
Investigation of the Anti-Methicillin-Resistant Staphylococcus aureus Activity of (+)-Tanikolide- and (+)-Malyngolide-Based Analogues Prepared by Asymmetric Synthesis

 and
and
Abstract
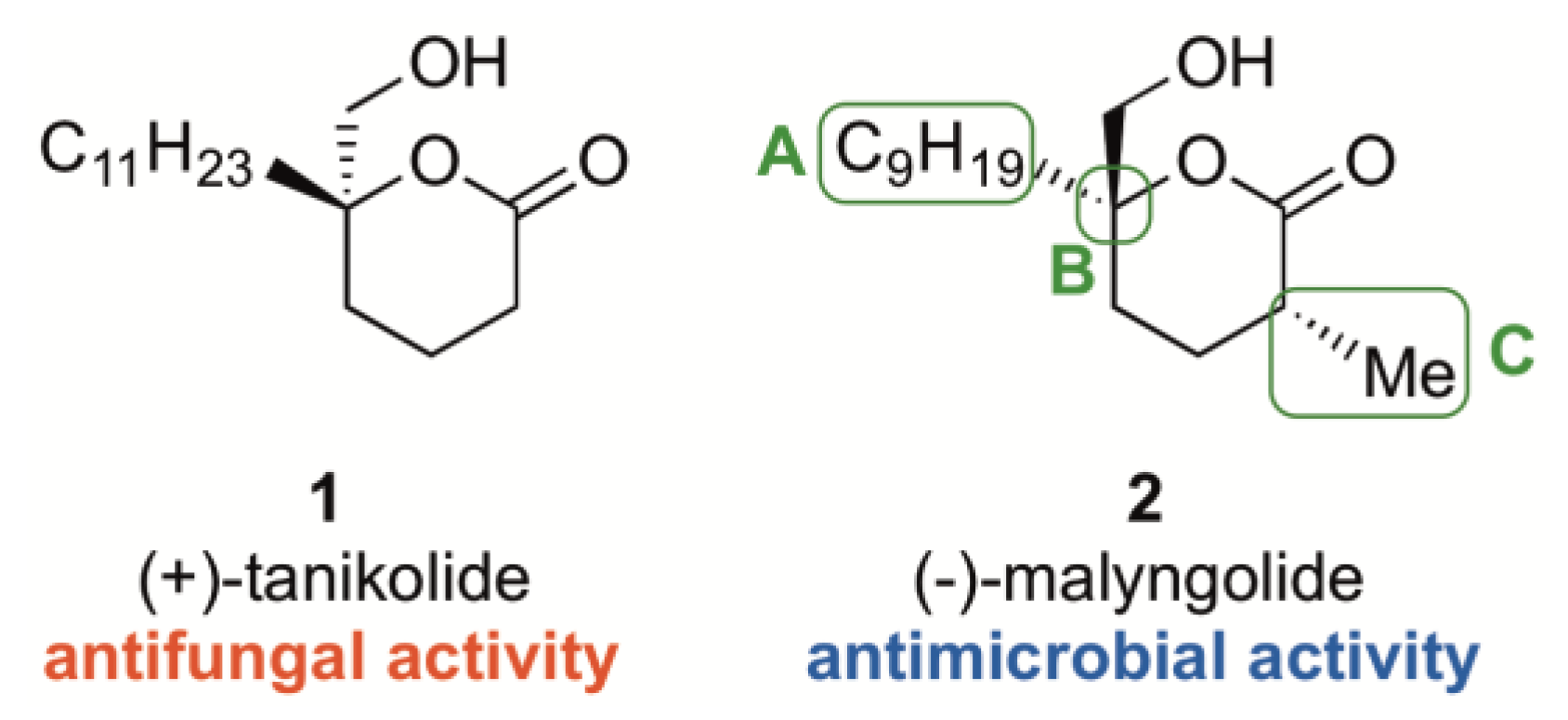
:1. Introduction
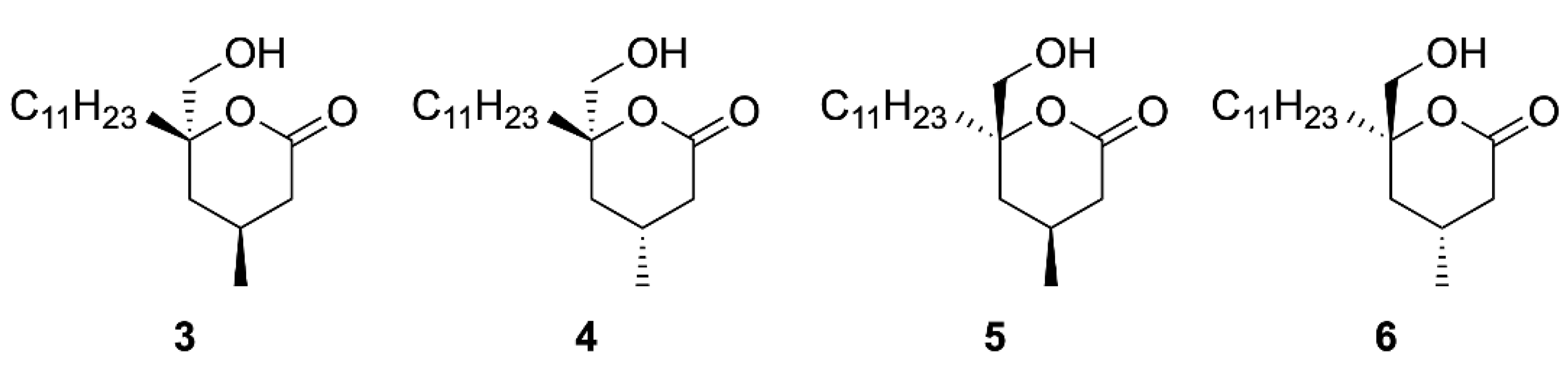
2. Results
3. Conclusions
4. Materials and Methods—Chemistry
4.1. Ethyl 2-(diethoxyphosphoryl)undecanoate (10)
4.2. Ethyl 2-methyleneundecanoate (11)
4.3. 2-Methyleneundecan-1-ol (12)
4.4. (R)-(2-Nonyloxiran-2-yl)methanol (13)
4.5. (S)-2-[Benzyloxy)methyl]-2-undecyloxirane (14)
4.6. 2-(2-Bromopropyl)-1,3-dioxane (15)
4.7. (2S,4S)-4-((Benzyloxy)methyl)-1-(1,3-dioxan-2-yl)-2-methyltridecan-4-ol ((2S,4S)-18) & (2R,4S)-4-((benzyloxy)methyl)-1-(1,3-dioxan-2-yl)-2-methyltridecan-4-ol ((2R,4S)-22)
4.8. (2S,4R)-2-((Benzyloxy)methyl)-6-methoxy-4-methyl-2-nonyltetrahydro-2H-pyran (23/24)
4.9. (2S,4S)-2-((Benzyloxy)methyl)-6-methoxy-4-methyl-2-nonyltetrahydro-2H-pyran (19/20)
4.10. (4R,6S)-6-((Benzyloxy)methyl)-4-methyl-6-nonyltetrahydro-2H-pyran-2-one (25)
4.11. (4.S,6S)-6-((Benzyloxy)methyl)-4-methyl-6-nonyltetrahydro-2H-pyran-2-one (21)
4.12. (4. R,6S)-4-Methylmalyngolide (8)
4.13. (4S,6S)-4-Methylmalyngolide (7)
5. Materials and Methods—Biological Testing
5.1. Preparation of Compounds
5.2. Antibacterial Activity Testing—Determination of Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singh, S.; Duffy, C.; Shah, S.T.A.; Guiry, P.J. ZrCl4 as an efficient catalyst for a novel one-pot protection/deprotection synthetic methodology. J. Org. Chem. 2008, 73, 6429–6432. [Google Scholar] [CrossRef]
- Singh, S.; Guiry, P.J. A facile synthesis of both enantiomers of 6-acetoxy-5-hexadecanolide, a major component of mosquito oviposition attractant pheromones. Eur. J. Org. Chem. 2009, 1896–1901. [Google Scholar] [CrossRef]
- Singh, S.; Guiry, P.J. Microwave-assisted synthesis of substituted tetrahydropyrans catalyzed by ZrCl4 and its application in the asymmetric synthesis of exo- and endo-brevicomin. J. Org. Chem. 2009, 74, 5758–5761. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Guiry, P.J. A short and efficient asymmetric synthesis of (−)-frontalin, (−)-exo-isobrevicomin and a volatile contributor of beer-aroma. Tetrahedron 2010, 66, 5701–5706. [Google Scholar] [CrossRef]
- Doran, R.; Duggan, L.; Singh, S.; Duffy, C.D.; Guiry, P.J. Asymmetric synthesis of (+)-tanikolide and the β-methyl-substituted analogues of (+)-tanikolide and (–)-malyngolide. Eur. J. Org. Chem. 2011, 7097–7106. [Google Scholar] [CrossRef]
- Singh, I.P.; Milligan, K.E.; Gerwick, W.H. Tanikolide, a toxic and antifungal lactone from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 1999, 62, 1333–1335. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Denning, D.W.; Gow, N.A.R.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Trans. Med. 2012, 4, 165rv13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giri, S.; Kindo, A.J. A review of Candida species causing blood stream infection. Indian J. Med. Microbiol. 2012, 30, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Rabes, A.; Zimmermann, S.; Reppe, K.; Lang, R.; Seeberger, P.H.; Suttopr, N.; Witzenrath, M.; Lepenies, B.; Opitz, B. The C-type lectin receptor mincle binds to streptococcus pneumoniae but plays a limited role in the anti-pneumococcal innate immune response. PLoS ONE 2015, 10, 0117022. [Google Scholar] [CrossRef]
- Odds, F.C.; Brown, A.J.P.; Gow, N.A.R. Antifungal agents: Mechanisms of action. Trends Microbiol. 2003, 11, 272–279. [Google Scholar] [CrossRef]
- Cardllina, J.H., II; Moore, R.E.; Arnold, E.V.; Clardy, J. Structure and absolute configuration of malyngolide, an antibiotic from the marine blue-green alga Lyngbya majuscula Gomont. J. Org. Chem. 1979, 44, 4039–4042. [Google Scholar] [CrossRef]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [Green Version]
- Klevens, R.M.; Morrison, M.A.; Nadle, J.; Petit, S.; Gershman, K.; Ray, S.; Harrison, L.H.; Lynfield, R.; Dumyati, G.; Townes, J.H.; et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 2007, 298, 1763–1771. [Google Scholar] [CrossRef] [Green Version]
- McDougal, L.K.; Carey, R.B.; Talan, D.A. Methicillin-resistant S. aureus infections among patients in the emergency department. N. Engl. J. Med. 2006, 355, 666–674. [Google Scholar] [CrossRef]
- Deresinski, S. Methicillin-resistant Staphylococcus aureus: An evolutionary, epidemiologic, and therapeutic odyssey. Clin. Infect. Dis. 2005, 40, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Walters, M.; Lonsway, D.; Rasheed, K.; Albrecht, V.; McAllister, S.; Limbago, B.; Kallen, A. Investigation and Control of Vancomycin-Resistant Staphylococcus aureus: A Guide for Health Departments and Infection Control Personnel. Atlanta, GA, USA, 2015. Available online: http://www.cdc.gov/hai/pdfs/VRSA-Investigation-Guide-05_12_2015.pdf (accessed on 10 June 2021).
- Gemmell, C.G. Susceptibility of a variety of clinical isolates to linezolid: A European inter-country comparison. J. Antimicrob. Chemother. 2001, 48, 47–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakito, Y.; Tanaka, S.; Asami, M.; Mukaiyama, T. An asymmetric total synthesis of a new marine antiobiotic-Malyngolide. Chem. Lett. 1980, 1223–1226. [Google Scholar] [CrossRef] [Green Version]
- Mukaiyama, T. Asymmetric synthesis based on chiral diamines having pyrrolidine ring. Tetrahedron 1981, 37, 4111–4119. [Google Scholar] [CrossRef]
- Kogure, T.; Eliel, E.L. A convergent asymmetric synthesis of (-)-malyngolide and its three stereoisomers. J. Org. Chem. 1984, 49, 576–578. [Google Scholar] [CrossRef]
- Guingant, A. An asymmetric synthesis of (R)-(+)-2-nonyl-2-(carbomethoxy) cyclopentanone, a known precursor of the antibiotic (−)-malyngolide. Tetrahedron Asymmetry 1991, 2, 415–418. [Google Scholar] [CrossRef]
- Enders, D.; Knopp, M. Novel asymmetric syntheses of (−)-malyngolide and (+)-epi-malyngolide. Tetrahedron 1996, 52, 5805–5818. [Google Scholar] [CrossRef]
- Maezaki, N.; Matsumori, Y.; Shogaki, T.; Soejima, M.; Tanaka, T.; Ohishi, H.; Iwata, C. Stereoselective synthesis of a chiral synthon, 2,2,5-trisubstituted tetrahydropyran, based on simultaneous 1,3- and 1,6-asymmetric induction via nucleophilic acetal cleavage reaction of the bicyclic acetal: A total synthesis of (-)-malyngolide. Chem. Commun. 1997, 1755–1756. [Google Scholar] [CrossRef]
- Winter, E.; Hoppe, D. A new route to the asymmetric synthesis of (−)-malyngolide and (−)-epi-malyngolide using N-sulfonyl-1,3-oxazolidines as chiral auxiliaries. Tetrahedron 1998, 54, 10329–10338. [Google Scholar] [CrossRef]
- Maezaki, N.; Matsumori, Y.; Shogaki, T.; Soejima, M.; Ohishi, H.; Tanaka, T.; Iwata, C. Stereoselective synthesis of a 2,2,5-trisubstituted tetrahydropyran chiron via 1,3- and 1,6-asymmetric induction: A total synthesis of (−)-malyngolide. Tetrahedron 1998, 54, 13087–13104. [Google Scholar] [CrossRef]
- Suziki, T.; Ohmori, K.; Suzuki, K. Pseudo-C3-symmetric tertiary alcohol building block via group-selective hydroalumination: A synthesis of (−)-malyngolide. Org. Lett. 2001, 3, 1741–1744. [Google Scholar] [CrossRef] [PubMed]
- Date, M.; Tamai, Y.; Hattori, T.; Takayama, H.; Kamikubo, Y.; Miyano, S. Efficient 1,8- and 1,9-asymmetric inductions in the Grignard reaction of δ- and ε-keto esters of 1,1′-binaphthalen-2-ols with an oligoether tether as the 2′-substituent: Application to the synthesis of (−)-malyngolide. J. Chem. Soc. Perkin Trans. 2001, 1, 645–653. [Google Scholar] [CrossRef]
- Pougny, J.-R.; Rollin, P.; Sinay, P. A synthesis of the marine antibiotic (-)-malyngolide from D-glucose. Tetrahedron Lett. 1982, 23, 4929–4932. [Google Scholar] [CrossRef]
- Ho, P.-T.; Wong, S. Branched-chain sugars in asymmetric synthesis. Total synthesis of marine antibiotic (−)-malyngolide. Can. J. Chem. 1985, 63, 2221–2224. [Google Scholar] [CrossRef]
- Tokunaga, Y.; Nagano, H.; Shiota, M. Synthesis of (+)-malyngolide from (+)-tartaric acid. J. Chem. Soc. Perkin Trans. 1986, 1, 581–584. [Google Scholar] [CrossRef]
- Trinh, M.-C.; Florent, J.-C.; Monneret, C. Total synthesis of (s)-(-) and (r)-(+)-frontalin and of (-)-malyngolide from the branched-chain sugar “α”-d-isosaccharino-lactone as chiral template. Tetrahedron 1988, 44, 6633–6644. [Google Scholar] [CrossRef]
- Honda, T.; Imai, M.; Keino, K.; Tsubuki, M. An enantiocontrolled synthesis of (–)-malyngolide. J. Chem. Soc. Perkin Trans. 1990, 1, 2677–2680. [Google Scholar] [CrossRef]
- Ichimoto, I.; Machiya, K.; Kirihata, M.; Ueda, H. Stereoselective Synthesis of Marine Antibiotic (−)-Malyngolide and Its Stereoisomers. Agric. Biol. Chem. 1990, 54, 657–662. [Google Scholar] [CrossRef]
- Matsuo, K.; Hasuike, Y.; Kado, H. Synthesis of (-)-Malyngolide from D-Lactose. Chem. Pharm. Bull. 1990, 38, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Nagano, H.; Ohno, M.; Miyamae, Y. Diastereoselective addition of grignard reagents to 3,4-O-Isopropylidene-1-O-triphenylmethyl-L-glycero-2-tetrulose and 1-O-Benzoyl-3,4-O-isopropylidene-L-glycero-2-tetrulose. Bull. Chem. Soc. Jpn. 1992, 65, 2814–2820. [Google Scholar] [CrossRef]
- Ohira, S.; Ida, T.; Moritani, M.; Hasegawa, T. Synthesis of (–)-malyngolide using reactions of alkylidenecarbenes. J. Chem. Soc. Perkin Trans. 1998, 1, 293–297. [Google Scholar] [CrossRef]
- Carda, M.; Castillo, E.; Rodriguez, S.; Marco, J.A. A stereoselective synthesis of (+)-malyngolide via a ring-closing olefin metathesis. Tetrahedron Lett. 2000, 41, 5511–5513. [Google Scholar] [CrossRef]
- Noda, Y.; Kikuchi, M. A convenient synthesis of (+)-Malyngolide. Synth. Commun. 1985, 15, 1245–1252. [Google Scholar] [CrossRef]
- Wan, Z.; Nelson, S.G. Optically active allenes from β-lactone templates: Asymmetric total synthesis of (−)-Malyngolide. J. Am. Chem. Soc. 2000, 122, 10370–10471. [Google Scholar] [CrossRef]
- Asaoka, M.; Hayashibe, S.; Sonoda, S.; Takei, H. New route to (−)-frontalin and (−)-malyngolide via epoxyketone rearrangement. Tetrahedron 1991, 47, 6967–6974. [Google Scholar] [CrossRef]
- Flörke, H.; Schaumann, E. Synthesis of (—)-Malyngolide. Liebigs Ann. 1996, 147–151. [Google Scholar] [CrossRef]
- Konno, H.; Hiroya, K.; Ogasawara, K. A new tactic for diastereo- and enantiocontrolled synthesis of (−)-malyngolide via catalytic meso-asymmetrization. Tetrahedron Lett. 1997, 38, 6023–6026. [Google Scholar] [CrossRef]
- Kanada, R.M.; Taniguchi, T.; Ogasawara, K. Asymmetric hydrogen transfer protocol for a synthesis of (+)-frontalin and (−)-malyngolide. Tetrahedron Lett. 2000, 41, 3631–3635. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Shirai, M. Asymmetric hetero Diels–Alder route to quaternary carbon centers: Synthesis of (−)-malyngolide. Tetrahedron Lett. 2001, 42, 6231–6233. [Google Scholar] [CrossRef]
- Miyamoto, H.; Iwamoto, M.; Nakada, M. A new asymmetric total synthesis of enantiopure (-)-Malyngolide. Heterocycles 2005, 66, 61–68. [Google Scholar] [CrossRef]
- Trost, B.M.; Tang, W.; Schulte, J.L. Asymmetric synthesis of quaternary centers. Total synthesis of (−)-Malyngolide. Org. Lett. 2000, 2, 4013–4015. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Maeno, H.; Noro, T.; Fujisawa, T. Asymmetric synthesis of (-)-Malyngolide and (-)-frontalin by utilizing bakers’ yeast reduction of (S)-Ethyl-2-cyclopentanonecarboxylthiolate. Chem. Lett. 1988, 17, 1739–1742. [Google Scholar] [CrossRef] [Green Version]
- Suemune, H.; Harabe, T.; Xie, Z.-F.; Sakai, K. Enzymatic hydrolysis of 2,2-Bis(acetoxymethyl)cycloalkanones, and its application to formal synthesis of (-)-Maylngolide. Chem. Pharm. Bull. 1988, 36, 4337–4344. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, N.; Reddy, B.V.S. Biocatalytic Approach for the total synthesis of (–)-Malyngolide and Its C(5)-Epimer. Helv. Chim. Acta 2016, 99, 267–272. [Google Scholar] [CrossRef]
- O’Sullivan, T.P.; Vallin, K.S.A.; Shah, S.T.A.; Fakhry, J.; Maderna, P.; Scannell, M.; Sampaio, A.L.F.; Perretti, M.; Godson, C.; Guiry, P.J. Aromatic lipoxin A4 and lipoxin B4 analogues display potent biological activities. J. Med. Chem. 2007, 50, 5894–5902. [Google Scholar] [CrossRef] [PubMed]
- Grieco, P.A.; Oguri, T.; Yokoyama, Y. One-step conversion of protected lactols into lactones. Tetrahedron Lett. 1978, 19, 419–420. [Google Scholar] [CrossRef]
- Wan, S.; Gunaydin, H.; Houk, K.N.; Floreancig, P.E. An experimental and computational approach to defining structure/reactivity relationships for intramolecular addition reactions to bicyclic epoxonium ions. J. Am. Chem. Soc. 2007, 129, 7915–7923. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Compound | E. coli 25922 | E. coli 4 | MRSA ATCC 43300 | MRSA 06/04 | ||||
|---|---|---|---|---|---|---|---|---|
| MIC [b] | MBC | MIC | MBC | MIC | MBC | MIC | MBC | |
| 3 | >100 | >100 | >100 | >100 | >100 | >100 | >100 * | >100 |
| 4 | >100 | >100 | >100 | >100 | >100 | >100 | >100 * | >100 |
| 5 | >100 | >100 | >100 | >100 | 12.5 | 12.5 | 12.5 | 50 |
| 6 | >100 | >100 | >100 | >100 | >100 | >100 | >100 * | >100 |
| Compound | E. coli 25922 | E. coli 4 | MRSA ATCC 43300 | MRSA 06/04 | ||||
|---|---|---|---|---|---|---|---|---|
| MIC [a] | MBC | MIC | MBC | MIC | MBC | MIC | MBC | |
| 7 | >100 | >100 | >100 | >100 | 50 | 50 | 50 | 50 |
| 8 | >100 | >100 | >100 | >100 | 50 | 100 | 50 | 50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breheny, J.; Kingston, C.; Doran, R.; Anes, J.; Martins, M.; Fanning, S.; Guiry, P.J. Investigation of the Anti-Methicillin-Resistant Staphylococcus aureus Activity of (+)-Tanikolide- and (+)-Malyngolide-Based Analogues Prepared by Asymmetric Synthesis. Int. J. Mol. Sci. 2021, 22, 6400. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126400
Breheny J, Kingston C, Doran R, Anes J, Martins M, Fanning S, Guiry PJ. Investigation of the Anti-Methicillin-Resistant Staphylococcus aureus Activity of (+)-Tanikolide- and (+)-Malyngolide-Based Analogues Prepared by Asymmetric Synthesis. International Journal of Molecular Sciences. 2021; 22(12):6400. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126400
Chicago/Turabian StyleBreheny, Joseph, Cian Kingston, Robert Doran, Joao Anes, Marta Martins, Séamus Fanning, and Patrick J. Guiry. 2021. "Investigation of the Anti-Methicillin-Resistant Staphylococcus aureus Activity of (+)-Tanikolide- and (+)-Malyngolide-Based Analogues Prepared by Asymmetric Synthesis" International Journal of Molecular Sciences 22, no. 12: 6400. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126400