werner, A.; Pieh, D.; Echchannaoui, H.; Rupp, J.; Rajalingam, K.; Theobald, M.; Closs, E.I.; Munder, M. Cationic Amino Acid Transporter-1-Mediated Arginine Uptake Is Essential for Chronic Lymphocytic Leukemia Cell Proliferation and Viability. Front. Oncol. 2019, 9, 1268. [Google Scholar] [CrossRef] [PubMed]
- Perkins, C.P.; Mar, V.; Shutter, J.R.; del Castillo, J.; Danilenko, D.M.; Medlock, E.S.; Ponting, I.L.; Graham, M.; Stark, K.L.; Zuo, Y.; et al. Anemia and perinatal death result from loss of the murine ecotropic retrovirus receptor mCAT-1. Genes Dev. 1997, 11, 914–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsueh, E.C.; Knebel, S.M.; Lo, W.H.; Leung, Y.C.; Cheng, P.N.; Hsueh, C.T. Deprivation of arginine by recombinant human arginase in prostate cancer cells. J. Hematol. Oncol. 2012, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, S.L.; Ho, M.K.; Lam, Y.M.; Chow, H.Y.; So, Y.H.; Leung, Y.C. PEGylated recombinant human arginase as a drug for breast cancer. Hong Kong Med. J. 2019, 25 (Suppl. 9), 28–31. [Google Scholar]
- Shen, W.; Zhang, X.; Fu, X.; Fan, J.; Luan, J.; Cao, Z.; Yang, P.; Xu, Z.; Ju, D. A novel and promising therapeutic approach for NSCLC: Recombinant human arginase alone or combined with autophagy inhibitor. Cell Death Dis. 2017, 8, e2720. [Google Scholar] [CrossRef]
- Coothankandaswamy, V.; Cao, S.; Xu, Y.; Prasad, P.D.; Singh, P.K.; Reynolds, C.P.; Yang, S.; Ogura, J.; Ganapathy, V.; Bhutia, Y.D. Amino acid transporter SLC6A14 is a novel and effective drug target for pancreatic cancer. Br. J. Pharmacol. 2016, 173, 3292–3306. [Google Scholar] [CrossRef] [Green Version]
- Babu, E.; Bhutia, Y.D.; Ramachandran, S.; Gnanaprakasam, J.P.; Prasad, P.D.; Thangaraju, M.; Ganapathy, V. Deletion of the amino acid transporter Slc6a14 suppresses tumour growth in spontaneous mouse models of breast cancer. BioChem. J. 2015, 469, 17–23. [Google Scholar] [CrossRef]
- Karunakaran, S.; Ramachandran, S.; Coothankandaswamy, V.; Elangovan, S.; Babu, E.; Periyasamy-Thandavan, S.; Gurav, A.; Gnanaprakasam, J.P.; Singh, N.; Schoenlein, P.V.; et al. SLC6A14 (ATB0,+) protein, a highly concentrative and broad specific amino acid transporter, is a novel and effective drug target for treatment of estrogen receptor-positive breast cancer. J. Biol. Chem. 2011, 286, 31830–31838. [Google Scholar] [CrossRef] [Green Version]
- Goberdhan, D.C.; Meredith, D.; Boyd, C.A.; Wilson, C. PAT-related amino acid transporters regulate growth via a novel mechanism that does not require bulk transport of amino acids. Development 2005, 132, 2365–2375. [Google Scholar] [CrossRef] [Green Version]
- Heublein, S.; Kazi, S.; Ogmundsdottir, M.H.; Attwood, E.V.; Kala, S.; Boyd, C.A.; Wilson, C.; Goberdhan, D.C. Proton-assisted amino-acid transporters are conserved regulators of proliferation and amino-acid-dependent mTORC1 activation. Oncogene 2010, 29, 4068–4079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, S.J.; Snell, C.; Turley, H.; Li, J.L.; McCormick, R.; Perera, S.M.; Heublein, S.; Kazi, S.; Azad, A.; Wilson, C.; et al. PAT4 levels control amino-acid sensitivity of rapamycin-resistant mTORC1 from the Golgi and affect clinical outcome in colorectal cancer. Oncogene 2016, 35, 3004–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goberdhan, D.C. Intracellular amino acid sensing and mTORC1-regulated growth: New ways to block an old target? Curr. Opin. Investig. Drugs 2010, 11, 1360–1367. [Google Scholar] [PubMed]
- Pinilla, J.; Aledo, J.C.; Cwiklinski, E.; Hyde, R.; Taylor, P.M.; Hundal, H.S. SNAT2 transceptor signalling via mTOR: A role in cell growth and proliferation? Front. BioSci. 2011, 3, 1289–1299. [Google Scholar] [CrossRef]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Ogmundsdottir, M.H.; Heublein, S.; Kazi, S.; Reynolds, B.; Visvalingam, S.M.; Shaw, M.K.; Goberdhan, D.C. Proton-assisted amino acid transporter PAT1 complexes with Rag GTPases and activates TORC1 on late endosomal and lysosomal membranes. PLoS ONE 2012, 7, e36616. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, A.; Bu, Y.; Qie, S.; Wrangle, J.; Camp, E.R.; Hazard, E.S.; Hardiman, G.; de Leeuw, R.; Knudsen, K.E.; Diehl, J.A. SLC36A1-mTORC1 signaling drives acquired resistance to CDK4/6 inhibitors. Sci. Adv. 2019, 5, eaax6352. [Google Scholar] [CrossRef] [Green Version]
- Andrzejewska, Z.; Nevo, N.; Thomas, L.; Chhuon, C.; Bailleux, A.; Chauvet, V.; Courtoy, P.J.; Chol, M.; Guerrera, I.C.; Antignac, C. Cystinosin is a Component of the Vacuolar H+-ATPase-Ragulator-Rag Complex Controlling Mammalian Target of Rapamycin Complex 1 Signaling. J. Am. Soc. Nephrol. 2016, 27, 1678–1688. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.; Sabatini, D.M. Ragulator and SLC38A9 activate the Rag GTPases through noncanonical GEF mechanisms. Proc. Natl. Acad. Sci. USA 2018, 115, 9545–9550. [Google Scholar] [CrossRef] [Green Version]
- Wyant, G.A.; Abu-Remaileh, M.; Wolfson, R.L.; Chen, W.W.; Freinkman, E.; Danai, L.V.; Vander Heiden, M.G.; Sabatini, D.M. mTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient. Cell 2017, 171, 642–654.e612. [Google Scholar] [CrossRef]
- Wang, S.; Tsun, Z.Y.; Wolfson, R.L.; Shen, K.; Wyant, G.A.; Plovanich, M.E.; Yuan, E.D.; Jones, T.D.; Chantranupong, L.; Comb, W.; et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 2015, 347, 188–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, R.E.; Fromm, S.A.; Fu, Y.; Yokom, A.L.; Kim, D.J.; Thelen, A.M.; Young, L.N.; Lim, C.Y.; Samelson, A.J.; Hurley, J.H.; et al. Structural mechanism of a Rag GTPase activation checkpoint by the lysosomal folliculin complex. Science 2019, 366, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Verdon, Q.; Boonen, M.; Ribes, C.; Jadot, M.; Gasnier, B.; Sagne, C. SNAT7 is the primary lysosomal glutamine exporter required for extracellular protein-dependent growth of cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E3602–E3611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milkereit, R.; Persaud, A.; Vanoaica, L.; Guetg, A.; Verrey, F.; Rotin, D. LAPTM4b recruits the LAT1-4F2hc Leu transporter to lysosomes and promotes mTORC1 activation. Nat. Commun. 2015, 6, 7250. [Google Scholar] [CrossRef] [Green Version]
- Weng, L.; Han, Y.P.; Enomoto, A.; Kitaura, Y.; Nagamori, S.; Kanai, Y.; Asai, N.; An, J.; Takagishi, M.; Asai, M.; et al. Negative regulation of amino acid signaling by MAPK-regulated 4F2hc/Girdin complex. PLoS Biol. 2018, 16, e2005090. [Google Scholar] [CrossRef] [Green Version]
- Beaumatin, F.; O’Prey, J.; Barthet, V.J.A.; Zunino, B.; Parvy, J.P.; Bachmann, A.M.; O’Prey, M.; Kania, E.; Gonzalez, P.S.; Macintosh, R.; et al. mTORC1 Activation Requires DRAM-1 by Facilitating Lysosomal Amino Acid Efflux. Mol. Cell 2019, 76, 163–176.e168. [Google Scholar] [CrossRef] [Green Version]
- Edinger, A.L.; Thompson, C.B. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol. Biol. Cell 2002, 13, 2276–2288. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. A Time for MYC: Metabolism and Therapy. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 79–83. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [Green Version]
- Csibi, A.; Lee, G.; Yoon, S.O.; Tong, H.; Ilter, D.; Elia, I.; Fendt, S.M.; Roberts, T.M.; Blenis, J. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr. Biol. 2014, 24, 2274–2280. [Google Scholar] [CrossRef] [Green Version]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Reyna-Neyra, A.; Philippe, L.; Thoreen, C.C. mTORC1 Balances Cellular Amino Acid Supply with Demand for Protein Synthesis through Post-transcriptional Control of ATF4. Cell Rep. 2017, 19, 1083–1090. [Google Scholar] [CrossRef] [Green Version]
- Torrence, M.E.; MacArthur, M.R.; Hosios, A.M.; Valvezan, A.J.; Asara, J.M.; Mitchell, J.R.; Manning, B.D. The mTORC1-mediated activation of ATF4 promotes protein and glutathione synthesis downstream of growth signals. bioRxiv 2020. [Google Scholar] [CrossRef]
- Li, C.; Chen, H.; Lan, Z.; He, S.; Chen, R.; Wang, F.; Liu, Z.; Li, K.; Cheng, L.; Liu, Y.; et al. mTOR-dependent upregulation of xCT blocks melanin synthesis and promotes tumorigenesis. Cell Death Differ. 2019, 26, 2015–2028. [Google Scholar] [CrossRef]
- Shay, K.P.; Michels, A.J.; Li, W.; Kong, A.N.; Hagen, T.M. Cap-independent Nrf2 translation is part of a lipoic acid-stimulated detoxification stress response. Biochim. Biophys. Acta 2012, 1823, 1102–1109. [Google Scholar] [CrossRef] [Green Version]
- Lerner, C.; Bitto, A.; Pulliam, D.; Nacarelli, T.; Konigsberg, M.; Van Remmen, H.; Torres, C.; Sell, C. Reduced mammalian target of rapamycin activity facilitates mitochondrial retrograde signaling and increases life span in normal human fibroblasts. Aging Cell 2013, 12, 966–977. [Google Scholar] [CrossRef] [Green Version]
- Netto, L.E.; de Oliveira, M.A.; Monteiro, G.; Demasi, A.P.; Cussiol, J.R.; Discola, K.F.; Demasi, M.; Silva, G.M.; Alves, S.V.; Faria, V.G.; et al. Reactive cysteine in proteins: Protein folding, antioxidant defense, redox signaling and more. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2007, 146, 180–193. [Google Scholar] [CrossRef]
- Marino, S.M.; Gladyshev, V.N. Cysteine function governs its conservation and degeneration and restricts its utilization on protein surfaces. J. Mol. Biol. 2010, 404, 902–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, Q.; Yan, X.; Kato, Y.; Tanaka, M.; Inokuchi, S.; Yoshizawa, T.; Morohashi, S.; Kijima, H. Podoplanin-mediated TGF-beta-induced epithelial-mesenchymal transition and its correlation with bHLH transcription factor DEC in TE-11 cells. Int. J. Oncol. 2016, 48, 2310–2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vucetic, M.; Daher, B.; Cassim, S.; Meira, W.; Pouyssegur, J. Together we stand, apart we fall: How cell-to-cell contact/interplay provides resistance to ferroptosis. Cell Death Dis. 2020, 11, 789. [Google Scholar] [CrossRef] [PubMed]
- Conlon, M.; Poltorack, C.; Forcina, G.C.; Wells, A.; Mallais, M.; Kahanu, A.; Magtanong, L.; Pratt, D.A.; Dixon, S.J. A Compendium of Kinetic Cell Death Modulatory Profiles Identifies Ferroptosis Regulators. bioRxiv 2019, 826925. [Google Scholar] [CrossRef]
- Lee, Y.; Wiriyasermkul, P.; Jin, C.; Quan, L.; Ohgaki, R.; Okuda, S.; Kusakizako, T.; Nishizawa, T.; Oda, K.; Ishitani, R.; et al. Cryo-EM structure of the human L-type amino acid transporter 1 in complex with glycoprotein CD98hc. Nat. Struct. Mol. Biol. 2019, 26, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Plotnikova, O.; Bonin, P.D.; Subashi, T.A.; McLellan, T.J.; Dumlao, D.; Che, Y.; Dong, Y.Y.; Carpenter, E.P.; West, G.M.; et al. Cryo-EM structures of the human glutamine transporter SLC1A5 (ASCT2) in the outward-facing conformation. Elife 2019, 8, e48120. [Google Scholar] [CrossRef]
- Oda, K.; Lee, Y.; Wiriyasermkul, P.; Tanaka, Y.; Takemoto, M.; Yamashita, K.; Nagamori, S.; Nishizawa, T.; Nureki, O. Consensus mutagenesis approach improves the thermal stability of system xc (-) transporter, xCT, and enables cryo-EM analyses. Protein Sci. 2020. [Google Scholar] [CrossRef]
- Gingras, A.C.; Raught, B.; Sonenberg, N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001, 15, 807–826. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palm, W.; Park, Y.; Wright, K.; Pavlova, N.N.; Tuveson, D.A.; Thompson, C.B. The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell 2015, 162, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cormerais, Y.; Vučetić, M.; Parks, S.K.; Pouyssegur, J. Amino Acid Transporters Are a Vital Focal Point in the Control of mTORC1 Signaling and Cancer. Int. J. Mol. Sci. 2021, 22, 23. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010023
Cormerais Y, Vučetić M, Parks SK, Pouyssegur J. Amino Acid Transporters Are a Vital Focal Point in the Control of mTORC1 Signaling and Cancer. International Journal of Molecular Sciences. 2021; 22(1):23. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010023
Chicago/Turabian StyleCormerais, Yann, Milica Vučetić, Scott K. Parks, and Jacques Pouyssegur. 2021. "Amino Acid Transporters Are a Vital Focal Point in the Control of mTORC1 Signaling and Cancer" International Journal of Molecular Sciences 22, no. 1: 23. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010023