Nephrotoxic Potential of Putative 3,5-Dichloroaniline (3,5-DCA) Metabolites and Biotransformation of 3,5-DCA in Isolated Kidney Cells from Fischer 344 Rats


Abstract
:1. Introduction
2. Results
2.1. Time and Concentration Cytotoxicity Relationships for 3,5-DCA Metabolites
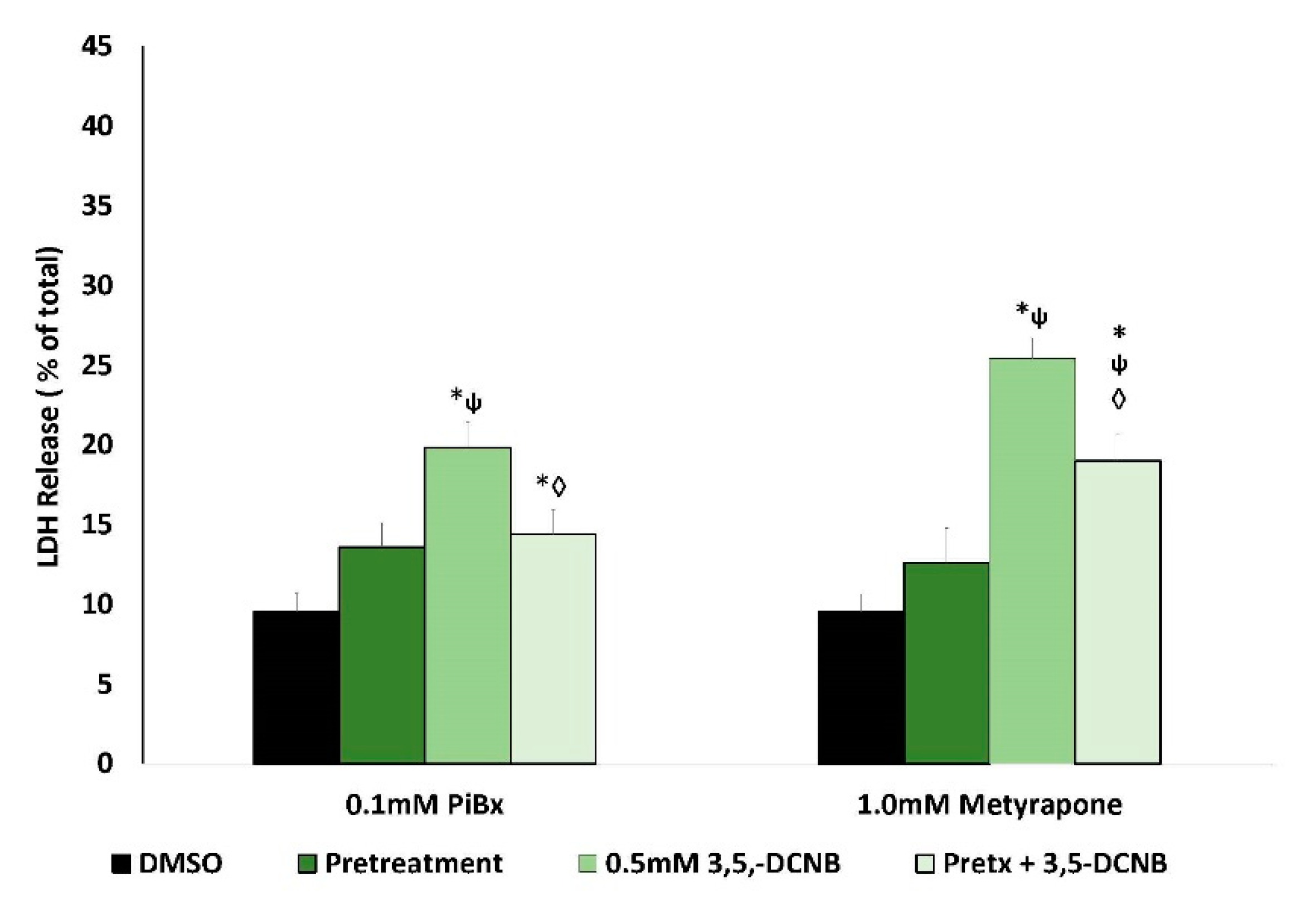
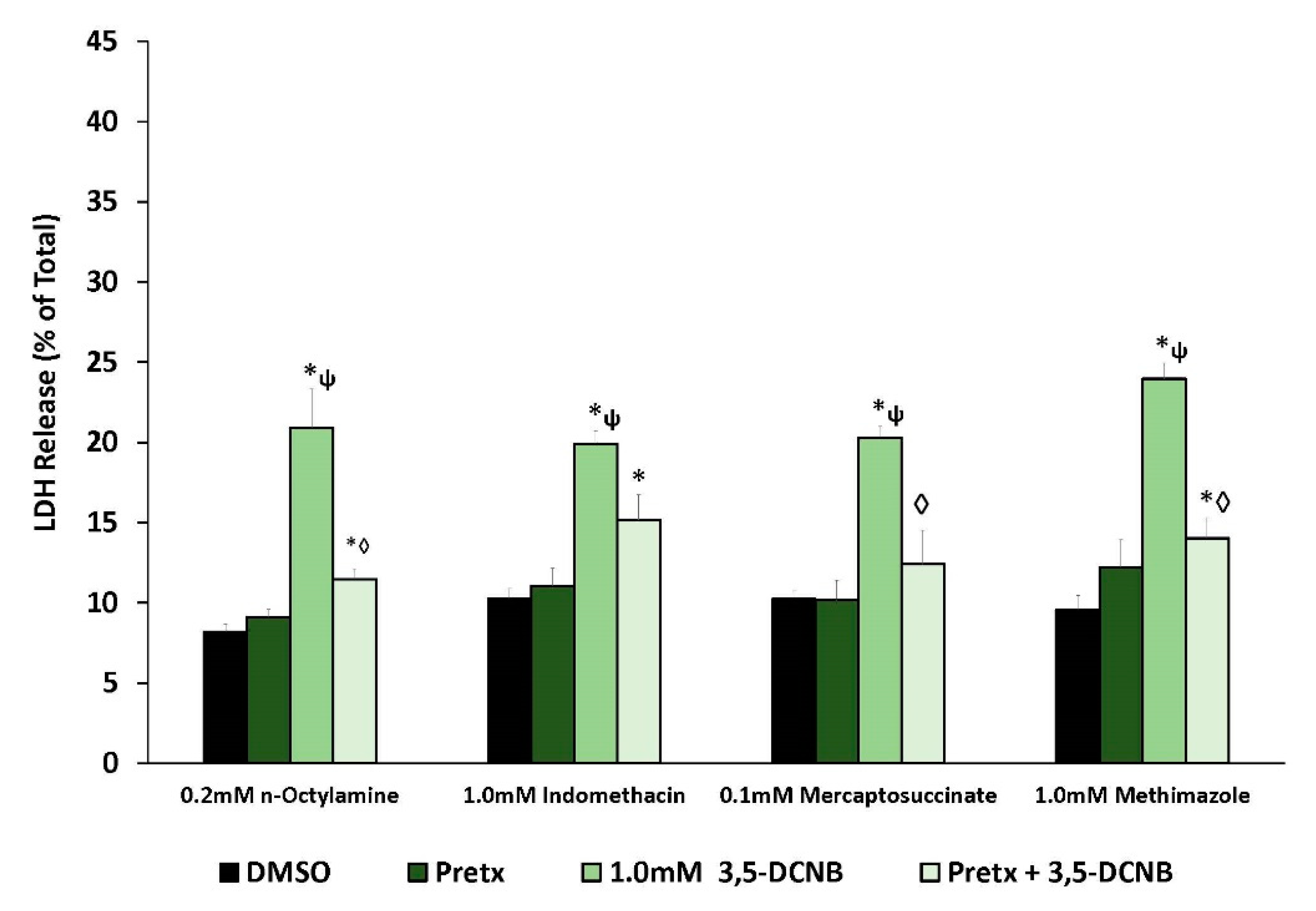
2.2. Renal Biotransformation and 3,5-DCNB Cytotoxicity
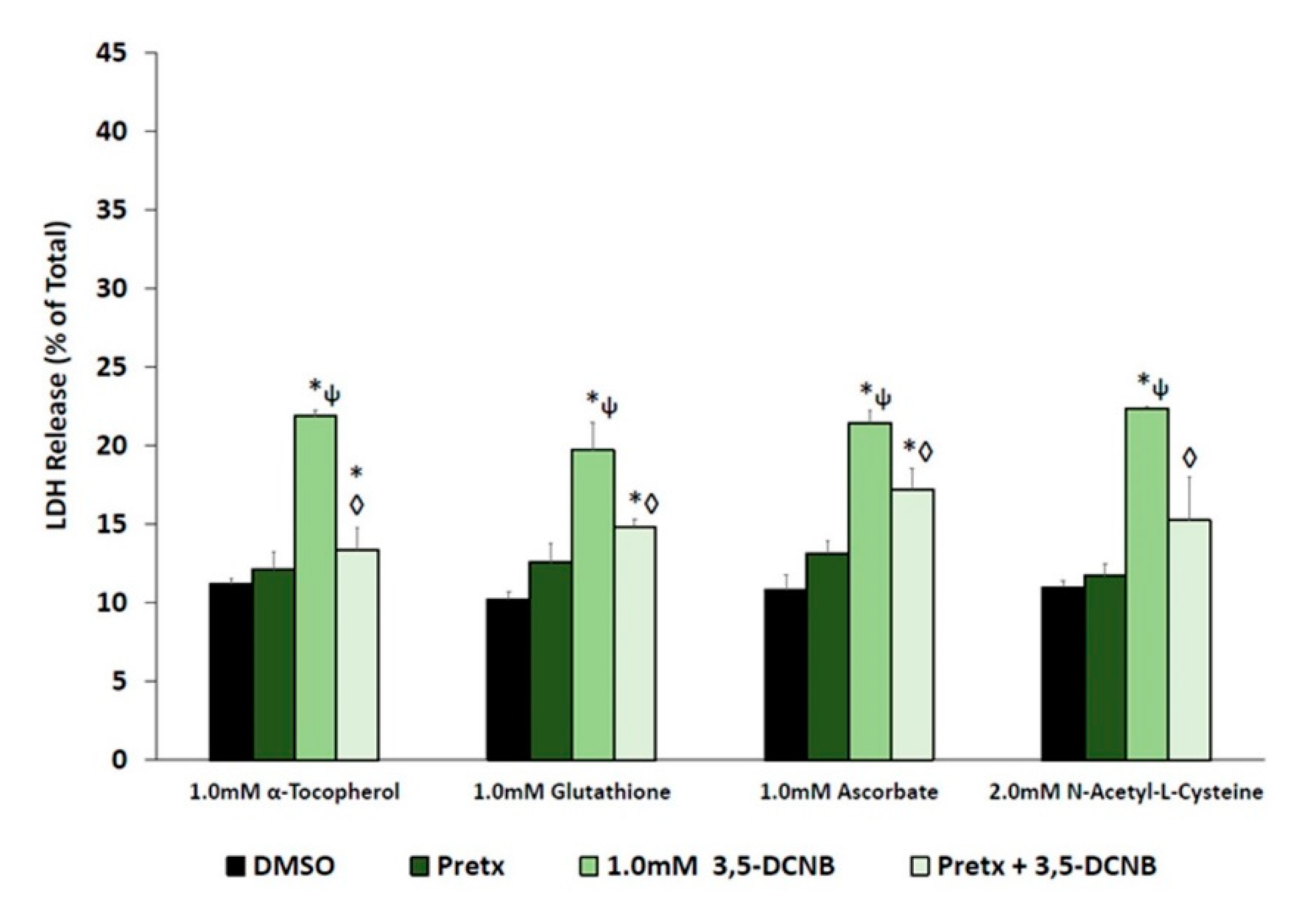
2.3. Antioxidants and 3,5-DCPHA Cytotoxicity
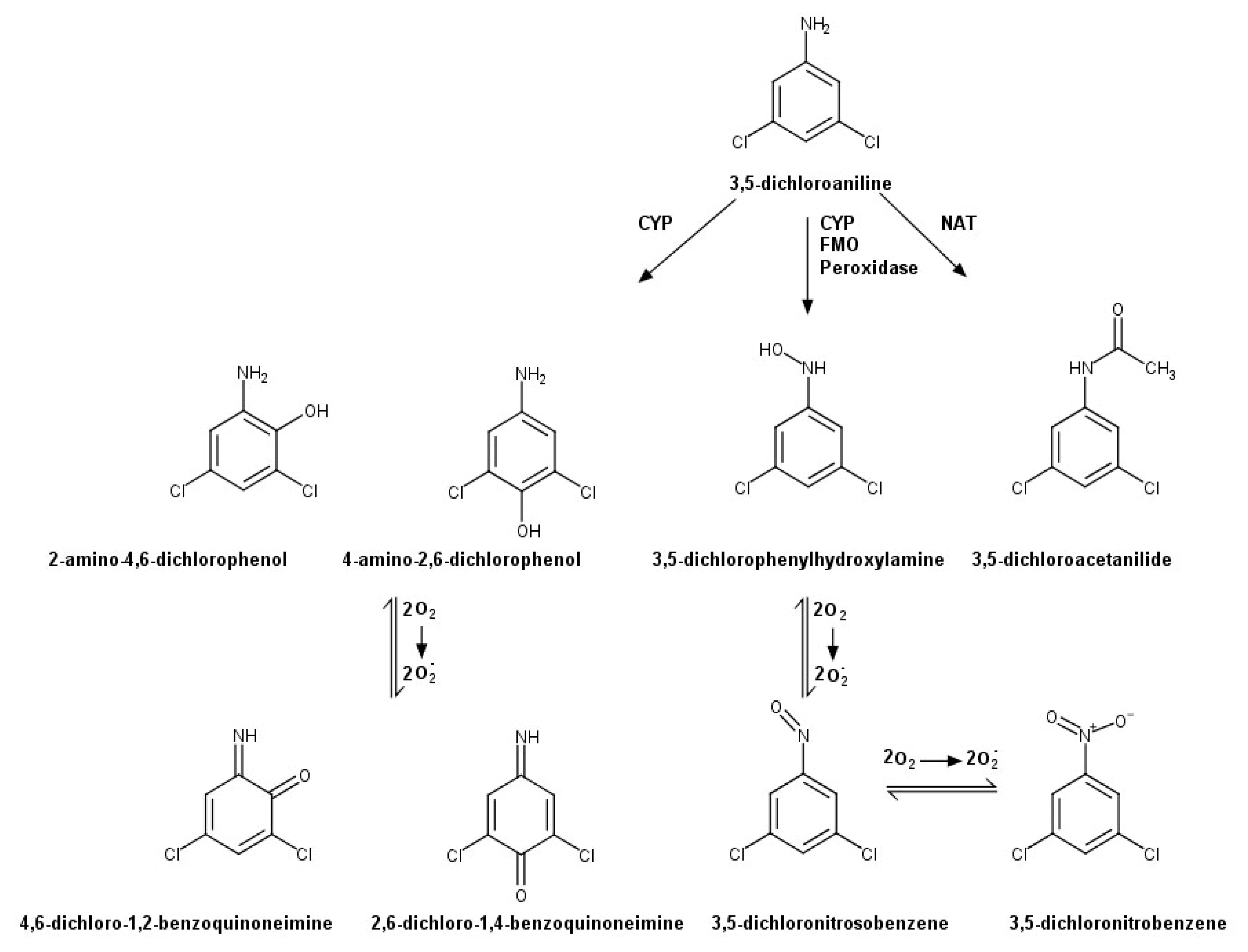
2.4. 3,5-DCA Metabolism by IKC
2.5. Effect of Diethyldithiocarbamic Acid (DEDTCA) on 3,5-DCA Metabolism
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Chemicals
4.3. Preparation and Treatment of Isolated Kidney Cells
4.4. IKC Metabolism of 3,5-DCA
4.4.1. IKC Incubations and Metabolite Isolation
4.4.2. Determination of Sulfate and Glucuronide Conjugates
4.4.3. Inhibition of CYP2C Metabolism by DEDCTA
4.4.4. HPLC Determination of Metabolites
4.5. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2-A-4,6-DCP | 2-Amino-4,6-dichlorophenol |
| 3,5-DCA | 3,5-Dichloroaniline |
| 3,5-DCAA | 3,5-Dichloroacetanilide |
| 3,5-DCNB | 3,5-Dichloronitrobenzene |
| 3,5-DCPHA | 3,5-Dichlorophenylhydroxylamine |
| 4-A-2,6-DCP | 4-Amino-2,6-dichlorophenol |
| ASC | Ascorbate |
| CYP | Cytochrome P450 |
| DEDTCA | Diethyldithiocarbamate |
| DMSO | Dimethyl sulfoxide |
| FMO | Flavin-containing monooxygenase |
| GSH | Glutathione |
| IKC | Isolated kidney cells |
| LDH | Lactate dehydrogenase |
| NAC | N-Acetyl-L-cysteine |
| PiBx | Piperonyl butoxide |
References
- Aizawa, H. Metabolic Maps of Pesticides; Academic Press, Inc.: London, UK, 1989; Volume 2. [Google Scholar]
- Ehlhardt, W.J. Metabolism and disposition of the anticancer agent sulfenur in mouse, rat, monkey, and human. Drug Metab. Dispos. 1991, 19, 370–375. [Google Scholar] [PubMed]
- Rickert, D.D.; Held, S.D. Metabolism of chloronitrobenzenes by isolated rat hepatocytes. Drug Metab. Dispos. 1990, 18, 5–9. [Google Scholar] [PubMed]
- Lee, J.B.; Sohn, H.Y.; Shin, K.S.; Kim, J.S.; Jo, M.S.; Jeon, C.P.; Jang, J.O.; Kim, J.E.; Kwon, G.S. Microbial biodegradation and toxicity of vinclozolin and its toxic metabolite 3,5-dichloroaniline. J. Microbiol. Biotechnol. 2008, 18, 343–349. [Google Scholar] [PubMed]
- Santos, T.C.R.; Rocha, J.C.; Alonsa, R.M.; Martinez, E.; Ibanez, C.; Barcelo, D. Rapid degradation of propanil in rice field crops. Environ. Sci. Technol. 1998, 32, 3479–3484. [Google Scholar] [CrossRef]
- Marlett, V.L.; Martyniuk, C.J. Biological responses to phenylurea herbicides in fish and amphibians; new directions for characterizing mechanisms of toxicity. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2017, 194, 9–21. [Google Scholar] [CrossRef]
- Lindh, C.H.; Littorin, M.; Amilon, A.; Jonsson, B.A. Analysis of 3,5-dichloroaniline as a biomarker of vinclozolin and iprodione in human urine using liquid chromatography/triple quadrupole mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 536–542. [Google Scholar] [CrossRef] [Green Version]
- Kutting, B.; Goen, T.; Schwegler, U.; Fromme, H.; Uter, W.; Angerer, J.; Drexler, H. Monoarylamines in the general population—A cross-sectional population-based study including 1004 Bavarian subjects. Int. J. Hyg. Environ. Health 2009, 212, 298–309. [Google Scholar] [CrossRef]
- Turci, R.; Barisano, A.; Balducci, C.; Colosio, C.; Minoia, C. Determination of dichloroanilines in human urine by gas chromatography/mass spectrometry: Validation protocol and establishment of Reference Values in a population group living in central Italy. Rapid Commun. Mass Spectrom. 2006, 20, 2621–2625. [Google Scholar] [CrossRef]
- Vitelli, N.; Chiodini, A.; Colosio, C.; De Paschale, G.; Somaruga, C.; Turci, R.; Minoia, C.; Brambilla, G.; Colombi, A. Occupational and environmental exposure to anilide and dicarboximide pesticides. G. Ital. Med. Lav. Ergon. 2007, 29 (Suppl. 3), 276–277. [Google Scholar]
- Chhabra, R.S.; Thompson, M.; Elwell, M.R.; Gerken, D.K. Toxicity of p-chloroaniline in rats and mice. Food Chem. Toxicol. 1990, 28, 717–722. [Google Scholar] [CrossRef]
- Guilhermino, L.; Soares, A.M.; Carvalho, A.P.; Lopes, M.C. Acute effects of 3,4-dichloroaniline on blood of male Wistar rats. Chemosphere 1998, 37, 619–632. [Google Scholar] [CrossRef]
- Pizon, A.F.; Schwartz, A.R.; Shum, L.M.; Rittenberger, J.C.; Lower, D.R.; Giannoutsos, S.; Virji, M.A.; Krasowski, M.D. Toxicology laboratory analysis and human exposure to p-chloroaniline. Clin. Toxicol. 2009, 47, 132–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenig, C.M.; Beevers, C.; Pant, K.; Young, R.R. Assessment of the mutagenic potential of para-chloroaniline and aniline in the liver, spleen, and bone marrow of Big Blue® rats with micronuclei analysis in peripheral blood. Environ. Mol. Mutagen. 2018, 59, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Valentovic, M.A.; Ball, J.G.; Anestis, D.K.; Beers, K.W.; Madan, E.; Hubbard, J.L.; Rankin, G.O. Acute renal and hepatic toxicity of 2-haloanilines in Fischer 344 rats. Toxicology 1992, 75, 121–131. [Google Scholar] [CrossRef]
- Valentovic, M.A.; Ball, J.G.; Anestis, D.K.; Rankin, G.O. Comparison of the in vitro toxicity of dichloroaniline structural isomers. Toxicol. In Vitro 1995, 9, 75–81. [Google Scholar] [CrossRef]
- Valentovic, M.A.; Lo, H.H.; Brown, P.I.; Rankin, G.O. 3,5-Dichloroaniline toxicity in Fischer 344 rats pretreated with inhibitors and inducers of cytochrome P450. Toxicol. Lett. 1995, 78, 207–214. [Google Scholar] [CrossRef]
- Bhuiyan, M.N.H.; Kang, H.; Kim, J.H.; Kim, S.; Kho, Y.; Choi, K. Endocrine disruption by several aniline derivatives and related mechanisms in a human adrenal H295R cell line and adult male zebrafish. Ecotoxicol. Environ. Saf. 2019, 180, 326–332. [Google Scholar] [CrossRef]
- Hong, S.K.; Anestis, D.K.; Henderson, T.T.; Rankin, G.O. Haloaniline-induced in vitro nephrotoxicity: Effects of 4-haloanilines and 3,5-dihaloanilines. Toxicol. Lett. 2000, 14, 125–133. [Google Scholar] [CrossRef]
- Racine, C.; Ward, D.; Anestis, D.K.; Ferguson, T.; Preston, D.; Rankin, G.O. 3,4,5-Trichloroaniline nephrotoxicity in vitro: Potential role of free radicals and renal biotransformation. Int. J. Mol. Sci. 2014, 15, 20900–20912. [Google Scholar] [CrossRef] [Green Version]
- Boehncke, A.; Kielhorn, J.; Konnecker, G.; Pohlenz-Michel, C.; Mangelsdorfer, I. Concise International Chemical Assessment Document 48—4-Chloroaniline; World Health Organization: Geneva, Switzerland, 2003. [Google Scholar]
- Vangnai, A.S.; Kataoka, N.; Soonglerdsongpha, S.; Kslsmbaheti, C.; Tajima, T.; Kato, J. Construction and application of an Escherichia coli bioreporter for aniline and chloroaniline detection. J. Ind. Microbiol. Biotechnol. 2012, 39, 1801–1810. [Google Scholar] [CrossRef]
- Lo, H.H.; Brown, P.I.; Rankin, G.O. Acute nephrotoxicity induced by isomeric dichloroanilines in Fischer 344 rats. Toxicology 1990, 63, 215–231. [Google Scholar] [CrossRef]
- Rankin, G.O.; Yang, D.J.; Teets, V.J.; Lo, H.H.; Brown, P.I. 3,5-Dichloroaniline-induced nephrotoxicity in the Sprague-Dawley rat. Toxicol. Lett. 1986, 30, 173–179. [Google Scholar] [CrossRef]
- Racine, C.R.; Ferguson, T.; Preston, D.; Ward, D.; Ball, J.; Anestis, D.; Valentovic, M.; Rankin, G.O. The role of biotransformation and oxidative stress in 3,5-dichloroaniline (3,5-DCA) induced nephrotoxicity in isolated renal cortical cells from male Fischer 344 rats. Toxicology 2016, 341–343, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.K.; Rankin, G.O. Biotransformation of 2-chloroaniline in the Fischer 344 rat: Identification of urinary metabolites. Xenobiotica 1998, 28, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Rankin, G.O.; Racine, C.; Sweeney, A.; Kraynie, A.; Anestis, D.K.; Barnett, J.B. In vitro nephrotoxicity induced by propanil. Environ. Toxicol. 2008, 23, 435–442. [Google Scholar] [CrossRef] [Green Version]
- Cnubben, N.H.P.; Peelen, S.; Borst, J.-W.; Vervoot, J.; Veeger, C.; Rietjens, I.M.C.M. Molecular orbital-based quantatative structure-activity relationship for the cytochrome P50-catalyzed 4-hydroxylation of halogenated anilines. Chem. Res. Toxicol. 1994, 7, 590–598. [Google Scholar] [CrossRef]
- McMillan, D.C.; Freeman, J.P.; Hinson, J.A. Metabolism of the arylamide herbicide propanil. I. Microsomal metabolism and in vitro methemoglobinemia. Toxicol. Appl. Pharmacol. 1990, 103, 90–101. [Google Scholar] [CrossRef]
- McMillan, D.C.; Bradshaw, T.P.; Hinson, J.A.; Jollow, D.J. Role of metabolites in propanil-induced hemolytic anemia. Toxicol. Appl. Pharmacol. 1991, 110, 70–78. [Google Scholar] [CrossRef]
- Rankin, G.O.; Valentovic, M.A.; Nicoll, D.W.; Ball, J.G.; Anestis, D.K.; Wang, R.T.; Brown, P.I. In vivo and in vitro 4-amino-2,6-dichlorophenol nephrotoxicity and hepatotoxicity in the Fischer 344 rat. Toxicology 1994, 90, 115–128. [Google Scholar] [CrossRef]
- Hong, S.K.; Anestis, D.K.; Ball, J.G.; Valentovic, M.A.; Brown, P.I.; Rankin, G.O. 4-Amino-2,6-dichlorophenol (ADCP) nephrotoxicity in the Fischer 344 rat: Protection by ascorbic acid, AT-125 and aminooxyacetic acid. Toxicol. Appl. Pharmacol. 1997, 147, 115–125. [Google Scholar] [CrossRef]
- Rankin, G.O.; Hong, S.-K.K.; Anestis, D.K.; Ball, J.G.; Valentovic, M.A. Mechanistic aspects of 4-amino-2,6-dichlorophenol-induced in vitro nephrotoxicity. Toxicology 2008, 245, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.K.; Anestis, D.K.; Ball, J.G.; Valentovic, M.A.; Rankin, G.O. In vitro nephrotoxicity induced by chloronitrobenzenes in renal cortical slices from Fischer 344 rats. Toxicol. Lett. 2002, 129, 133–141. [Google Scholar] [CrossRef]
- Rankin, G.O.; Valentovic, M.A.; Beers, K.W.; Nicoll, D.W.; Ball, J.G.; Anestis, D.K.; Brown, P.I.; Hubbard, J.L. Renal and hepatic toxicity of monochloroacetanilides in the Fischer 344 rat. Toxicology 1993, 79, 181–193. [Google Scholar] [CrossRef]
- Rankin, G.O.; Valentovic, M.A.; Hong, S.K.; Anestis, D.K.; Ball, J.G.; Dial, L.D. In vivo and in vitro renal effects induced by 2-chloro-4-hydroxyacetanilide and 4-chloro-2-hydroxyacetanilide in the Fischer 344 rat. Toxic Subst. Mech. 1995, 14, 93–109. [Google Scholar]
- Valentovic, M.A.; Ball, J.G. 2-Aminophenol and 4-aminophenol toxicity in renal cortical slices from Sprague-Dawley and Fischer 344 rats. J. Toxicol. Environ. Health Part A 1998, 55, 225–240. [Google Scholar]
- Valentovic, M.A.; Ball, J.G.; Hong, S.K.; Rogers, B.A.; Meadows, M.K.; Harmon, R.C.; Rankin, G.O. In vitro toxicity of 2- and 4-chloroaniline: Comparisons with 4-amino-3-chlorophenol, 2-amino-5-chlorophenol and aminophenols. Toxicol. In Vitro 1996, 10, 713–720. [Google Scholar] [CrossRef]
- Harrison, J.H., Jr.; Jollow, D.J. Contribution of aniline metabolites to aniline-induced methemoglobinemia. Mol. Pharmacol. 1987, 32, 423–431. [Google Scholar]
- Valentovic, M.A.; Rogers, B.A.; Meadows, M.K.; Conner, J.T.; Williams, E.; Hong, S.K.; Rankin, G.O. Characterization of methemoglobin formation by 3,5-dichloroaniline, 4-amino-2,6-dichlorophenol and 3,5-dichlorophenylhydroxylamine. Toxicology 1997, 118, 23–36. [Google Scholar] [CrossRef]
- McMillan, D.C.; McRae, T.A.; Hinson, J.A. Propanil-induced methemoglobinemia and hemoglobin binding in the rat. Toxicol. Appl. Pharmacol. 1990, 105, 503–507. [Google Scholar] [CrossRef]
- Eyer, P.; Ascherl, M. Reactions of para-substituted nitrosobenzenes with human hemoglobin. Biol. Chem. Hoppe Seyler 1987, 368, 285–294. [Google Scholar] [CrossRef]
- Cheng, L.; Stewart, B.J.; You, Q.; Petersen, D.R.; Ware, J.A.; Picotti, J.R.; Kawabata, T.T.; Ju, C. Covalent binding of the nitrso metabolite of sulfamethoxazole is important in induction of drug-specific T-cell responses in vivo. Mol. Pharmacol. 2008, 73, 1769–1775. [Google Scholar] [CrossRef] [PubMed]
- McLean, S.; Starmer, G.A.; Thomas, J. Methaemoglobinemia formation by aromatic amines. J. Pharm. Pharmacol. 1969, 21, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Radomski, J.L. The primary aromatic amines: Their biological properties and structure-activity relationships. Ann. Rev. Pharmacol. Toxicol. 1979, 19, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Rankin, G.O.; Sweeney, A.; Racine, C.; Ferguson, T.; Preston, D.; Anestis, D.K. 4-Amino-2-chlorophenol: Comparative in vitro nephrotoxicity and mechanisms of bioactivation. Chemico-Biol. Interact. 2014, 222, 126–132. [Google Scholar] [CrossRef] [Green Version]
- Racine, C.R. 3,5-Dichloroaniline: Biotransformation and Mechanistic Aspects of Nephrotoxicity In Vitro. Ph.D. Thesis, Marshall University, Huntington, WV, USA, December 2016. [Google Scholar]
- Gorrod, J.W.; Damani, L.A. Biological Oxidation of Nitrogen in Organic MOLECULES: Chemistry, Toxicology and Pharmacology; Ellis Horwood Ltd.: Chichester, UK, 1985. [Google Scholar]
- Burstyn, J.N.; Iskandar, M.; Brady, J.F.; Fukuto, J.M.; Cho, A.K. Comparative studies of N-hydroxylation and N-demethylation by microsomal cytochrome P-450. Chem. Res. Toxicol. 1991, 4, 70–76. [Google Scholar] [CrossRef]
- Hlavica, P.; Golly, I.; Mietaschk, J. Comparative studies on the cumene hydroperoxide- and NADPH-supported N-oxidation of 4-chloroaniline by cytochrome P-450. Biochem. J. 1983, 212, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Golly, I.; Hlavica, P. N-Oxidation of 4-chloroaniline by prostaglandin synthase. Biochem. J. 1985, 260, 803–809. [Google Scholar] [CrossRef] [Green Version]
- Travlos, G.S.; Mahler, J.; Ragan, H.A.; Choy, B.J.; Bucher, J.R. Thirteen-week inhalation study of 2- and 4-chloronitrobenzene in F344/N rats and B6C3F1 mice. Fundam. Appl. Toxicol. 1996, 30, 75–92. [Google Scholar] [CrossRef]
- Yamazaki, K.; Aiso, S.; Matsumoto, M.; Arito, H.; Nagano, K.; Yamamoto, S.; Matsushima, T. Thirteen week oral toxicity study of 1,4-dichloro-2-nitrobenzene in rats and mice. Ind. Health 2005, 43, 597–610. [Google Scholar] [CrossRef] [Green Version]
- Rondestvedt, C.S., Jr.; Johnston, T.A. Rapid preparation of chloroarylhydroxylamine by hydrazine-palladium reduction of chloronitrobenzene. Synthesis 1977, 12, 850–851. [Google Scholar] [CrossRef]
- Valentovic, M.; Ball, J.G.; Stoll, S.; Rankin, G.O. 3,4-Dichlorophenylhydroxylamine cytotoxicity in renal cortical slices from Fischer 344 rats. Toxicology 2001, 162, 149–156. [Google Scholar] [CrossRef]
- Newell, M.; Argus, M.; Ray, F.E. Routes of metabolism of [36Cl] ring-substituted monochloroacetanilides. Biochem. Pharmacol. 1960, 5, 30–38. [Google Scholar] [CrossRef]
- McMillan, D.C.; Leaky, J.E.; Arlotto, M.P.; McMillan, J.M.; Hinson, J.A. Metabolism of the arylamide herbicide propanil. II. Effects of propanil and its derivatives on hepatic microsomal drug metabolizing enzymes in the rat. Toxicol. Appl. Pharmacol. 1990, 103, 102–112. [Google Scholar] [CrossRef]
- Jones, D.P.; Sundby, G.B.; Ormstad, K.; Orrenius, S. Use of isolated kidney cells for study of drug metabolism. Biochem. Pharmacol. 1979, 28, 929–935. [Google Scholar] [CrossRef]
- Hagen, T.M.; Aw, T.Y.; Jones, D.P. Glutathione uptake and protection against oxidative injury in isolated kidney cells. Kidney Int. 1988, 34, 74–81. [Google Scholar] [CrossRef] [Green Version]
- Lash, L.H. Isolated kidney cells in the study of chemical toxicity. In In Vitro Toxicology: Model Systems and Methods; McQueen, C.A., Ed.; Teleford Press: Caldwell, NJ, USA, 1989; pp. 231–262. [Google Scholar]
- Lash, L.H. Nephrotoxicity studies with freshly isolated cells from rat kidney. In In Vitro Methods of Toxciology; Watson, R.R., Ed.; CRC Press: Boca Raton, FL, USA, 1992; pp. 115–122. [Google Scholar]
- Henesey, C.M.; Harvison, P.J. Potential metabolism and cytotoxicity of N-(3,5-dichlorophenyl)succinimide and its hepatic metabolites in isolated rat renal cortical tubule cells. Toxicology 1995, 104, 9–16. [Google Scholar] [CrossRef]
- Lash, L.H. In vitro methods of assessing renal damage. Toxicol. Pathol. 1998, 26, 33–42. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Retention Time (Min) | Limit of Detection (µg) | Extraction Efficiency (%) |
|---|---|---|---|
| 4-A-2,6-DCP | 3.8 | 0.018 | 75.8 |
| 2-A-4,6-DCP | 13.8 | 0.018 | 97.8 |
| 3,5-DCPHA | 24.6 | 17.8 | 0.0 * |
| 3,5-DCA | 29.4 | 0.002 | 89.5 |
| 3,5-DCAA | 42.6 | 0.002 | 96.1 |
| 3,5-DCNB | 57.7 | 0.002 | 79.7 |
| Compound | 0.5 mM | 1.0 mM | ||
|---|---|---|---|---|
| Percentage of Dose (%) | ||||
| Media | Cells | Media | Cells | |
| 4-A-2,6-DCP | ND | ND | ND | ND |
| 2-A-4,6-DCP | ND | ND | ND | ND |
| 3,5-DCPHA | ND | ND | ND | ND |
| 3,5-DCA | 89.31 ± 6.32 | 6.82 ± 3.33 | 87.13 ± 4.56 | 13.19 ± 2.12 |
| 3,5-DCAA | 1.86 ± 0.42 | 0.07 ^ | 0.56 ± 0.07 | ND |
| 3,5-DCNB | 2.80 ^ | 0.41 ^ | 1.18 ± 0.57 # | 0.34 ± 0.19 # |
| Total | 93.97 ± 5.52 | 7.30 ± 3.32 | 88.83 ± 5.14 | 13.53 ± 5.14 |
| Compound | −DEDTCA | +DEDTCA | ||
|---|---|---|---|---|
| Percent of Dose (%) | ||||
| Media | Cells | Media | Cells | |
| 3,5-DCA | 89.43 ± 5.66 | 16.01 ± 2.02 | 91.14 ± 3.04 | 14.67 ± 1.78 |
| 3,5-DCAA | 0.50 ± 0.03 | 0.02 ^ | 0.30 ± 0.05 | ND |
| 3,5-DCNB | 0.31 ± 0.19 # | 0.13 ^ | 0.09 ^ | 0.06 ^ |
| Pretreatment | Concentration | Pretreatment Time | Mechanism of Action |
|---|---|---|---|
| (mM) | (Min) | or Enzyme Inhibited | |
| α-Tocopherol | 1 | 5 | Antioxidant |
| Ascorbate | 1.0 or 2.0 | 5 | Antioxidant |
| Glutathione | 1 | 30 | Antioxidant |
| N-Acetyl-L-Cysteine | 2 | 30 | Antioxidant |
| Piperonyl Butoxide | 1 | 15 | CYP General |
| Metyrapone | 1 | 5 | CYP General |
| n-Octylamine | 2 | 5 | FMO |
| Methimazole | 1 | 30 | FMO |
| Indomethacin | 1 | 15 | Cyclooxygenaase |
| Mercaptosuccinate | 0.1 | 15 | Peroxidase |
| DEDTCA | 0.1 | 30 | CYP2C > CYP2E |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rankin, G.O.; Racine, C.R.; Valentovic, M.A.; Anestis, D.K. Nephrotoxic Potential of Putative 3,5-Dichloroaniline (3,5-DCA) Metabolites and Biotransformation of 3,5-DCA in Isolated Kidney Cells from Fischer 344 Rats. Int. J. Mol. Sci. 2021, 22, 292. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010292
Rankin GO, Racine CR, Valentovic MA, Anestis DK. Nephrotoxic Potential of Putative 3,5-Dichloroaniline (3,5-DCA) Metabolites and Biotransformation of 3,5-DCA in Isolated Kidney Cells from Fischer 344 Rats. International Journal of Molecular Sciences. 2021; 22(1):292. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010292
Chicago/Turabian StyleRankin, Gary O., Christopher R. Racine, Monica A. Valentovic, and Dianne K. Anestis. 2021. "Nephrotoxic Potential of Putative 3,5-Dichloroaniline (3,5-DCA) Metabolites and Biotransformation of 3,5-DCA in Isolated Kidney Cells from Fischer 344 Rats" International Journal of Molecular Sciences 22, no. 1: 292. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010292