
Tandem Repeats in Bacillus: Unique Features and Taxonomic Distribution

Department of Computer Science, Polytechnic University of Catalonia, 08034 Barcelona, Spain
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(10), 5373; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105373
Submission received: 20 April 2021
/
Revised: 14 May 2021
/
Accepted: 18 May 2021
/
Published: 20 May 2021
(This article belongs to the Special Issue Repetitive DNA Sequences in Eukaryotic Genomes)
Abstract
:Little is known about DNA tandem repeats across prokaryotes. We have recently described an enigmatic group of tandem repeats in bacterial genomes with a constant repeat size but variable sequence. These findings strongly suggest that tandem repeat size in some bacteria is under strong selective constraints. Here, we extend these studies and describe tandem repeats in a large set of Bacillus. Some species have very few repeats, while other species have a large number. Most tandem repeats have repeats with a constant size (either 52 or 20–21 nt), but a variable sequence. We characterize in detail these intriguing tandem repeats. Individual species have several families of tandem repeats with the same repeat length and different sequence. This result is in strong contrast with eukaryotes, where tandem repeats of many sizes are found in any species. We discuss the possibility that they are transcribed as small RNA molecules. They may also be involved in the stabilization of the nucleoid through interaction with proteins. We also show that the distribution of tandem repeats in different species has a taxonomic significance. The data we present for all tandem repeats and their families in these bacterial species will be useful for further genomic studies.
1. Introduction
Bacterial genomes contain repetitive sequences, some of which have been studied in great detail [1]. Many of these repeats are identical or inverted sequences of different sizes interspersed throughout the genome. Some of these repeats do not have any adaptive value, while others influence gene expression. Another large group of repetitive sequences are tandem repeats or satellites. Many of them are polymorphic and present a variable number of tandem repeats in bacterial populations. These differences allow the characterization of different strains of a given bacterial species. A database covering tandem repeats suitable for this application was developed by Denoeud and Vergnaud [2].
In the previous work, we have studied the distribution of tandem repeats in 1241 bacterial genomes which had been fully sequenced and contain more than 19 tandem repeats per genome [3]. Among them we found species with an enigmatic group of tandem repeats, with a constant repeat size but variable sequence. Surprisingly, only two repeat sizes were found, either 20–21 or 52 nt. The constant size of repeats in many distantly related species strongly suggests that these sizes are important for their function. Tandem 20–23 nt repeats are found in individual species in many bacterial groups, whereas those with 52 nt repeats are only found in Firmicutes, mainly in Bacillus. For this reason, we decided to study in greater detail the tandem repeats in Bacillus.
In this paper, we provide a catalogue of all the DNA tandem repeats present in the 176 genomes of Bacillus recently studied by Khurana et al. [4]. The parameters of our search have been adjusted to obtain all tandem repeats with a minimum of four repeats and repeat length 10–270 nt. We have determined the families of tandem repeats found in individual and related species. We include all species, whereas in our previous work [3] we only studied species with more than 20 tandem repeats, which excluded a large proportion of bacterial genomes. We have also analyzed in greater detail the sequences of tandem repeats and found that in many species short characteristic nucleotide sequences are present throughout all the tandem repeats of the same species.
A detailed analysis of the tandem repeats present in different species of Bacillus will allow determining if they have any phylogenetic significance. The taxonomy of Bacillus is controversial [5] and different approaches yield conflicting results [4,5,6,7]. Our detailed analysis of tandem repeats demonstrates that different taxonomic groups have a unique distribution of tandem repeats in their genomes. Finally, we explore the possibility that transcribed tandem repeats play a role as small RNAs. We also compare the results obtained with Bacillus and those found in eukaryotes.
2. Materials and Methods
2.1. Detection of Tandem Repeats
We have chosen to analyze the set of 176 Bacillus genomes studied by Khurana et al. [4], downloaded from the NCBI website [8]. All these genomes have been fully sequenced, but many of them have not been aligned. A complete list is given in Supplementary Table S1, including genome size, number of tandem repeats, and CG% for each species. For comparison, we have also used some of the fully assembled genomes we studied in our previous paper [3]. Tandem repeats have been searched with the SATFIND program, which is available in our website [9] and is described in great detail elsewhere [10,11]. The program determines the localization of clusters of any short sequence of a prefixed size without internal repetitions and repeated a minimum number of times in regions with a fixed size. Repeats of 1–5 nt are automatically eliminated. The minimum length of a repeat is taken as 10 nt in our search. As a result, short repeats, for example 7 nt, will appear as repeats with a double size, 14 nt. Once a tandem repeat is located, the program continues its search along the genome until no further neighboring repeats are detected, with no upper limit for the number of repeats. This program allows a precise definition of tandem repeats (repeat size, number of repeats, and internal regularity). We have adjusted the parameters in order to capture short tandem repeats with at least four repeats in a genome region of 800 nt. In order to eliminate the most irregular tandem repeats, we have only accepted those which have at least 60% of their repeats with an identical length (±1 nt). In this way, most irregular tandem repeats are eliminated, although with these parameters some tandem repeats with only four repeats may still be irregular. Occasionally, we have also changed the parameters of the program to detect additional tandem repeats with a decreased regularity by requiring that only 10% of them had an identical length.
Each tandem repeat has also been characterized by a similarity score obtained upon alignment of all its repeats which have an identical size, thus excluding all repeats with indels. Each tandem repeat may also be characterized by a homogeneity parameter which gives the proportion of repeats with the same length in each tandem repeat. This parameter varies between 0.6 (60%) and 1.0 (100%), since tandem repeats with low homogeneity have not been accepted, as mentioned above. The regularity of each tandem repeat is thus characterized by two parameters. Ni gives the number of repeats in the tandem repeat which have an identical length and an alignment score is calculated for these Ni repeats.
A limitation of our study is due to the difficulty to determine accurately the complete sequence of tandem repeats due to common sequencing errors [12,13]. However, when sequences of the same genome reported by different authors are compared [12], all of the found tandem repeats are in the same positions, although their length may differ due to limitations of the sequencing methodologies. Often when different strains of the same species are compared, fewer tandem repeats are found in the genomes that have not been fully assembled, an indication that some tandem repeats may be lost. For this reason, we have studied in more detail tandem repeats of Bacillus coagulans, for which we have complete sequences of several strains.
2.2. Identification of Tandem Repeat Families
In order to detect related tandem repeats, we have used MALIG, a progressive multiple sequence alignment algorithm, which we have developed to align tandem repeats and identify families with a related sequence, available on our website [9]. It has been described in detail elsewhere [10]. The program considers reverse sequences as well, normalizes the alignment score to the maximum possible value, and selects the cyclic permutation with the highest score. Then, the progressive multi-alignment is applied to the matrix of pairwise alignment scores. The process finishes when the score is smaller than a similarity threshold (input parameter) which we set to 0.6.
We have searched for tandem repeat families in the whole set of genomes studied. Each family is characterized by three values: Fam_a_b_c. The order in the list of families is given by a, starting with those families with the largest number of members. The second value b gives the size of the consensus repeat, and c gives the number of members in the family. The consensus sequence of the repeat in each family is calculated taking into account the circularly permuted sequence of all repeats. Individual families may contain tandem repeats with slightly different repeat lengths (±1 nt). The b parameter should be interpreted with caution. When the sequences of the tandem repeats which are compared to build a family have indels, their average repeat length may not coincide with b. For example, a tandem repeat family with b = 53 may have most of its tandem repeats with a shorter length of 51–52 nt.
The use of all the tandem repeats from all the Bacillus genomes together gives families which either include tandem repeats of several species or families which are only present in a single species. In the list of families, tandem repeats which have unique repeats, appear as families with a single member, c = 1.
2.3. Genome Alignments
We have determined the correspondence of genomes of different strains of Bacillus coagulans. We have used the multiple genome comparison and alignment tool (M-GCAT), a multiple genome alignment tool based on the search of maximal unique matches (MUMs) between genomes on both strands [14]. First, a set of anchor MUMs is found where those MUMs shorter than a specific parameter (minimum anchor length) or randomly found (shorter than log base 4 on the length of the genome) are discarded. These sets of anchor MUMs divide the genomes in several short parts in which a recursive search of MUMs is made. This recursive search is made until the length of the part is shorter that a given parameter (100 nucleotides in our case). Finally, close consecutive MUMs, separated by less than a given parameter (in our case 2000 nucleotides), are grouped in clusters. The program provides a numerical and a graphical representation of the alignment.
3. Results
3.1. General Features of Tandem Repeats
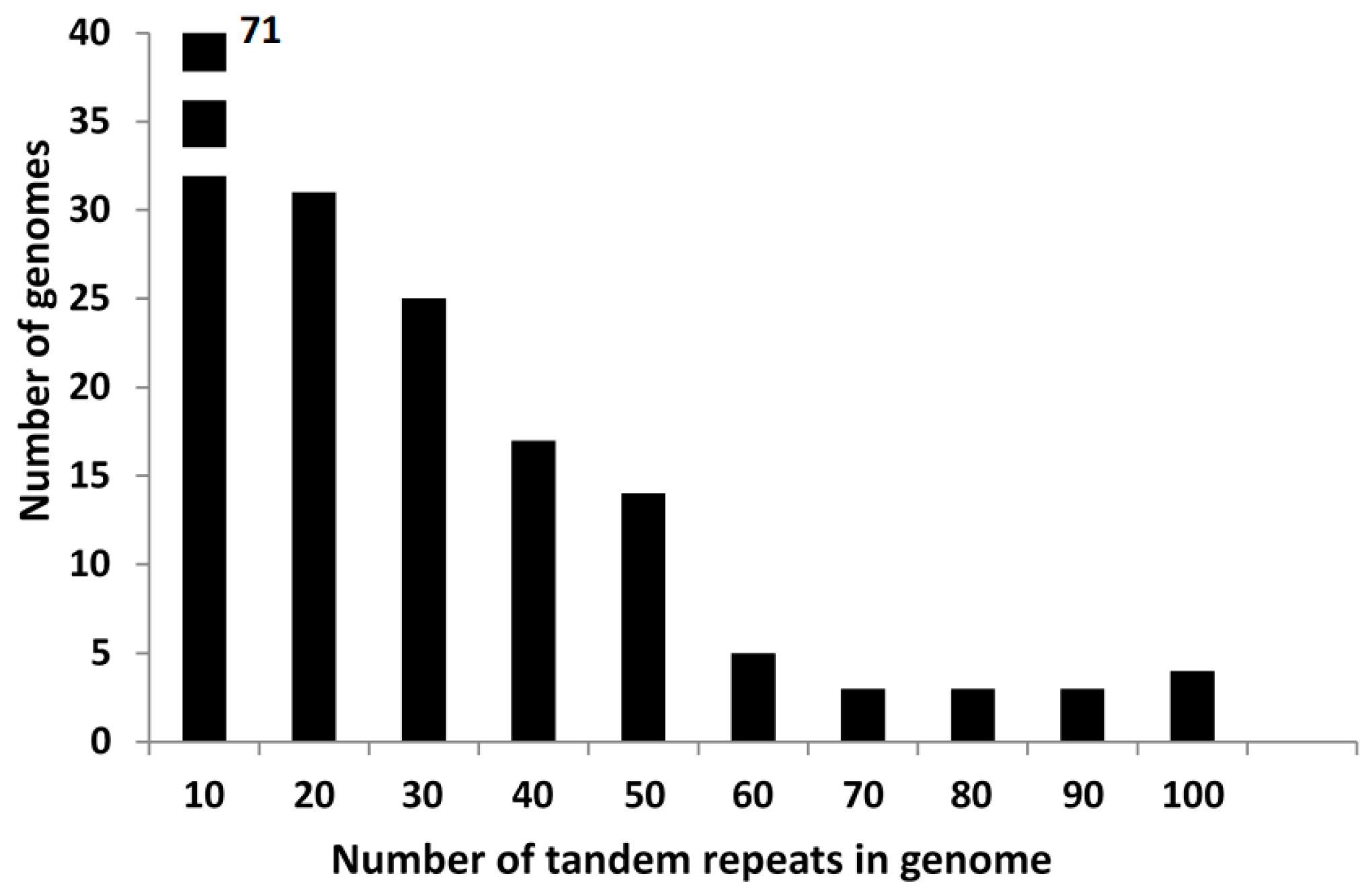
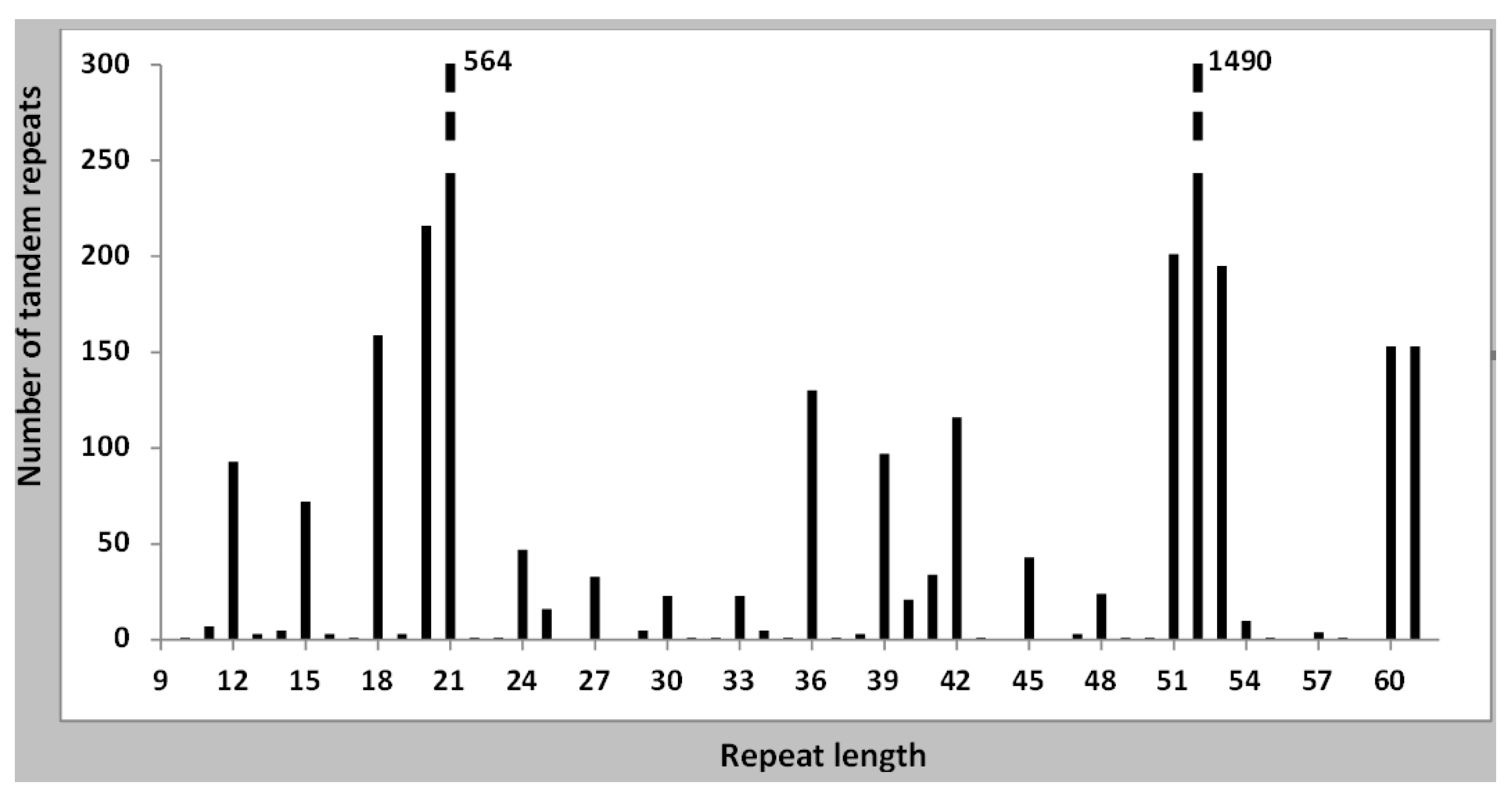
We have searched all the tandem repeats found in the 176 genomes which we have downloaded from GenBank, and detected 4029 tandem repeats in them, an average of 22.8 tandem repeats per genome. A complete list is given in Supplementary Table S2. For the study of the different types of tandem repeats we have prepared a list in which the size of all tandem repeats for each individual genome is given (Supplementary Table S1). A large number of species has very few tandem repeats, as shown in Figure 1. B. subtilis and the related species are found in this group. The overall distribution of tandem repeats, as a function of repeat size is presented in Figure 2. They fall into two main classes, with either a 20–21 repeat (781 cases) or a 51–53 repeat (1886 cases). Together they represent 66.2% of all tandem repeats. A third class is formed by those tandem repeats which have a repeat which is a multiple of three. We have next determined the tandem repeat families. A complete list is given in Supplementary Table S3, including all tandem repeats in each family. A summary of all families is also given in Supplementary Table S4. In the following, we will analyze separately each class of tandem repeats.
3.2. Tandem Repeats with a 52 nt Repeat
These tandem repeats offer a particular interest, since they have a very constant repeat length, while their composition is very variable. This is the most abundant class of tandem repeats in Bacillus (Figure 2), but they are only present in about 30% of the species we have studied. The repeat length is not a multiple of three, therefore these tandem repeats will not be able to code for amino acid repeats. A complete list of the species which have these tandem repeats is given in Supplementary Table S1. Each of them has a different group of tandem repeat families and unique tandem repeats, all with a constant repeat size but a different sequence. Occasionally, the repeat size may vary by one nucleotide (51 or 53 nt). The constant repeat length indicates that it is required for their function. The sequences of the repeats found in the species with a larger number of tandem repeats are given in Table 1. They are clearly species specific, with a few exceptions of closely related species. An additional feature of this group of species is the surprisingly small proportion of tandem repeats with different repeat lengths, as it is also apparent in Table 1. In most cases, when a species acquires 52 nt tandem repeats, other tandem repeats are absent, even those which might code for amino acid repeats in proteins. Most of these tandem repeats are rather short, with 4–10 repeats, but in all species a few longer tandem repeats are found. Histograms for a few species are presented in Figure S1. A particular case is B. cellulosilyticus, which has two long tandem repeats, with 25 and 37 repeats, the last one with over 2 Kb in length.
Each genome contains several families of these tandem repeats, but a close inspection demonstrates that in many cases all 52 nt repeats in a genome present conserved short sequences. A few examples are given in Table 2, note that each species has different sequences. Often these characteristic short sequences and their reverse are present in the same repeat. A list of all tandem repeats in the species shown in Table 2 is given in Supplementary Table S5. This nucleotide structure is reminiscent of the CRISPR repeats, which also present constant and variable sequence regions. However, the length of the sequences is completely different and, most important, the 52 nt tandem repeats are very abundant. CRISPR sequences are only present once or a few times in any bacterial genome [15]. Furthermore, there is no evidence for a protein similar to Cas which might interact specifically with the 52 nt repeats.
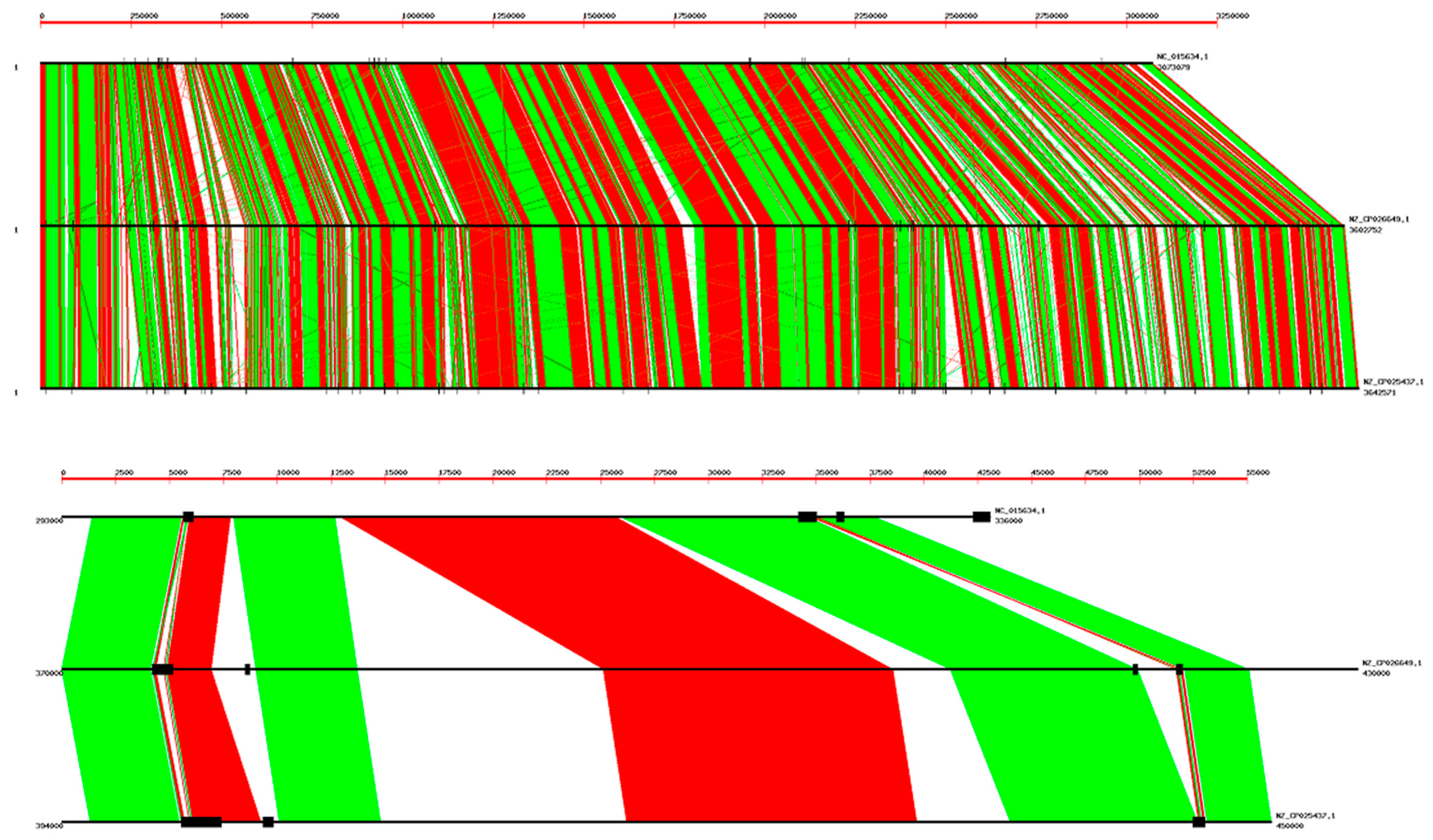
The next question is to find out if there is any turnover or specific position of the 52 nt tandem repeats in the genome. For this purpose, we have carried out a comparison of these tandem repeats in the genome of three strains of B. coagulans, as shown in Figure 3. As it is apparent, there is only a partial conservation of tandem repeats in different strains, in particular the length of tandem repeats is seldom conserved. More examples are presented in Figure S2.
In summary, several tandem repeat families with the 52 nt repeat are found in many Bacillus species. Their sequence is species specific. In different strains of the same species they are only partially conserved, each strain contains tandem repeats not found in other strains. All the 52 nt repeats in a particular species present a limited number (2–5) of characteristic short nucleotide sequences, which may be important for their function. There is no obvious explanation for the conservation of a constant 52 nt repeat in these groups of tandem repeats. It appears that the 52 nt length is required for whatever function these tandem repeats might have.
3.3. Tandem Repeats with a 20–21 nt Repeat
Another group of Bacillus species presents several families of tandem repeats with a repeat of 20–21 nt which are very abundant (Figure 1). In general, they do not code for amino acid repeats, a list is given in Supplementary Table S1. This type of tandem repeats is not exclusive of Bacillus, it is found in many other bacterial groups, including Cyanobacteria, Actinobacteria, etc. [3]. Some of the Bacillus species also present 52 nt tandem repeats. Each genome in this group has species-specific families of related tandem repeats, with the exception of B. simplex and B. muralis. These two species share several families of related tandem repeats, in agreement with their close taxonomic relationship. When all tandem repeats of a large family belong to the same species, it is clear that they are not part of protein coding genes. Practically all families with more than four tandem repeats are in this class. For example, all tandem repeats in the 13_21_15, 93_21_6, and 135_21_5 families, found in B. weihaiensis, are also not part of protein coding genes. On the other hand, when all members of a tandem repeat family belong to different species, it is likely that the tandem repeats correspond to part of a related gene with the same protein repeat: The 62_21_8 tandem repeat family is the largest example, with a repeat of seven amino acids found in the “choice-of-anchor A family protein” of B. cereus and several related species.
Tandem repeats in this group also present a characteristic short nucleotide sequence conserved in all repeats of most tandem repeats. Its sequence is given in Table 2 for B. weihaiensis and its position in all tandem repeats is found in the complete list of tandem repeats of this species, presented in Supplementary Table S5. Thus, this group of tandem repeats has features similar to the 52 nt tandem repeats: They are also species specific. However, it is not clear if their role in the genome is related in some way.
3.4. Tandem Repeats with Repeat Length Multiple of Three
Most Bacillus genomes contain a few of these tandem repeats. They usually correspond to regions coding for amino acid repeats in proteins. In some cases, they form families which include related genomes. Many of these families correspond to B. cereus and related species, the largest ones are 21_36_13 and 27_39_11. A few other species also have abundant tandem repeats of this type. For example, all tandem repeats in the 54_114_8 family are found in B. indicus, which contains several proteins with identical 38 amino acid repeats in its genome. A striking example is the 1_60_41 family, which is the largest family of Bacillus tandem repeats that we have detected. Tandem repeats in this family are part of genes coding for a general stress protein, which has four or five twenty amino acid repeats. This protein is found in several Bacillus species and in many other bacteria, including Escherichia coli. A few examples are given in Table 3. A striking case is B. subtilis, whose single tandem repeat belongs to this family. On the other hand, no species of the B. cereus group presents tandem repeats in this family.
These tandem repeats are found as part of the coding region for a general stress protein. This gene is found in many bacterial species, but the 60 nt repeat is only present in some cases. In B. coagulans, a tandem repeat is only found in one strain. In the other strain shown in the Table, the corresponding gene is heavily mutated and does not appear as a tandem repeat.
3.5. Taxonomic Distribution of Tandem Repeats
The distribution of tandem repeats in different species allows their classification in different groups, as shown in Table 4. Species which contain tandem repeats with either 20–21 or 52 nt repeats do not appear to correspond to a particular taxonomic group, they are found in several unrelated species. In order to ascertain whether tandem repeats have any taxonomic significance, we have compared our results with the taxonomic classifications proposed by different authors [4,5,6,7], as shown in detail in Supplementary Table S6. Such comparison allows us to include tandem repeats as a taxonomic feature of Bacillus, as summarized in Table 5. We have tried to include the genomes we have analyzed in the groups suggested by Secaira-Morocho et al. [7], which in most cases agree with other suggestions [5,6]. Nevertheless, some Bacillus species do not fit in any of these groups. The clusters suggested by Khurana et al. [4] only show a partial agreement with the classifications suggested by the other authors [5,6,7]. Only genomes which are covered by at least two groups have been analyzed, which limits our analysis to only 66 of the 176 genomes we have studied. When the taxonomic placement of a species suggested by different authors does not coincide, we have placed the species in a MISCELLANEOUS group.
The CEREUS group is very homogeneous, with similar genome sizes and CG%. A moderate amount of tandem repeats (25.7 average) is found. Practically all these tandem repeats have repeats with a size multiple of three, which correspond to amino acid repeats in proteins, other tandem repeats are practically absent. Tandem repeat families contain tandem repeats from different species: They correspond to conserved genes. In fact, the boundaries between members of this group are difficult to define [5], so the genomes in this group might be considered as different strains of a single species. We have not detected any species outside this group with such an abundance of genes containing tandem repeats coding for amino acid strings.
A characteristic feature of several groups is the presence of very few tandem repeats: SUBTILIS, PUMILUS, and MEGATERIUM. The HALODURANS group does not appear to be homogeneous and probably should be subdivided: Some species have many tandem repeats (group A), while other species have few tandem repeats (group B). Genome size and base composition are also different. The same situation is found in the METHANOLICUS group, which may also be divided in several groups, as suggested by Gupta et al. [16,17].
The SIMPLEX group has a considerable amount of tandem repeats, most of them with a 21 nt repeat, a characteristic feature of this group. We have detected other genomes with this feature (Supplementary Table S1), but we have been unable to place them in any of the groups shown in Table 5. We should also note that some authors combine MEGATERIUM and SIMPLEX in a single group [5]. Tandem repeats with a 52 nt repeat are a characteristic feature of the COAGULANS and HALODURANS-A groups, also found in some genomes in the METHANOLICUS group.
In summary, the distribution of tandem repeats among Bacillus species is not random. Each of the phylogenetic groups shown in Table 5 has a characteristic distribution of tandem repeats, which may help complement other phylogenetic studies. For example, the HALODURANS and METHANOLICUS groups may be clearly divided, depending on the presence or absence of 52 nt tandem repeats. In any case, it is clear that further work is required for a complete taxonomic classification, many species do not fit in any group: The genus Bacillus is in need of taxonomic revision [18]. In fact, Gupta et al. have recently suggested a division of Bacillus in 25 different groups [16,17].
4. Discussion
4.1. Unique Features of Tandem Repeats
An unexpected result of our study is the strict length conservation of tandem repeats. We have only detected families of tandem repeats of two repeat lengths (20–21 and 52 nt), with no sequence conservation. These tandem repeats have appeared in different groups of Bacillus and also in other Firmicutes. They are particularly abundant in the genus Paenibacillus [3], which is a clearly separate group from Bacillus [19]. The constant size of the repeats suggests a biological role for these tandem repeats, but there is no obvious function for them. In addition to our bioinformatic analysis, further experimental studies will be useful to answer this question. A possible explanation is that there is a protein or group of proteins which recognize some feature of the sequence and require a constant repeat size to polymerize on the DNA. Such protein-DNA complex might be involved in the stabilization of the bacterial nucleoid, as we have discussed in detail in our previous publication [3]. The presence of characteristic short sequences in all repeats (Table 2) gives support to this interpretation. An alternative is that these tandem repeats are transcribed as short RNAs, a hypothesis that we will discuss below.
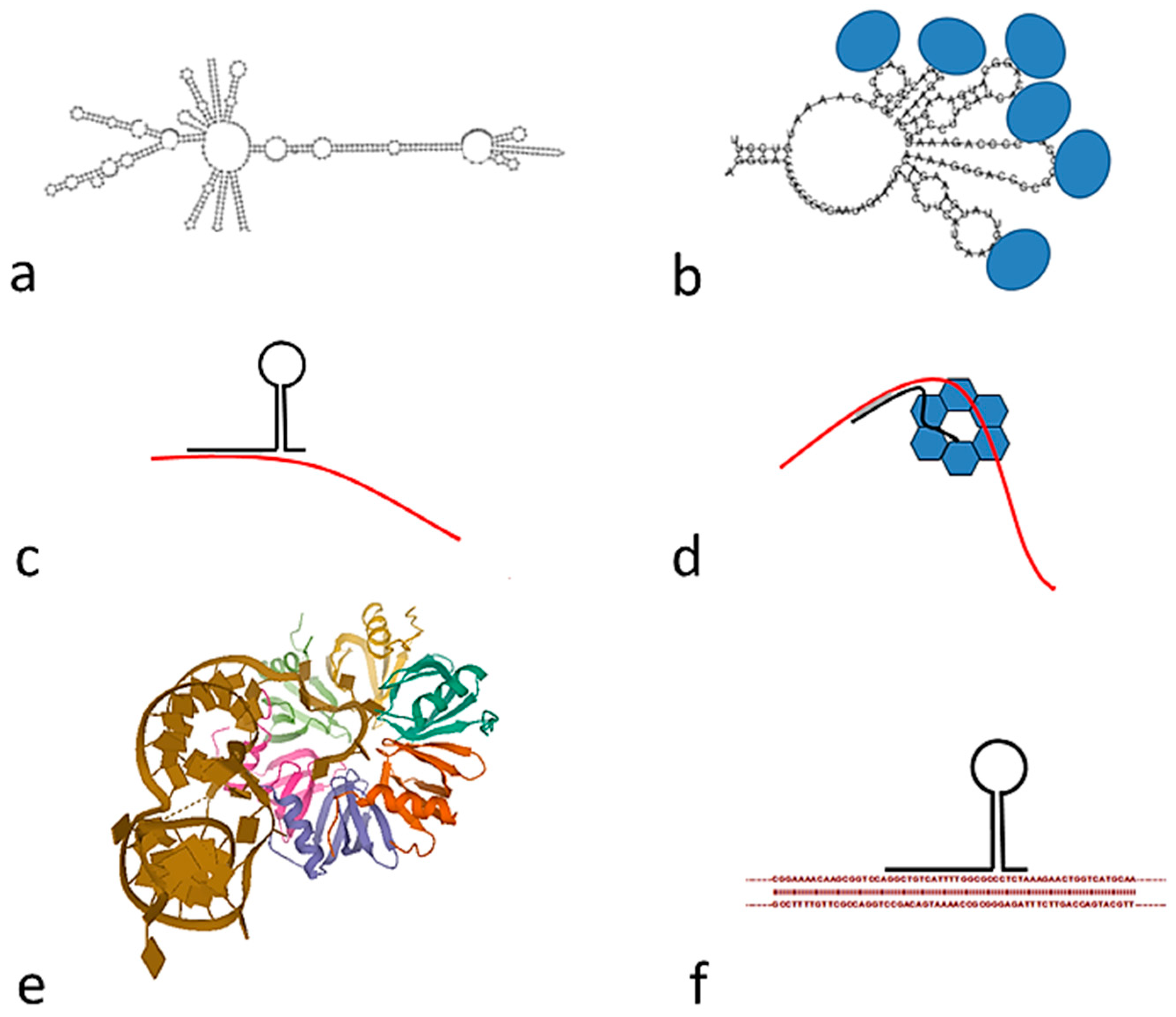
Bacteria contain hundreds of short RNAs with different lengths. They display many distinct mechanisms of action, usually through effects on target mRNA translation [20]. We wonder if tandem repeats may be transcribed and included in the pool of short RNAs. An analysis of the RNA-seq studies of B. coagulans [21] shows that the 52 nt tandem repeats are indeed transcribed. There are many RNA-seq studies of other Bacillus species, but we have not found any study with other species which contain 52 nt tandem repeats. However, it is generally accepted that most of the DNA in the genome of bacteria may be transcribed, including regions not coding for proteins. The 52 nt tandem repeats might be either completely or partially transcribed. Their structure may be either preserved or degraded as small RNAs [22].After transcription they may fold in different ways, as determined with RNAfold [23]; a substantial number of double-helical regions are found, as shown in Figure 4.
Tandem repeat RNAs may interact directly with mRNA (Figure 4c). Many small non-coding RNAs exert their regulatory function by directly base pairing with mRNA targets to alter their stability and/or affect their translation [24]. This RNA-RNA interaction is often facilitated by the hexameric Hfq protein [25], as it is schematically shown in Figure 4d. Hfq is an abundant bacterial RNA binding protein, present in most bacterial species. It has many important physiological roles that are usually mediated by interacting with Hfq binding small RNAs. Hfq is now recognized as an RNA chaperone that interacts with many RNAs and plays crucial roles in riboregulation [26]. A crystallographic model of the interaction of an RNA fragment with Hfq [27] is shown in Figure 4e. This is not the only type of interaction, RNA may interact with the Hfq hexamer in multiple regions, involving both faces and the sides of the hexamer [26]. Thus, tandem repeat RNA might bind in multiple places, but there is no evidence for a requirement of a 52 nt repeat. Furthermore, Hfq may also bind directly to specific DNA sequences [28], we cannot exclude that it directly recognizes tandem repeat DNA.
Other mechanisms of action are possible for small RNAs, which will influence gene transcription [20]. They may have a role as an RNA sponge (Figure 4b), sequestering either Hfq or other proteins usually associated with mRNA [25]. They may also interact directly with double stranded DNA, as shown in Figure 4f.
4.2. Origin of Tandem Repeats and Comparison with Eukaryotes
The most general feature in bacteria is the very low number of tandem repeats in most species [3]. Bacillus genomes are exceptional, since many species present a significant number of tandem repeats (Figure 1). The number of repeats per Mb in these species is similar to that found in many eukaryotes. We will compare the different types of tandem repeats we have found in Bacillus with similar features in eukaryotic tandem repeats.
Tandem repeats coding for amino acid repeats are found in a small proportion in all species, but the prominent number found in the B. cereus group is unique. These species probably benefit from this feature to easily change the details of protein structure, this may help them invade different hosts and ecological niches. Other cases, similar to the tandem repeats coding for the 20 amino acid repeats we have described (Table 3), are also found in eukaryotes. The abundant tandem repeats with a repeat of 84 nucleotides found in mammals [10] are a clear example: They code for 28 amino acid repeats in zinc-finger proteins [30].
Tandem 20–21 nt repeats are present in a small proportion in many bacteria [3], including Bacillus (Supplementary Table S1). Unexpectedly, B. simplex and a few other species present a large proportion of these repeats. We have found no similar case in eukaryotes. The constant length of this repeat in different species indicates that they play some specific role. We can only note that this length corresponds to two turns of the DNA helix, which might indicate some function in either DNA replication or transcription.
The most unusual case we have detected are the constant length 52 nt repeats found in some Bacillus and in a few other Firmicutes. In spite of their constant size, they do not show any sequence conservation. They may play an important role associated to their constant size and may appear by chance in species in which their presence improves some of the functions we have discussed. The horizontal DNA transfer between closely related species [31] may also help spread these tandem repeats. Their sequence may then change by adapting to the new invaded genome. The horizontal gene transfer between closely related Bacillus species has been demonstrated [7,32]. A search of the B. coagulans genome indeed shows that it contains the gene for the ComEA protein required for the horizontal gene transfer [33]. In eukaryotes, we also find abundant tandem repeats of specific sizes, for example, in Caenorhabditis elegans, we find a predominance tandem repeat with a 35 nt repeat, not found in the related species [10].
We may also ask if the role played by tandem repeats is similar in eukaryotes and in Bacillus. Although the general role played by tandem repeats is not clear for any species, some similarities are worth mentioning. In the case of human tandem repeats [34], their role is only clear in some cases: The abundant alpha tandem repeats required in mitosis [35] are a clear example. In C. elegans, we also find the tandem repeats required for mitosis [10] and for meiotic chromosome pairing [36]. In a similar way, some bacterial tandem repeats may play a role in nucleoid structure and function [3]. Another related feature is the transcription of tandem repeats to RNA we have suggested in Bacillus (Figure 4), which has been clearly demonstrated in Drosophila [37]. It is likely that the transcription of tandem repeats may be found in other species, a question which deserves further investigation.
5. Conclusions
We have determined the tandem repeats and their families in 176 genomes of Bacillus. These genomes only present three types of tandem repeats: Repeats of either 20–21 or 52 nt and coding tandem repeats which are part of genes.
We describe in detail a unique group of tandem repeats with a repeat of 52 nt, with a strict conservation of length and variable sequence. We analyze their possible role as transcribed small RNA molecules.
The distribution of tandem repeats in different species of Bacillus is variable and differs substantially from eukaryotes: Some groups have very few tandem repeats, while other groups have a large proportion of tandem repeats.
Cereus group genomes are unique: They have a large proportion of tandem repeats which correspond to parts of the genes coding for repeats of amino acid stretches.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/ijms22105373/s1. Figure S1: Histograms of tandem repeat lengths; Figure S2: Comparison of genome regions; Supplementary Table S1: Genome properties. Distribution of repeat sizes; Supplementary Table S2: Sequence of all tandem repeats; Supplementary Table S3: Tandem repeat families, including all tandem repeats; Supplementary Table S4: List of Bacillus tandem repeat families; Supplementary Table S5: Examples of complete tandem repeat sequences; Supplementary Table S6: Bacillus Phylogeny.
Author Contributions
Conceptualization, visualization, writing—original draft, J.A.S.; data curation, formal analysis, software, X.M.; funding acquisition, investigation, methodology, project administration, resources, supervision, validation, writing—review and editing, J.A.S. and X.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Ministerio de Ciencia e Innovación –Agencia Estatal de Investigación, Spain (Project RTI2018-094403-B-C33) and FEDER. The funders had no role in the study design, data collection and interpretation or the decision to submit the work for publication.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The sequence of all tandem repeats and a list of all tandem repeat families and their members are available in the Supplementary materials.
Acknowledgments
We thank J. Lourdes Campos for help with the figures and M. Mar Albà for the insightful discussion and suggestions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Treangen, T.J.; Abraham, A.; Touchon, M.; Rocha, E.P.C. Genesis, effects and fates of repeats in prokaryotic genomes. FEMS Microbiol. Rev. 2009, 33, 539–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denoeud, F.; Vernaud, G. Identification of polymorphic tandem repeats by direct comparison of genome sequence from different bacterial strains: A web-based resource. BMC Bioinform. 2004, 5, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subirana, J.A.; Messeguer, X. Unique features of tandem repeats in bacteria. J. Bacteriol. 2020, 202, e00229-20. [Google Scholar] [CrossRef] [PubMed]
- Khurana, H.; Sharma, M.; Verma, H.; Lopes, B.S.; Lal, R.; Negi, R.N. Genomic insights into the phylogeny of Bacillus strains and elucidation of their secondary metabolic potential. Genomics 2020, 112, 3191–3200. [Google Scholar] [CrossRef]
- Maughan, H.; Van der Auwera, G. Bacillus taxonomy in the genomic era finds phenotypes to be essential though often misleading. Infect. Gen. Evol. 2011, 11, 789–797. [Google Scholar] [CrossRef]
- Hernández-González, I.L.; Moreno-Hagelsieb, G.; Olmedo-Álvarez, G. Environmentally-driven gene content convergence and the Bacillus phylogeny. BMC Evol. Biol. 2018, 18, 148. [Google Scholar] [CrossRef]
- Secaira-Morocho, H.; Castillo, J.A.; Driks, A. Diversity and evolutionary dynamics of spore-coat proteins in spore-forming species of Bacillales. Microb. Genom. 2020, 6, 000451. [Google Scholar] [CrossRef]
- NCBI. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/genome/browse#!/prokaryotes/ (accessed on April 2021).
- ALGGEN Algorísmica I Genètica. Available online: http://alggen.lsi.upc.edu (accessed on April 2021).
- Subirana, J.A.; Albà, M.M.; Messeguer, X. High evolutionary turnover of tandem repeat families in Caenorhabditis. BMC Evol. Biol. 2015, 15, 218. [Google Scholar] [CrossRef] [Green Version]
- Subirana, J.A.; Messeguer, X. Evolution of Tandem Repeat Satellite Sequences in Two Closely Related Caenorhabditis Species. Diminution of Satellites in Hermaphrodites. Genes 2017, 8, 351. [Google Scholar] [CrossRef] [Green Version]
- Subirana, J.A.; Messeguer, X. How Long Are Long Tandem Repeats? A Challenge for Current Methods of Whole-Genome Sequence Assembly: The Case of Satellites in Caenorhabditis elegans. Genes 2018, 9, 500. [Google Scholar] [CrossRef] [Green Version]
- Tørresen, O.K.; Star, B.; Mier, B.; Andrade-Navarro, M.A.; Bateman, A.; Jarnot, P.; Gruca, A.; Grynberg, M.; Kajava, A.V.; Promponas, V.J.; et al. Tandem repeats lead to sequence assembly errors and impose multi-level challenges for genome and protein databases. Nucleic Acids Res. 2019, 47, 10994–11006. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T.J.; Messeguer, X. M-GCAT: Interactively and efficiently constructing large-scale multiple genome comparison frameworks in closely related species. BMC Bioinform. 2006, 7, 433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishino, Y.; Krupovic, M.; Forterre, P. History of CRISPR-Cas from Encounter with a Mysterious Repeated Sequence to Genome Editing Technology. J. Bacteriol. 2018, 200, e00580-17. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Gupta, R.S. A phylogenomic and comparative genomic framework for resolving the polyphyly of the genus Bacillus: Proposal for six new genera of Bacillus species, Peribacillus gen.nov., Cytobacillus gen. nov., Mesobacillus gen. nov., Neobacillus gen. nov., Metabacillus gen. nov. and Alkalihalobacillus gen. nov. Int. J. Syst. Evol. Microbiol. 2020, 70, 406–438. [Google Scholar] [PubMed]
- Gupta, R.S.; Patel, S.; Saini, N.; Chen, S. Robust demarcation of 17 distinct Bacillus species clades, proposed as novel Bacillaceae genera, by phylogenomics and comparative genomic analyses: Description of Robertmurraya kyonggiensis sp. nov. and proposal for an emended genus Bacillus limiting it only to the members of the Subtilis and Cereus clades of species. Int. J. Syst. Evol. Microbiol. 2020, 70, 5753–5798, 6531–6533. [Google Scholar]
- Barco, R.A.; Garrity, G.M.; Scott, J.J.; Amend, J.P.; Nealson, K.H.; Emerson, D. A Genus Definition for Bacteria and Archaea Based on a Standard Genome Relatedness Index. mBio 2020, 11, e02475-19. [Google Scholar] [CrossRef] [Green Version]
- Ash, C.; Priest, F.G.; Collins, M.D. Molecular identification of rRNA group 3 bacilli (Ash, Farrow, Wallbanks and Collins) using a PCR probe test. Antonie Van Leeuwenhoek 1993, 64, 253–260. [Google Scholar] [CrossRef]
- Wagner, G.H.; Romby, P. Small RNAs in Bacteria and Archaea: Who They Are, What They Do, and How They Do It. In Advances in Genetics; Friedmann, T., Dunlap, J., Goodwin, S.F., Eds.; Elsevier Science Direct: Amsterdam, The Netherlands, 2015; Volume 90, pp. 133–208. ISBN 978-0-12-803694-5. [Google Scholar]
- Qin, J.; Wang, X.; Wang, L.; Zhu, B.; Zhang, X.; Yao, Q.; Xu, P. Comparative transcriptome analysis reveals different molecular mechanisms of Bacillus coagulans 2-6 response to sodium lactate and calcium lactate during lactic acid production. PLoS ONE 2015, 10, e0124316. [Google Scholar] [CrossRef] [Green Version]
- Bechhofer, D.H.; Deutscher, M.P. Bacterial ribonucleases and their roles in RNA metabolism. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 242–300. [Google Scholar] [CrossRef]
- RNAfold web server. Available online: http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi (accessed on April 2021).
- Peer, A.; Margalit, H. Accessibility and Evolutionary Conservation Mark Bacterial Small-RNA Target-Binding Regions. J. Bacteriol. 2011, 193, 1690–1701. [Google Scholar] [CrossRef] [Green Version]
- Quendera, A.P.; Seixas, A.F.; dos Santos, R.F.; Santos, I.; Silva, J.P.N.; Arraiano, C.M.; Andrade, J.M. RNA-Binding Proteins Driving the Regulatory Activity of Small Non-coding RNAs in Bacteria. Front. Mol. Biosci. 2020, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Updegrove, T.B.; Zhang, A.; Storz, G. Hfq: The flexible RNA matchmaker. Curr. Opin. Microbiol. 2016, 30, 133–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimastrogiovanni, D.; Fröhlich, K.S.; Bandyra, K.J.; Bruce, H.A.; Hohensee, S.; Vogel, J.; Luisi, B.F. Recognition of the small regulatory RNA RydC by the bacterial Hfq protein. eLife 2014, 3, e05375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orans, J.; Kovach, A.R.; Hoff, K.E.; Horstmann, N.M.; Brennan, R.G. Crystal structure of an Escherichia coli Hfq Core(residues 2–69)–DNA complex reveals multifunctional nucleic acid binding sites. Nucleic Acids Res. 2020, 38, 3987–3997. [Google Scholar] [CrossRef]
- PDB, Protein Data Bank. Available online: https://www.rcsb.org/3d-view/4V2S/1 (accessed on April 2021).
- Castresana, J.; Guigó, R.; Albà, M.M. Clustering of genes coding for DNA binding proteins in a region of atypical evolution of the human genome. J. Mol. Evol. 2004, 59, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Kirchberger, P.C.; Schmidt, M.L.; Ochman, H. The Ingenuity of Bacterial Genomes. Annu. Rev. Microbiol. 2020, 74, 815–834. [Google Scholar] [CrossRef]
- Slomka, S.; Françoise, I.; Hornung, G.; Asraf, O.; Biniashvili, T.; Pilpel, Y.; Dahan, O. Experimental Evolution of Bacillus subtilis Reveals the Evolutionary Dynamics of Horizontal Gene Transfer and Suggests Adaptive and Neutral Effects. Genetics 2020, 216, 543–558. [Google Scholar] [CrossRef]
- Dubnau, D.; Blokesch, M. Mechanisms of DNA Uptake by Naturally Competent Bacteria. Annu. Rev. Genet. 2019, 53, 217–237. [Google Scholar] [CrossRef]
- Liehr, T. Repetitive Elements in Humans. Int. J. Mol. Sci. 2021, 22, 2072. [Google Scholar] [CrossRef]
- Miga, K.H. Centromeric satellite DNAs: Hidden sequence variation in the human population. Genes 2019, 10, 352. [Google Scholar] [CrossRef] [Green Version]
- Phillips, C.M.; Meng, X.; Zhang, L.; Chretien, J.H.; Urnov, F.D.; Dernburg, A.F. Identification of chromosome sequence motifs that mediate meiotic pairing and synapsis in C. elegans. Nat. Cell Biol. 2009, 11, 934–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shatskikh, A.S.; Kotov, A.A.; Adashev, V.E.; Bazylev, S.S.; Olenina, L.V. Functional Significance of Satellite DNAs: Insights from Drosophila. Front. Cell Dev. Biol. 2020, 8, 312. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Number of genomes as a function of the number of tandem repeats in bins of 10 tandem repeats. Genomes with few tandem repeats (0–10) represent 40% of the total genomes analyzed.
Figure 1.
Number of genomes as a function of the number of tandem repeats in bins of 10 tandem repeats. Genomes with few tandem repeats (0–10) represent 40% of the total genomes analyzed.

Figure 2.
The number of tandem repeats with each repeat length is shown. The entirety of all tandem repeats of all Bacillus genomes has been used. The bar at 61 nt includes all lengths over 60 nt. The distribution is clearly non-random, three types of repeat lengths predominate: 20–21, 51–53, and multiples of three nucleotides. A few repeats of size 40–41 nt are also present, which are related to the 20–21 nt tandem repeats.
Figure 2.
The number of tandem repeats with each repeat length is shown. The entirety of all tandem repeats of all Bacillus genomes has been used. The bar at 61 nt includes all lengths over 60 nt. The distribution is clearly non-random, three types of repeat lengths predominate: 20–21, 51–53, and multiples of three nucleotides. A few repeats of size 40–41 nt are also present, which are related to the 20–21 nt tandem repeats.

Figure 3.
Alignment of three B. coagulans genomes; NCBI codes: NC_015634.1, NZ_CP026649.1, and NZ_CP025437.1. Tandem repeats are plotted as vertical black lines with a thickness proportional to the tandem repeat length. The whole genomes are presented in the upper frame; there is an extensive overall alignment, but many small gaps are apparent. The lower frame shows a small amplified region (50 Kb). Further examples are given in Figure S2. The correspondence of tandem repeats in different genomes is only approximate.
Figure 3.
Alignment of three B. coagulans genomes; NCBI codes: NC_015634.1, NZ_CP026649.1, and NZ_CP025437.1. Tandem repeats are plotted as vertical black lines with a thickness proportional to the tandem repeat length. The whole genomes are presented in the upper frame; there is an extensive overall alignment, but many small gaps are apparent. The lower frame shows a small amplified region (50 Kb). Further examples are given in Figure S2. The correspondence of tandem repeats in different genomes is only approximate.

Figure 4.
Models of transcribed RNA tandem repeats: (a) Model of an RNA with nine repeats of 52 nt, prepared with RNA; (b) RNA sponge: Fragment of folded RNA (five repeats) interacting with several proteins; (c) interaction of tandem repeat RNA (black) with messenger RNA (red); (d) interaction of tandem repeat RNA (black) with messenger RNA (red), facilitated by interaction with the Hfq protein hexamer; (e) model of the complex of a partially doubly stranded RNA and the Hfq protein, PDB code 4V2S [29]; (f) direct interaction of an RNA tandem repeat with DNA.
Figure 4.
Models of transcribed RNA tandem repeats: (a) Model of an RNA with nine repeats of 52 nt, prepared with RNA; (b) RNA sponge: Fragment of folded RNA (five repeats) interacting with several proteins; (c) interaction of tandem repeat RNA (black) with messenger RNA (red); (d) interaction of tandem repeat RNA (black) with messenger RNA (red), facilitated by interaction with the Hfq protein hexamer; (e) model of the complex of a partially doubly stranded RNA and the Hfq protein, PDB code 4V2S [29]; (f) direct interaction of an RNA tandem repeat with DNA.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Main Bacillus 52 nt tandem repeat families. Sequence of repeats.
| Bacillus Species | Number of Tandem Repeats | % 52 nt Repeat | Family Code | Consensus Repeat |
|---|---|---|---|---|
| cellulosilyticus | 64 | 73.4 | 37_52_10 | gTGTaTCATACgaaggCAATGACACgtGAgAAAGtaGaaGaaacgnAATAAa |
| 61_52_6 | CAcTCAACGAAGGTcATCATAAGcAAGCAATGCTaCCCCAAAACCAAAcCcn | |||
| coagulans | 53av | 90.8 | 1_52_139 | GTgAAGgAAGgcCnTCnTTTTTcCncGCTTcCTTAACGTAGACGcgCTCTAT |
| 2_52_35 | TTTTGTCCTTTTGaCaGcTTCAAAAnGACATTTCGgGCCCgGATgCAgCntG | |||
| 8_52_18 | TGTCCTTCATaagggtGATGAAaGACAAAACaCnGGcCgggaAAcGgCgAAt | |||
| horikoshii | 64 | 71.9 | 22_52_12 | ATGAAGACaTcAGTGAcGAGaAAtcAGGAGgAGAGAaGTCcTCATcGncGTt |
| kochii | 59 | 88.1 | 52_51_7 | TTCnTTCTGAGTGACtTGCtAatCCcTTTTGCGAAGCgCTCAnCtCtgGtt |
| 92_54_4 | CTTTTaTTAGTcGCGaTTcTcacTTTTgCcCTACTtATCttnTcacTnatTCTT | |||
| litoralis | 80 | 73.7 | 42_52_9 | TGTCGTTCATaAGggtgATGAACGACAAAAGtGnTnaGAAAAGagngnGtag |
| 77_52_5 | AATCGGGACAGAAAAaaGAgcgaGCAGtgaAAnTgaGTCTCGATAgTgggng | |||
| oceanisediminis | 38 | 84.2 | 79_52_5 | nCgCcaAcTTCGGACTCatTCtCtCggTTTTccgcttCTtCTGTCCGAAgtn |
| 96_54_4 | cGATTAACTACCATTTTnCctTncaCCnnccTcaTTTTCGtCGTtAatcCaTcc | |||
| thermoamylovorans | 70 | 78.6 | 4_52_21 | AAAAtGacGACGAGAAnnGGTCTCGTCGCCAAAAAatGGAGTTTTcCGgctc |
| 5_52_21 | TTGgCGACGAGaCcnatTCTCGTCaCCaTTTTgaGGtGAAAAAnGCtCnaTT | |||
| 30_53_11 | TGTCCAATAGAACGGcTCTCgTGGACAAAATnGAGgnnTCAATCAGgAAAAAc | |||
| New species | ||||
| pseudalcaliphilus | 103 | 92.2 | 3_50_31 | gAATCnCGGGGTTGCGaGCnGAAAAAGaGGAGAAAgCCCGAGCAAAcGcn |
| 5_52_25 | GTTAATGTGaAGATACgGAgGCcAAACcttGgAGTAtCTGCACAAAGAGggG | |||
| 26_53_11 | nGtTGGTCGACATGATCAtGgGnAAAAAAGGncaAGAACcTGTCGATGAaGGn | |||
| alkalitelluris | 91 | 74.7 | 4_52_27 | AAgGGAATCAaACAAcgCtTTTCaTTCCCTTTgttggGCTTTtGGCATgAGA |
| 71_52_7 | TGTCTGAAGTaGCCTnGnGTTCGGACAGcTTTgaTtgtTTnnaaGcnAAAGC | |||
| 72_52_7 | CATAGgCctTCTATGATtcAGTTGCcgaaGCgAAAACAAGGAGnAAGTgAAT | |||
| indicus | 90av | 96.6 | 7_52_18 | ATCGTACCCTcgnAAACcGaaAAaCGATnTgggAGGGTAAGCAAanGcnnGA |
| alcalophilus | 103 | 86.4 | 9_52_16 | CCTtTGATTCCCTTTncGGCTtTtATTcaatgGCTTTtGgcaTCATTGcnGn |
| 11_51_15 | TTTTCATTACcTATtcCnntTTaTTCGCACCcTAATtcnncaCAgCnCgGc | |||
| 41_51_9 | TTTTTCATTACcTATcCncnnTTTTTCGCACcnTAATTTggcCtgCtcggC | |||
| sp.m3-13 | 72 | 73.6 | 20_52_13 | ATGAAGACnTcAGTGACgAggAAaaAGGAGgAGAGAaGTCCTCATcGccGTt |
| sp.SG-1 | 77 | 81.8 | 42_53_9 | CanaCCAACAtCcCTcncAtAATcCatTCTCaTTGGnctGaTTActCCcTTTT |
| selenatarsenatis | 82 | 59.8 | Many | |
| mesonae | 95 | 88.4 | Many |
In this table, the main families with the 52 nt repeat in each species are shown. We also include the results obtained in our previous study [3]. New species are those added in the present work. Species which contain many small tandem repeat families are indicated by “many”. Tandem repeats in B. selenatarsenatis are shared with two closely related species: B. boroniphilus and B. subterraneus.
Table 2.
Characteristic signals in tandem repeats.
| Bacillus Species | NCBI Code | Repeat (nt) | Characteristic Signals |
|---|---|---|---|
| oagulans | NZ_CP026649.1 | 52 | TCTAYG AARGACA |
| cellulosilyticus | NC_014829.1 | 52 | GGTCATCAT CAATGCT ACGAAGG ATGACAC |
| alkalitelluris | NZ_KV917374.1 | 52 | AAAgGGAAT AAAGCTGTC AATCATAG |
| mesonae | NZ_KV440949.1 | 52 | TTTTC TTCAT |
| weihaiensis | NZ_CP016020.1 | 21 | TCGCGG |
Table 3.
Coding features of Bacillus tandem repeats with a repeat length of 60 nt.
| NCBI Code | Bacillus Species | Tandem Repeat | Protein Gene | |||
|---|---|---|---|---|---|---|
| Start | Length | Start | Length | NCBI Code | ||
| NC_015634.1 | coagulans | 2718394 | 301 | 2718351 | 447 | WP_013860576 |
| NZ_CP026649.1 | coagulans | Heavily mutated | 3215973 | 387 | WP_035183339 | |
| NZ_LT603683.1 | glycinifermentans | 580499 | 301 | 580438 | 438 | WP_065894177 |
| NC_006582.1 | clausii | 3644328 | 241 | 3644228 | 402 | WP_011248345 |
| NC_017190.1 | amyloliquefaciens | 438961 | 301 | 438923 | 369 | WP_014471456 |
| NC_014551.1 | amyloliquefaciens | 456884 | 241 | 456846 | 309 | WP_013351072 |
| NC_006322.1 | licheniformis | 540638 | 301 | 540531 | 441 | WP_011197566 |
| NZ_CP007640.1 | atrophaeus | 4062283 | 301 | 4062244 | 375 | WP_010789649 |
| NC_000964 | subtilis | 494545 | 301 | 494506 | 372 | WP_003246542 |
Table 4.
Distribution of tandem repeats in different groups of Bacillus.
| NCBI Code | Bacillus Species | Genome Size (Mb) | GC% | Number of Tandem Repeats with a Given Repeat Size | |||||
|---|---|---|---|---|---|---|---|---|---|
| Total | 10–19 | 20–21 | 22–50 | 51–53 | >53 | ||||
| Rich in 52 nt repeat | |||||||||
| NZ_KV917374.1 | alkalitelluris | 5.43 | 36.4 | 91 | 7 | 7 | 3 | 69 | 5 |
| NC_014829.1 | cellulosilyticus | 4.68 | 36.5 | 64 | 7 | 0 | 2 | 47 | 8 |
| NC_015634.1 | coagulans | 3.07 | 47.3 | 26 | 0 | 0 | 1 | 21 | 4 |
| NC_016023.1 | coagulans | 3.55 | 46.5 | 63 | 0 | 1 | 1 | 57 | 4 |
| NZ_CP023704.1 | thermoamylovorans | 4.02 | 37.5 | 70 | 2 | 4 | 7 | 55 | 2 |
| Intermediate | |||||||||
| NC_022524.1 | infantis | 4.88 | 46 | 50 | 2 | 12 | 8 | 27 | 1 |
| NZ_BASE01000145 | selenatarsenitis | 4.76 | 42.1 | 82 | 1 | 30 | 1 | 49 | 1 |
| Rich in 21 nt repeat | |||||||||
| NZ_CP016020.1 | weihaiensis | 4.36 | 36.5 | 32 | 2 | 29 | 0 | 0 | 1 |
| NZ_CP011008.1 | simplex | 5.52 | 39.8 | 40 | 3 | 27 | 9 | 0 | 1 |
| NZ_CP017080.1 | muralis | 5.01 | 42.3 | 38 | 1 | 22 | 13 | 0 | 2 |
| Cereus group | |||||||||
| NC_004722.1 | cereus | 5.51 | 35.3 | 25 | 9 | 1 | 15 | 0 | 0 |
| NZ_CP018931.1 | cereus | 5.24 | 35.4 | 31 | 5 | 3 | 21 | 0 | 2 |
| NZ_CP007512.1 | bombysepticus | 5.88 | 35.0 | 31 | 9 | 4 | 15 | 0 | 3 |
| NC_003997.3 | anthracis | 5.23 | 35.4 | 19 | 3 | 5 | 11 | 0 | 0 |
| NZ_CP009692.1 | mycoides | 5.64 | 35.4 | 26 | 7 | 2 | 15 | 0 | 2 |
| Tandem repeat poor | |||||||||
| NC_014103.1 | megaterium | 5.1 | 38.1 | 8 | 7 | 1 | 0 | 0 | 0 |
| NC_017138.1 | megaterium | 5.08 | 38.1 | 5 | 2 | 2 | 0 | 0 | 1 |
| NZ_CP011007.1 | pumilus | 3.88 | 41.5 | 4 | 0 | 0 | 4 | 0 | 0 |
| NC_014551.1 | amyloliquefaciens | 3.98 | 46.1 | 3 | 1 | 0 | 0 | 0 | 2 |
| NC_000964.3 | subtilis | 4.22 | 43.5 | 1 | 0 | 0 | 0 | 0 | 1 |
| NZ_CP012024.1 | smithii | 3.38 | 40.8 | 0 | |||||
| NZ_CP012502.1 | beveridgei | 3.58 | 46.1 | 0 | |||||
| NZ_CP017786.1 | xiamenensis | 3.64 | 41.5 | 0 | |||||
Only a few examples of each group are shown. The characteristic feature of each group is enhanced in bold.
Table 5.
Tandem repeats in different phylogenetic groups of Bacillus.
| Group | Genome (Mb) | CG% | Number of Tandem Repeats | 52 nt Tandem Repeats |
|---|---|---|---|---|
| CEREUS | 5.5 | 35.4 | 25.7 | NO |
| SUBTILIS | 4.2 | 44.9 | 3.8 | NO |
| PUMILUS | 3.8 | 41.3 | 3.2 | NO |
| METHANOLICUS | 3.3–6.4 | 36–42 | 12–54 | Variable |
| MEGATERIUM | 3.9–5.5 | 35–38 | 2–7 | NO |
| SIMPLEX | 4.6–5.5 | 39–42 | 12–40 | NO |
| HALODURANS-A | 4.6 | 38.7 | 80.3 | YES |
| HALODURANS-B | 4.1 | 42.3 | 4 | NO |
| COAGULANS | 3.6 | 37–46 | 56 | YES |
| MISCELLANEOUS | 3.2–5.3 | 33–45 | 0–49 | Variable |
Average values are given when the group is homogeneous. Details are given in Supplementary Table S6. The SIMPLEX group is the only group characterized by the presence of abundant tandem repeats with a 21 nt repeat.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Subirana, J.A.; Messeguer, X. Tandem Repeats in Bacillus: Unique Features and Taxonomic Distribution. Int. J. Mol. Sci. 2021, 22, 5373. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105373
AMA Style
Subirana JA, Messeguer X. Tandem Repeats in Bacillus: Unique Features and Taxonomic Distribution. International Journal of Molecular Sciences. 2021; 22(10):5373. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105373
Chicago/Turabian StyleSubirana, Juan A., and Xavier Messeguer. 2021. "Tandem Repeats in Bacillus: Unique Features and Taxonomic Distribution" International Journal of Molecular Sciences 22, no. 10: 5373. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105373
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.