BION-2: Predicting Positions of Non-Specifically Bound Ions on Protein Surface by a Gaussian-Based Treatment of Electrostatics




Abstract
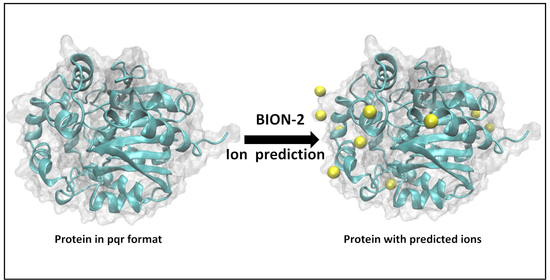
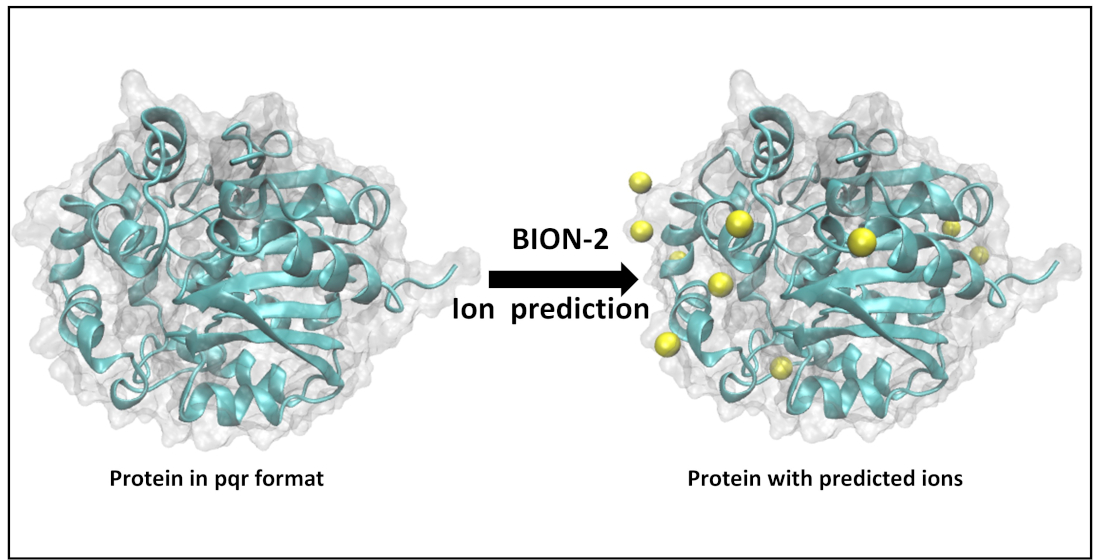
:
1. Introduction
2. Results and Discussion
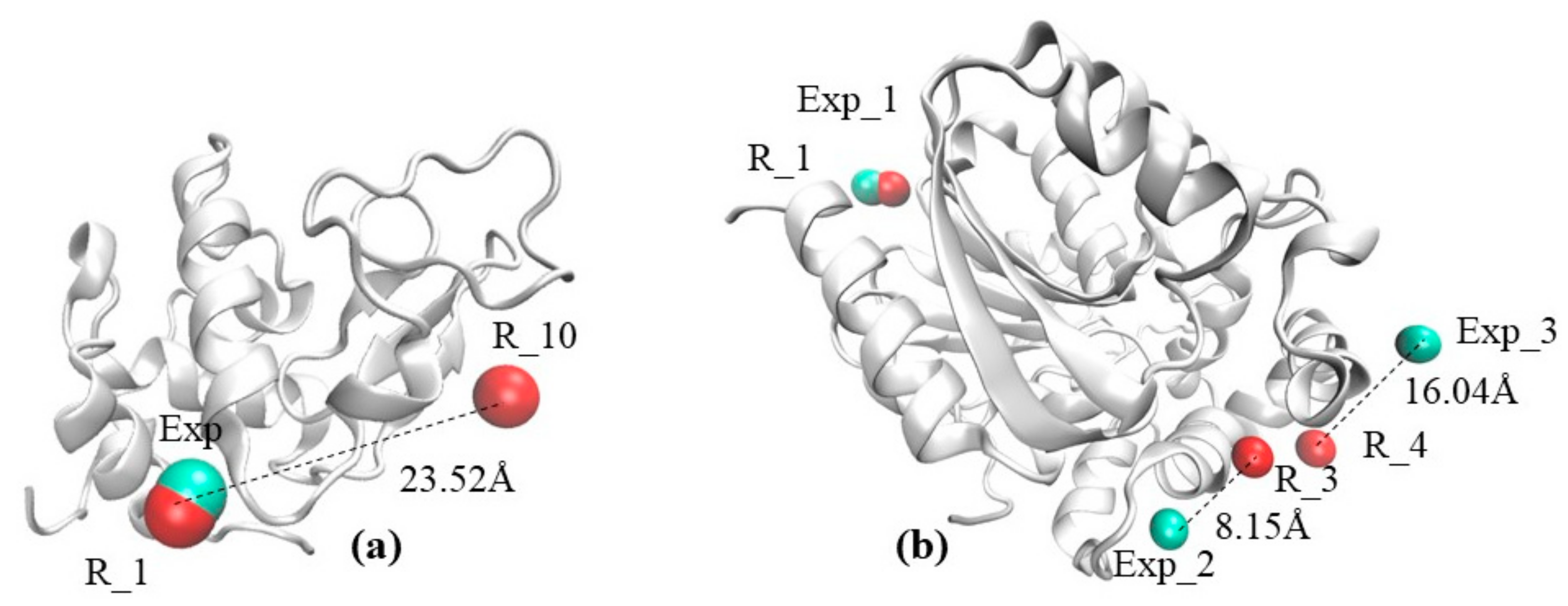
2.1. The Visual Example Section Outlines Two Cases
2.2. Benchmarking of BION-2 Performance
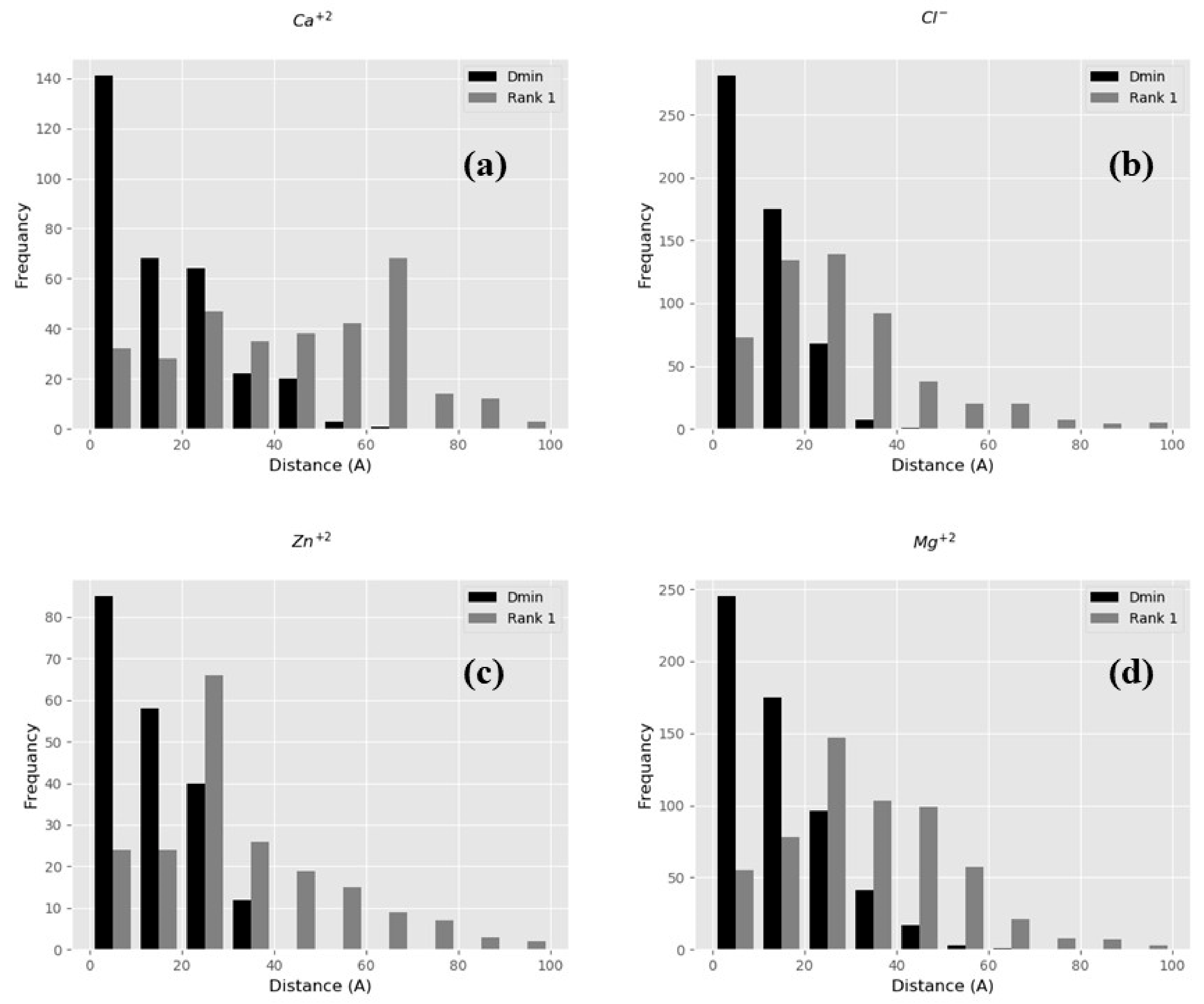
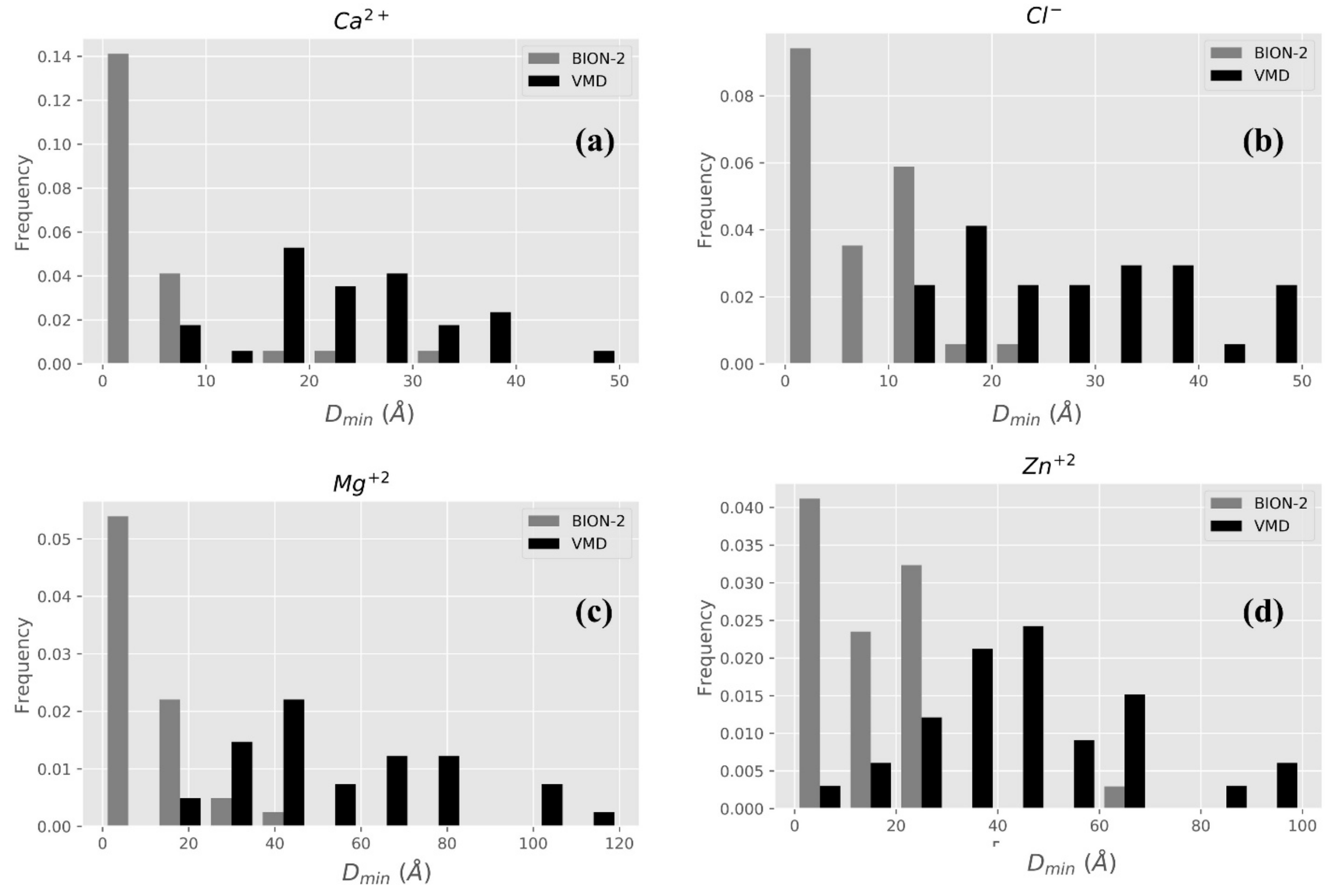
2.2.1. BION-2 vs. VMD
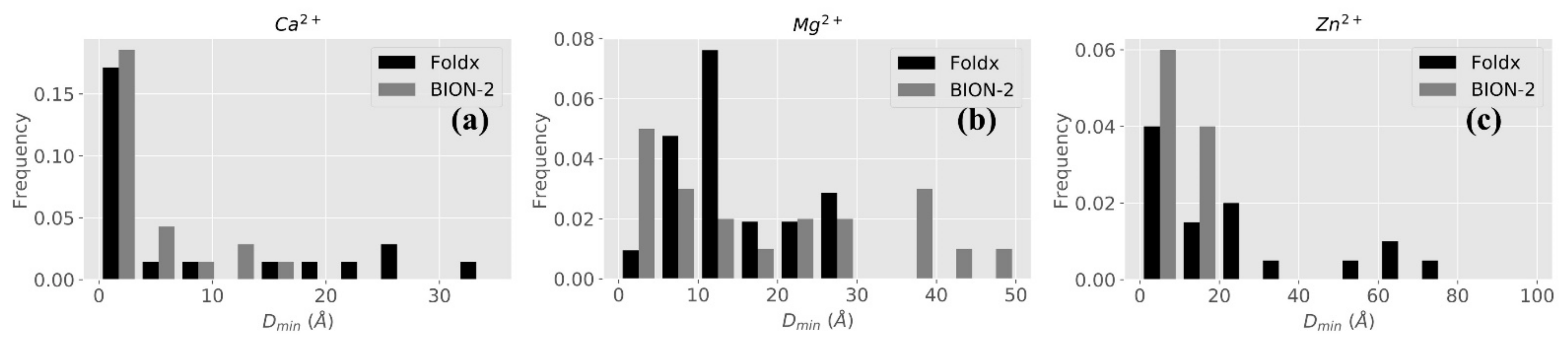
2.2.2. BION-2 vs. FoldX
2.2.3. Computational Efficiency
2.2.4. BION-2 Webserver
3. Materials and Methods
3.1. Database of Protein Structures
3.2. Ions’ Treatment in the Framework of Gaussian-Based Smooth Dielectric Function
3.3. Electrostatic Potential Map Calculations
3.4. Algorithm for Predicting Ion’s Position
3.5. Using VMD to Place Ions
3.6. Using FoldX to Predict Ions’ Positions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BION | bound ion prediction method |
| Zeta | Zeta potential |
| PDB | Protein data bank |
| DelPhi | Poisson–Boltzmann solver |
| VMD | visual molecular dynamics |
| PBE | Poisson–Boltzmann equation |
| vdW | van der Waals |
| PQR | position, charge, radius format |
| NMR | nucleic magnetic resonance |
References
- Chang, C.J. Chemical biology: Ions illuminated. Nature 2007, 448, 654–655. [Google Scholar] [CrossRef] [PubMed]
- Downard, K.M. Ions of the interactome: The role of MS in the study of protein interactions in proteomics and structural biology. Proteomics 2006, 6, 5374–5384. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, D.H.; Green, N.M. Structural biology. Pumping ions. Nature 2000, 405, 633–634. [Google Scholar] [CrossRef] [PubMed]
- Bresme, F.; Robotham, O.; Chio, W.K.; Gonzalez, M.A.; Kornyshev, A. Debye screening, overscreening and specific adsorption in solutions of organic ions. Phys. Chem. Chem. Phys. 2018, 20, 27684–27693. [Google Scholar] [CrossRef] [Green Version]
- Jordan, E.; Roosen-Runge, F.; Leibfarth, S.; Zhang, F.; Sztucki, M.; Hildebrandt, A.; Kohlbacher, O.; Schreiber, F. Competing salt effects on phase behavior of protein solutions: Tailoring of protein interaction by the binding of multivalent ions and charge screening. J. Phys. Chem. B 2014, 118, 11365–11374. [Google Scholar] [CrossRef]
- Beraudi, A.; Bianconi, E.; Catalani, S.; Canaider, S.; De Pasquale, D.; Apostoli, P.; Bordini, B.; Stea, S.; Toni, A.; Facchin, F. In vivo response of heme-oxygenase-1 to metal ions released from metal-on-metal hip prostheses. Mol. Med. Rep. 2016, 14, 474–480. [Google Scholar] [CrossRef] [Green Version]
- Choe, C.; Yang, L.; Lv, Z.; Mo, W.; Chen, Z.; Li, G.; Yin, G. Redox-inactive metal ions promoted the catalytic reactivity of non-heme manganese complexes towards oxygen atom transfer. Dalton Trans. 2015, 44, 9182–9192. [Google Scholar] [CrossRef]
- De Petris, A.; Chiavarino, B.; Crestoni, M.E.; Coletti, C.; Re, N.; Fornarini, S. Exploring the conformational variability in the heme b propionic acid side chains through the effect of a biological probe: A study of the isolated ions. J. Phys. Chem. B 2015, 119, 1919–1929. [Google Scholar] [CrossRef]
- Petukh, M.; Zhang, M.; Alexov, E. Statistical investigation of surface bound ions and further development of BION server to include pH and salt dependence. J. Comput. Chem. 2015, 36, 2381–2393. [Google Scholar] [CrossRef]
- Chiu, T.K.; Kaczor-Grzeskowiak, M.; Dickerson, R.E. Absence of Minor Groove Monovalent Cations in the Crosslinked Dodecamer C-G-C-G-A-A-T-T-C-G-C-G. J. Mol. Biol. 1999, 292, 589–608. [Google Scholar] [CrossRef]
- Huang, H.W.; Li, D.; Cowan, J.A. Biostructural chemistry of magnesium. Regulation of mithramycin-DNA interactions by Mg2+ coordination. Biochimie 1995, 77, 729–738. [Google Scholar] [CrossRef]
- Giambasu, G.M.; Case, D.A.; York, D.M. Predicting Site-Binding Modes of Ions and Water to Nucleic Acids Using Molecular Solvation Theory. J. Am. Chem. Soc. 2019, 141, 2435–2445. [Google Scholar] [CrossRef] [PubMed]
- Sreedhara, A.; Cowan, J.A. Structural and catalytic roles for divalent magnesium in nucleic acid biochemistry. Biometals 2002, 15, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Laing, L.G.; Gluick, T.C.; Draper, D.E. Stabilization of RNA Structure by Mg Ions: Specific and Non-specific Effects. J. Mol. Biol. 1994, 237, 577–587. [Google Scholar] [CrossRef]
- Wang, L.; Witham, S.; Zhang, Z.; Li, L.; Hodsdon, M.E.; Alexov, E. In silico investigation of pH-dependence of prolactin and human growth hormone binding to human prolactin receptor. Commun. Comput. Phys. 2013, 13, 207–222. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, L.; Li, L.; Alexov, E.; Sanabria, H.; Waxham, M.N. Cytoskeletal-like Filaments of Ca(2+)-Calmodulin-Dependent Protein Kinase II Are Formed in a Regulated and Zn(2+)-Dependent Manner. Biochemistry 2017, 56, 2149–2160. [Google Scholar] [CrossRef]
- Matzke, A.J.M.; Lin, W.D.; Matzke, M. Evidence That Ion-Based Signaling Initiating at the Cell Surface Can Potentially Influence Chromatin Dynamics and Chromatin-Bound Proteins in the Nucleus. Front. Plant Sci. 2019, 10, 1267. [Google Scholar] [CrossRef]
- Kirby, B.J.; Hasselbrink, E.F., Jr. Zeta potential of microfluidic substrates: 2. Data for polymers. Electrophoresis 2004, 25, 203–213. [Google Scholar] [CrossRef]
- Kirby, B.J.; Hasselbrink, E.F., Jr. Zeta potential of microfluidic substrates: 1. Theory, experimental techniques, and effects on separations. Electrophoresis 2004, 25, 187–202. [Google Scholar] [CrossRef]
- Chakravorty, A.; Jia, Z.; Li, L.; Alexov, E. A New DelPhi Feature for Modeling Electrostatic Potential around Proteins: Role of Bound Ions and Implications for Zeta-Potential. Langmuir 2017, 33, 2283–2295. [Google Scholar] [CrossRef]
- Petukh, M.; Alexov, E. Ion binding to biological macromolecules. Asian J. Phys. 2014, 23, 735–744. [Google Scholar] [PubMed]
- Petukh, M.; Kimmet, T.; Alexov, E. BION web server: Predicting non-specifically bound surface ions. Bioinformatics 2013, 29, 805–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Dong, Q.; Yang, J.; Zhang, Y. Recognizing metal and acid radical ion-binding sites by integrating ab initio modeling with template-based transferals. Bioinformatics 2016, 32, 3260–3269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.-F.; Cheng, C.-W.; Shih, C.-S.; Hwang, J.-K.; Yu, C.-S.; Lu, C.-H. MIB: Metal Ion-Binding Site Prediction and Docking Server. J. Chem. Inf. Model. 2016, 56, 2287–2291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravorty, A.; Jia, Z.; Peng, Y.; Tajielyato, N.; Wang, L.; Alexov, E. Gaussian-Based Smooth Dielectric Function: A Surface-Free Approach for Modeling Macromolecular Binding in Solvents. Front. Mol. Biosci. 2018, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravorty, A.; Panday, S.; Pahari, S.; Zhao, S.; Alexov, E. Capturing the Effects of Explicit Waters in Implicit Electrostatics Modeling: Qualitative Justification of Gaussian-Based Dielectric Models in DelPhi. J Chem. Inf. Model. 2020, 60, 2229–2246. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Li, L.; Chakravorty, A.; Alexov, E. Treating ion distribution with Gaussian-based smooth dielectric function in DelPhi. J. Comput. Chem. 2017, 38, 1974–1979. [Google Scholar] [CrossRef]
- Li, L.; Li, C.; Alexov, E. On the Modeling of Polar Component of Solvation Energy using Smooth Gaussian-Based Dielectric Function. J. Theor. Comput. Chem. 2014, 13, 1440002. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Li, C.; Zhang, Z.; Alexov, E. On the Dielectric “Constant” of Proteins: Smooth Dielectric Function for Macromolecular Modeling and Its Implementation in DelPhi. J. Chem. Theor. Comput. 2013, 9, 2126–2136. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, J.A.; Pickup, B.T.; Sykes, M.J.; Kitchen, C.A.; Nicholls, A. The Gaussian Generalized Born model: Application to small molecules. Phys. Chem. Chem. Phys. 2007, 9, 4913–4922. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB | FoldX(s) | BION-2 (s) | No. of Residues | Ion Type |
|---|---|---|---|---|
| 1L9A | 5.0 | 3.0 | 87 | Mg+2 |
| 1QGW | 33.0 | 9.0 | 176 | Mg+2 |
| 1E2D | 23.0 | 6.0 | 215 | Mg+2 |
| 1NG1 | 19.0 | 14.0 | 294 | Mg+2 |
| 1LR0 | 9.0 | 6.7 | 125 | Zn+2 |
| 2CEI | 27.7 | 9.3 | 183 | Zn+2 |
| 2AS9 | 72.7 | 12.0 | 210 | Zn+2 |
| 1ET5 | 54.0 | 16.0 | 341 | Zn+2 |
| 1TY3 | 22.0 | 14.0 | 357 | Zn+2 |
| 3HK5 | 205.0 | 65.0 | 427 | Zn+2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shashikala, H.B.M.; Chakravorty, A.; Panday, S.K.; Alexov, E. BION-2: Predicting Positions of Non-Specifically Bound Ions on Protein Surface by a Gaussian-Based Treatment of Electrostatics. Int. J. Mol. Sci. 2021, 22, 272. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010272
Shashikala HBM, Chakravorty A, Panday SK, Alexov E. BION-2: Predicting Positions of Non-Specifically Bound Ions on Protein Surface by a Gaussian-Based Treatment of Electrostatics. International Journal of Molecular Sciences. 2021; 22(1):272. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010272
Chicago/Turabian StyleShashikala, H. B. Mihiri, Arghya Chakravorty, Shailesh Kumar Panday, and Emil Alexov. 2021. "BION-2: Predicting Positions of Non-Specifically Bound Ions on Protein Surface by a Gaussian-Based Treatment of Electrostatics" International Journal of Molecular Sciences 22, no. 1: 272. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010272