
Osteocyte Dysfunction in Joint Homeostasis and Osteoarthritis


1
Department of Biomedical Engineering, Faculty of Engineering, The Hong Kong Polytechnic University, Hong Kong 999077, China
2
Research Institute for Smart Ageing, The Hong Kong Polytechnic University, Hong Kong 999077, China
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(12), 6522; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126522
Submission received: 19 May 2021
/
Revised: 13 June 2021
/
Accepted: 16 June 2021
/
Published: 17 June 2021
(This article belongs to the Special Issue Novel Osteoarthritis Pathogenesis and Management)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Structural disturbances of the subchondral bone are a hallmark of osteoarthritis (OA), including sclerotic changes, cystic lesions, and osteophyte formation. Osteocytes act as mechanosensory units for the micro-cracks in response to mechanical loading. Once stimulated, osteocytes initiate the reparative process by recruiting bone-resorbing cells and bone-forming cells to maintain bone homeostasis. Osteocyte-expressed sclerostin is known as a negative regulator of bone formation through Wnt signaling and the RANKL pathway. In this review, we will summarize current understandings of osteocytes at the crossroad of allometry and mechanobiology to exploit the relationship between osteocyte morphology and function in the context of joint aging and osteoarthritis. We also aimed to summarize the osteocyte dysfunction and its link with structural and functional disturbances of the osteoarthritic subchondral bone at the molecular level. Compared with normal bones, the osteoarthritic subchondral bone is characterized by a higher bone volume fraction, a larger trabecular bone number in the load-bearing region, and an increase in thickness of pre-existing trabeculae. This may relate to the aberrant expressions of sclerostin, periostin, dentin matrix protein 1, matrix extracellular phosphoglycoprotein, insulin-like growth factor 1, and transforming growth factor-beta, among others. The number of osteocyte lacunae embedded in OA bone is also significantly higher, yet the volume of individual lacuna is relatively smaller, which could suggest abnormal metabolism in association with allometry. The remarkably lower percentage of sclerostin-positive osteocytes, together with clustering of Runx-2 positive pre-osteoblasts, may suggest altered regulation of osteoblast differentiation and osteoblast-osteocyte transformation affected by both signaling molecules and the extracellular matrix. Aberrant osteocyte morphology and function, along with anomalies in molecular signaling mechanisms, might explain in part, if not all, the pre-osteoblast clustering and the uncoupled bone remodeling in OA subchondral bone.
1. Introduction
Osteoarthritis (OA) is the most common form of arthritic disease, mainly afflicting the load-bearing joints, e.g., knee and hip. It is also recognized as a major cause of joint pain and disability, contributing to a compromised quality of life in older adults [1,2]. Articular cartilage, subchondral bone, and synovium are among the tissues that may display abnormalities in OA. The radiological features of osteoarthritic subchondral bone include sclerosis, cyst and osteophyte formation on plain x-radiograph, and bone marrow lesions (BMLs) on magnetic resonance imaging, all of which have been proven to underline the anomalies of bone mineralization [3,4]. Such impaired mineralization was noted from all parts of the trabecular bone, along with compositional changes of the bone matrix, e.g., a low ratio of mineral/collagen content [5,6]. Sclerotic changes and osteophyte formation are both believed to arise from elevated bone turnover with an increase in osteoblastic over osteoclastic activities [7,8]. BMLs and sclerosis are likely associated with abnormal signaling related to sclerostin, periostin, and dentin matrix protein 1 (DMP-1) [9,10,11]. Meanwhile, cysts surrounded by less mineralized bone and osteoid formation uncoupled with mineralization may suggest spatially differential regulation of osteoblasts and osteoclasts by Wnt/β-catenin signaling and the OPG/RANKL/RANK pathway [12,13]. Modulation of bone remodeling was able to reduce osteophyte formation and alleviate cartilage degeneration in experimental models of OA and helped with restricting BMLs in human OA [14,15,16]. However, the pathomechanism of osteoarthritic bone remodeling and the resultant structural disturbances has not been fully understood yet.
Bone resorption and formation are tightly controlled and precisely coordinated by bone cells during the bone remodeling process to maintain the homeostasis of adult bone metabolism [17]. The structural and functional disturbances of the osteoarthritic bone are generally attributed to primary osteoblastic dysfunction [18]. Osteoarthritic osteoblasts showed blunted responses to the stimulus of cytokines [19,20] and an aberrant ratio of mineral and collagen production [6]. Meanwhile, osteocyte dysfunction and abnormal osteoblast-osteocyte differentiation are also recognized as the possible causes of OA [21]. Osteocytes compose 90–95% of bone cells and are responsible for mechano-transduction. It is known that osteocytes may respond to mechanical stimuli like micro-cracks with death. Following the deaths of osteocytes, bone remodeling is initiated via activation of osteoclastogenesis and bone resorption [17]. Osteocytes also act as the key regulators of osteoblast differentiation through the secretion of sclerostin. Through suppressing Wnt signaling, sclerostin can inhibit osteoblastic activity and terminate the cycle of bone remodeling [17,22,23]. There is growing evidence suggesting that osteocytes act as a central regulator of bone remodeling to maintain bone homeostasis and integrity [24]. However, the exact pathophysiology of osteocytes and their contributions to the structural and functional disturbances of the osteoarthritic bone remain unelucidated. Hereby, this review aimed to discuss the morphology and function of osteocytes and their association with subchondral bone remodeling in joint homeostasis and OA, along with the molecular pathways potentially involved in pathogenesis.
2. Disturbances of Subchondral Bone
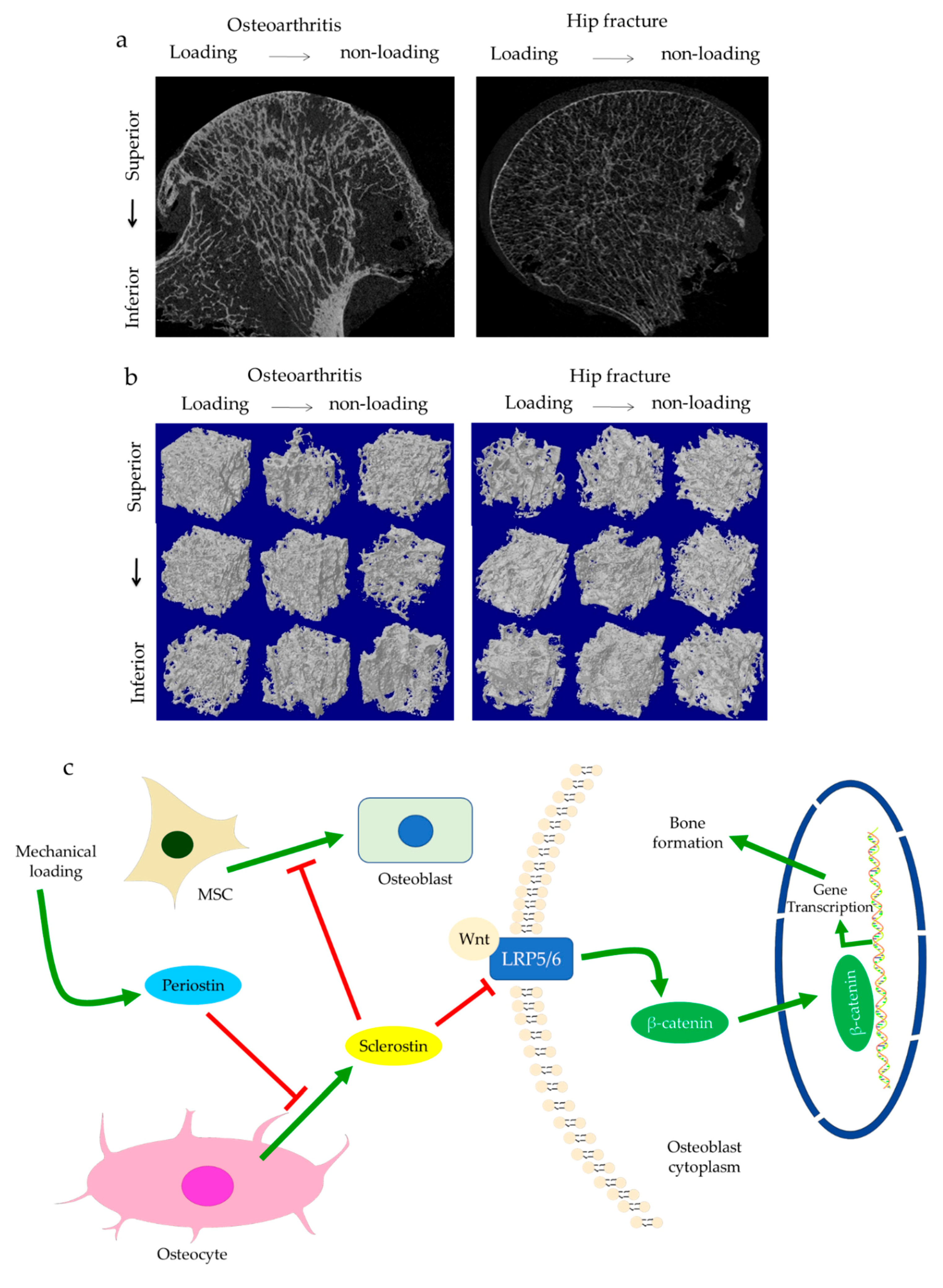
The role of subchondral bone in OA progression has been well documented. Disturbances of the subchondral bone are characterized by anomalous bone morphology at the tissue level, irregular osteocyte activities at the cellular level, and altered protein expressions at the molecular level. The Wnt/β-catenin pathway, which is responsible for osteogenesis and bone remodeling, is enriched in osteoarthritic subchondral bones [12]. Sclerostin, a major Wnt inhibitor, is expressed in calcified cartilage and subchondral bone. Its deficiency is often associated with OA development, likely through activating Wnt signaling [25]. In both OA and sclerostin-deficient models, an increase in bone volume fraction and trabecular bone thickness were observed in the subchondral bone, along with aberrant distribution of bone mineral density (BMD) [25,26,27,28]. This change is believed to be load-dependent, as higher bone mass is usually found excessively concentrated in the load-bearing region, with relatively lower bone mass in the less or non-loading region. For instance, our data suggested that osteoarthritic bone exhibited unevenly distributed bone mass from the load-bearing to non-load-bearing regions, such as in the superior portion of femoral heads underneath the articular cartilage. In contrast, there was no significant difference in the bone mass organization of the femoral heads from the control group (Figure 1a,b). In accordance with structural changes, sclerostin levels are also believed to be modulated by loading. Expression of sclerostin by osteocytes was downregulated by mechanical loading while upregulated by unloading in mice, suggesting a role in mechano-transduction [29,30]. Overexpression of SOST, the gene encoding sclerostin, was found to inactivate Wnt signaling, attenuate bone formation, and reduce mineralization in response to mechanical stimulation (Figure 1c). Suppressed bone remodeling then resulted in little to no difference in load-bearing and non-loading regions [30]. Hence, changes in sclerostin levels likely underlie the occurrence of sclerotic lesions preferentially in load-bearing regions. It might thus be considered as an adaption to support an abnormal level of compressive stress, such as when cartilage tissues are severely eroded [26,31] or when repetitive loading is applied [32].
Subchondral trabeculae are generally well-aligned with the direction of principal compressive stress, suggesting adaptation to loading conditions [26,27]. Periostin, a matricellular protein produced by osteocytes and osteoblasts, may participate in the regulation of bone formation and resorption associated with sclerostin. Increased periostin expression was observed upon mechanical stimulation, which was followed by a decreased sclerostin expression (Figure 1c) [10,33]. Periostin expression was enhanced in OA in subchondral bone, synovial fluid, and cartilage [34,35,36]. It was thought to exacerbate OA by promoting the expressions of pro-inflammatory cytokines, such as interleukin (IL)-6 and -8, and matrix metalloproteinase (MMP) production, such as through nuclear factor kappa B (NFκB) signaling [35]. High periostin level was linked with decreased bone surface/bone volume ratio, increased trabecular number, low structural model index (SMI), and high stiffness [10,11]. Such changes echoed the findings in OA, specifically the drop in bone surface area, the elevated number of trabeculae bones and their plate-like microarchitecture, as well as the stiffening of subchondral bones [26,27]. Plate-like trabecular bones were positively correlated with OA severity [37] and were generally able to sustain higher mechanical stress [38]. Meanwhile, stiffening of subchondral bone could cause ineffective load transfer from cartilage to bone. This was proposed as a possible source of cartilage damage and might explain the exacerbation of OA under excessive loading [39]. Thus, periostin might also induce cartilage degeneration by disturbing the underlying subchondral bone through sclerostin-Wnt signaling [11,40]. Interestingly, other characteristics of osteoarthritic bones, such as decreased trabeculae connectivity and BMD, were identified in periostin-deficient models [10,11]. Likewise, inhibition of sclerosing activities through antibodies resulted in higher connectivity density and BMD, which appeared to contradict the findings in OA [41,42]. Nevertheless, it should be noted that hyper-mineralization of the subchondral bone was also observed in early-stage OA [43]. Therefore, periostin and sclerostin might first affect bone mineralization in early-stage OA, while impaired mineralization in end-stage OA could relate to cellular dysfunction. Other signaling pathways might also be responsible for these sclerotic changes [44], such as transforming growth factor-beta (TGF-β) signaling [45], whose inhibition increased connectivity density and attenuated OA, DMP-1, whose deletion in cartilage led to an osteoarthritic phenotype [9,46].
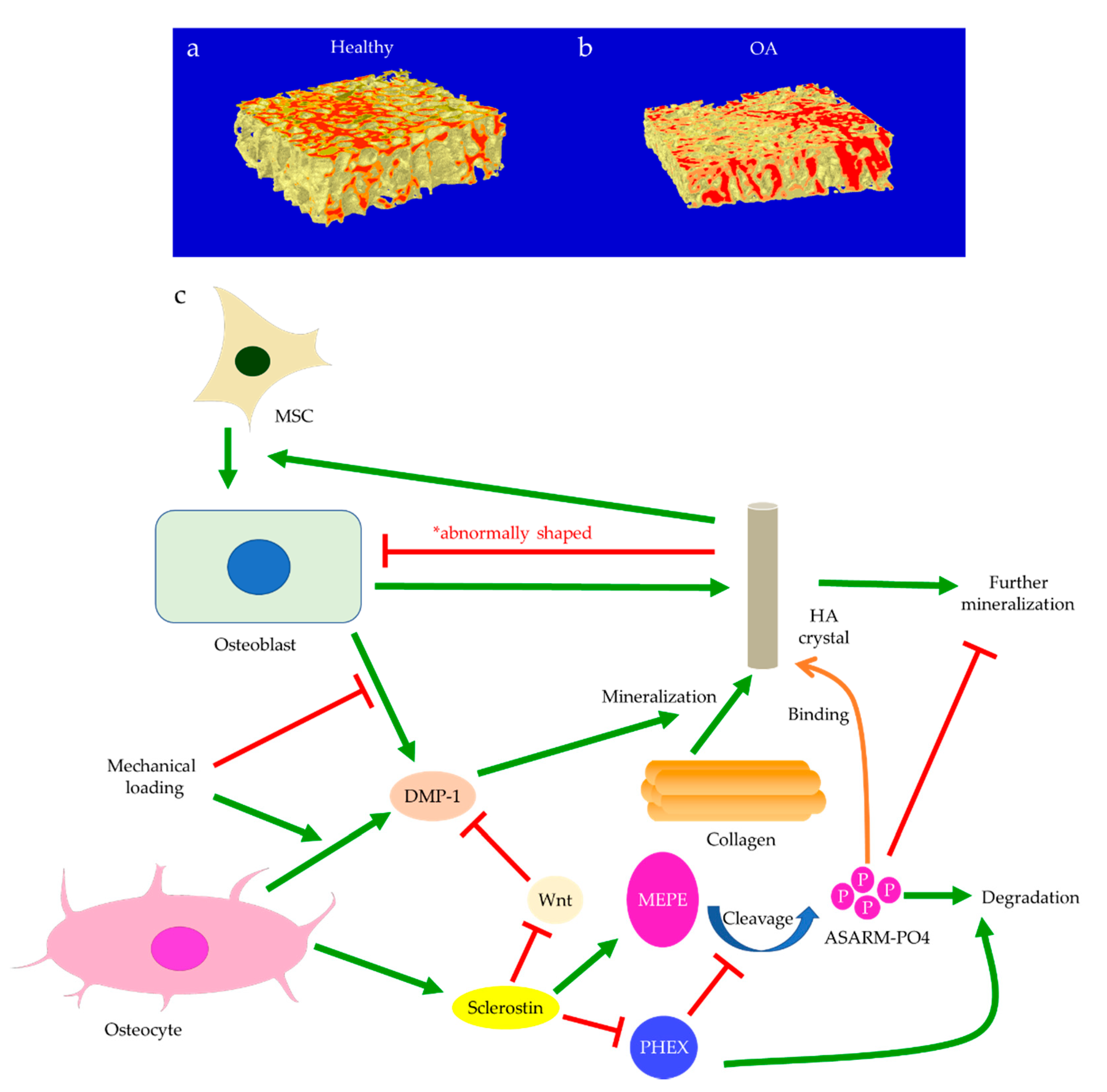
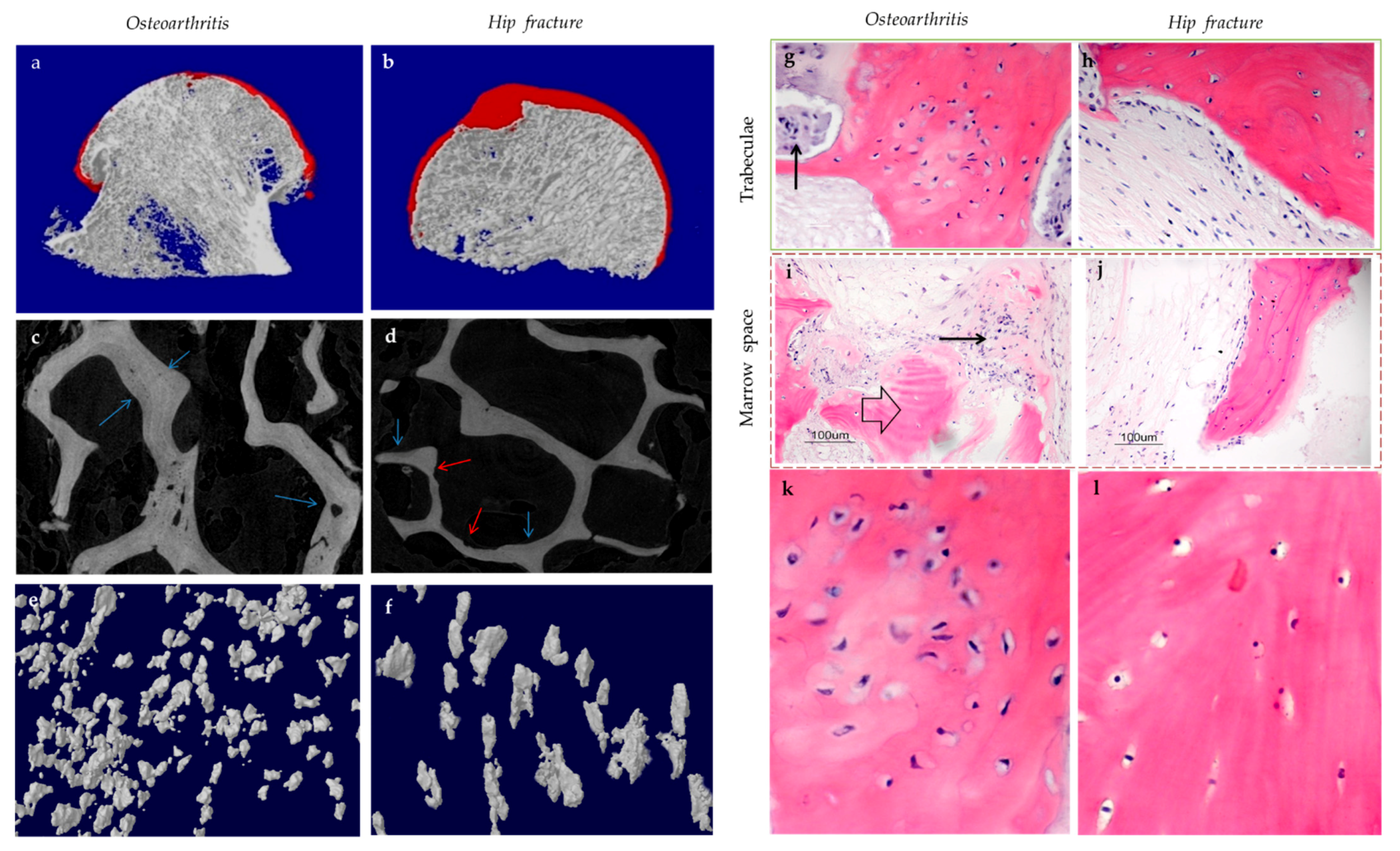
Subchondral bone marrow lesions (BMLs) have been characterized by sclerotic bones with less mineralization [3] and are believed to predict the progression of OA [47,48,49,50]. Our data also confirmed that the inorganic content of bone plugs from knee OA specimens was significantly lower (Figure 2a,b). We have also observed osteoid formation near trabeculae or in the marrow space of OA bone (Figure 3g,i), which were compatible with that of BMLs. Aberrant mineralization is likely associated with DMP-1. DMP-1 is responsible for the mineralization of collagen content, as it is critical for mineral nucleation [51]. Decreased DMP-1 expression and disrupted mineralization of the extracellular matrix (ECM) is accompanied by the activation of Wnt/β-catenin signaling, along with sclerostin deficiency [52]. Altered DMP-1 signaling might explain why OA bone exhibited higher mass yet with decreased structural connectivity and reduced mineral density. However, DMP-1 expression by osteocytes was higher in human OA samples [28]. Meanwhile, mechanical loading appeared to upregulate DMP-1 expression by osteocytes in both bone formation and resorption sites, while osteoblasts expressed less DMP-1 in the early stage of induction [53]. Hence, decreased DMP-1 expression in osteoblasts was proposed as a possible cause of low BMD in OA. This was supported by the results of our research, which showed the formation of osteoid-like tissues in marrow space together with the clustering of bone cells in OA (Figure 3i). Low BMD and poor connectivity were associated with decreased strength. Despite being plate-like and well-aligned, osteoarthritic bones were also weaker than normal bones with similar morphology [54,55]. Therefore, although the initial attempt was to increase BMD in response to mechanical loading, bone remodeling in OA might eventually render the subchondral bone mechanically inferior rather than superior.
Crosstalk with Cartilage Degeneration
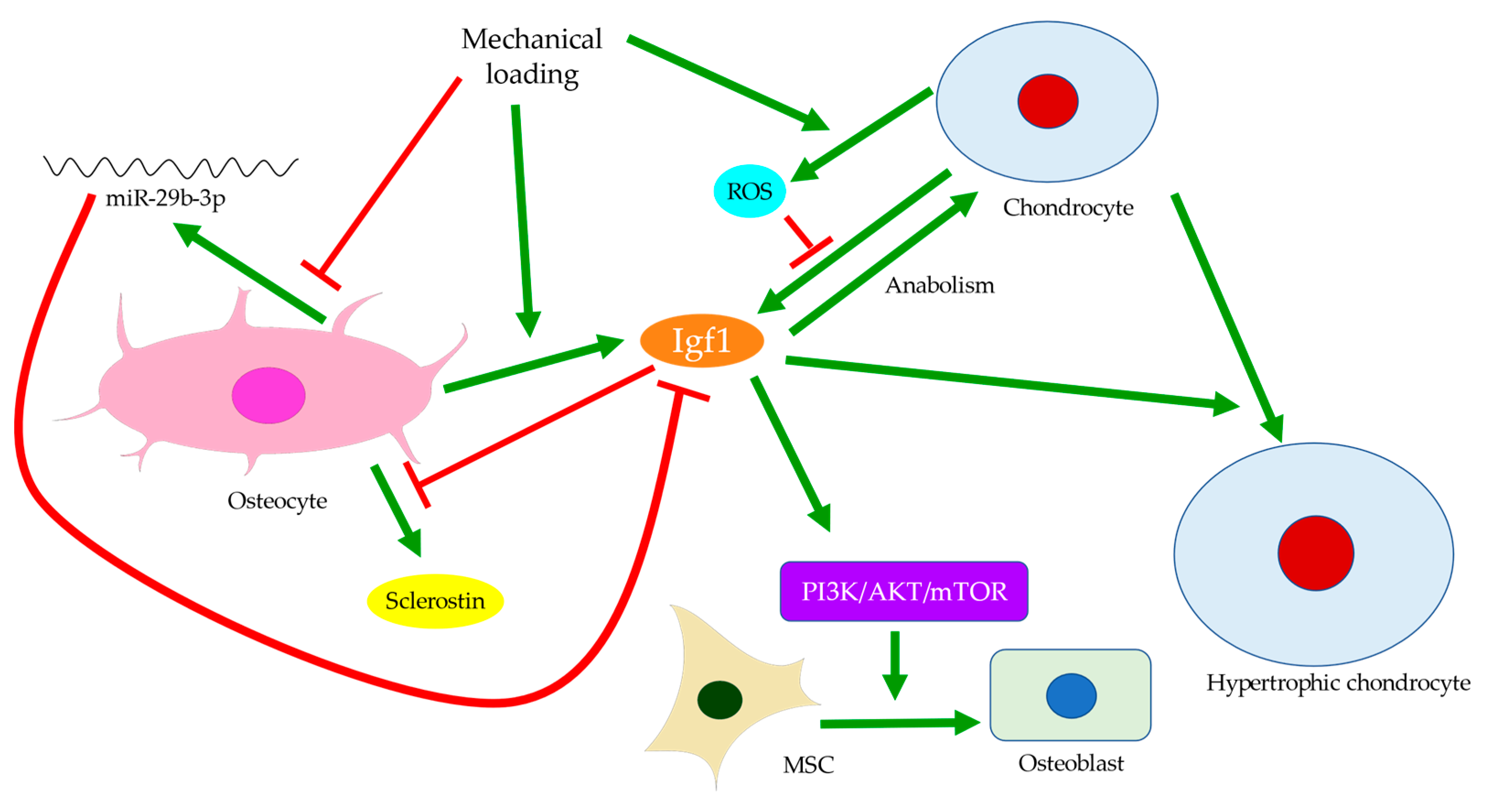
Disturbances of the subchondral bone have been closely associated with cartilage degeneration. For example, the size of the tibial subchondral bone might predict the overlying articular cartilage defect and OA severity [56]. Insulin-like growth factor 1 (Igf1) deficiency was linked with decreased bone formation, increased osteoid formation, and delayed and diminished mineral deposition in the subchondral bone (Figure 4) [57]. In OA, Igf1 expression by osteoblasts was upregulated [19,36]. Similarly, the mechanical strain was believed to increase Igf1 levels in osteocytes [58,59]. Hence, Igf1 was thought to facilitate the drop in sclerostin level upon mechanical loading and thus modulate bone remodeling [60]. In contrast, Igf1 expression by osteoarthritic chondrocytes was reduced [61]. This might be attributed to the presence of stressors, such as reactive oxygen species (ROS) [62], a well-identified cause of cartilage damage [63]. The addition of Igf1 to articular cartilage could increase matrix biosynthesis or enhance the stimulating effects of dynamic mechanical loading [64]. Likewise, matrix degradation in response to static loading could also be rescued by Igf1, which might suggest a role of Igf1 in maintaining the balance of anabolism and catabolism [65]. Cartilage degeneration is thus likely linked with both decreased anabolic activities partly due to Igf1 deficiency and increased catabolism associated with elevated expressions of degrative enzymes, such as MMPs [28].
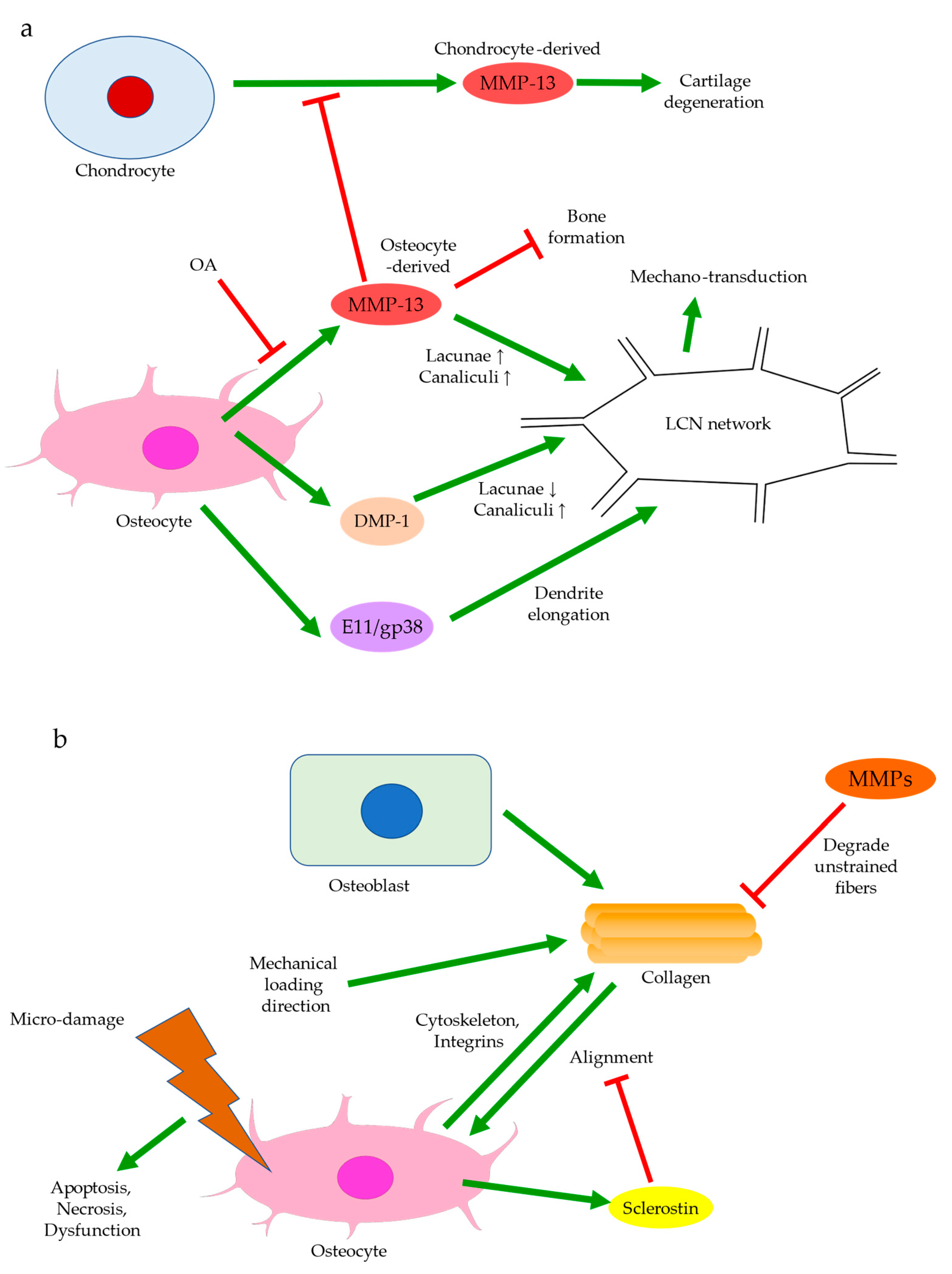
Bone remodeling mediated by Wnt signaling is believed to stiffen trabecular bones and alter their micro-architecture. For instance, in spontaneous OA models of guinea pigs, changes from rod-like to plate-like morphology were thought to precede or occur simultaneously with cartilage degeneration [66,67]. Past experiments also observed subchondral bone remodeling beneath intact cartilage in human OA, but only significant when cartilage tissues were damaged [26,66]. Ineffective load transfer due to structural disturbances of the subchondral bone might increase the strain endured by cartilage. High mechanical strain could then increase ROS production by chondrocytes, presumably because of mitochondrial deformation (Figure 4) [68,69]. Therefore, differential expressions of Igf1 in bone and cartilage, for instance, could be involved in the progression of OA. Similarly, while TGF-β could promote chondrocyte proliferation and exhibited protective effects in articular cartilage, its elevation in the subchondral bone was associated with cartilage loss and OA development [45]. The crosstalk between subchondral bone remodeling and cartilage degeneration was also believed to be regulated by MMP-13, which was less expressed by osteoarthritic osteocytes. Insufficient expression of MMP-13 was assumed to increase trabecular bone volume and cause cartilage degeneration independent of other stimuli. A reduction in osteocyte-derived MMP-13 was associated with disrupted lacuno-canalicular networks (LCN), which likely affected mechano-transduction and bone remodeling in early-stage OA. Meanwhile, expressions of proteins responsible for metabolism or matrix degradation, such as MMP-13 expression by chondrocytes, were also induced by insufficient osteocytic MMP-13 [43]. Hence, bone remodeling might start before cartilage degeneration and contribute to the aberrant metabolism in cartilage. With accumulated micro-damage, such as when an excessive load was applied or when cartilage was lost [66], mechanically inferior bones would be formed predominantly, and OA exacerbated. The above findings suggested that osteocytes could regulate trabecular bone morphology, bone mineralization, and ultimately bone and cartilage homeostasis in response to mechanical loading. Hence, the interplay between cartilage and subchondral bone is worth further investigation and should be extended to cellular and protein levels.
3. Osteocyte Dysfunction
Bone remodeling is a complex but precisely coordinated biological process involving osteoblasts, osteoclasts, and mesenchymal stem cells (MSCs), all of which are likely mediated by osteocytes [17]. Osteoclasts dissolve the minerals and collagens of the bone matrix; MSCs are recruited to bone formation sites and then differentiate into osteoblasts for collagen deposition and mineralization. Osteocytes are the major source of sclerostin, DMP-1, and matrix extracellular phosphoglycoprotein (MEPE) in bone [70], which designates its role in osteogenesis, mechano-transduction [22,71,72], and bone mineralization [9,73,74]. The mechano-sensitivity of osteocytes is facilitated by integrins, cytoskeleton, stretch-activated ion channels, gap junctions, and the LCN [21,75]. Integrins and focal adhesions are essential for mediating the response of gap junctions to mechanical stimulation and for the activation of Wnt signaling and other pathways in osteocytes. It has been proposed that mechanical deformations can induce ionic fluxes in LCN. The ionic currents can both amplify tissue strains and produce shear stress to activate osteocytes [13,21]. Osteocytes can sense the strain on the cytoskeleton. Upon stimulation, they will secrete signaling molecules to regulate osteoclastic and osteoblastic activities and modulate the recruitment of mesenchymal progenitors, which eventually results in bone remodeling. Disruption of microtubules resulted in a blunted decrease in sclerostin expression upon mechanical loading [76]. Similarly, Kindlin-2, a protein responsible for cytoskeleton organization and the formation of focal adhesions, was essential to the suppression of sclerostin expression through inhibiting Smad2/3 signaling [77]. These findings again confirm the role of the cytoskeleton in mechanical sensing. In OA, osteocyte dysfunction is characterized by altered lacuna morphology and protein expressions, both of which may be the response to excessive mechanical loading. The morphological and functional aberrations then cause dysregulation of bone cells, disruption of bone and cartilage homeostasis, and OA exacerbation [21,78,79]. Hence, the cellular and molecular mechanisms behind such anomalies should be elucidated.
3.1. Osteocyte Morphology
Osteocytes can actively change their microenvironment, notably the ECM and the lacunae where they reside. An altered extracellular environment can facilitate differential responses to mechanical stress. The morphology and alignment of osteocyte lacunae are assumed to be correlated with mechano-transduction. Notably, changes in the LCN network may affect the sensitivity of individual osteocytes to matrix deformation [21]. A decrease in LCN area seen in early-stage OA, for instance, might suggest that MMP-13 deficiency in osteocytes contributed to the reduced mechano-sensitivity of lacunae (Figure 5a) [43]. The uCT images obtained by the authors’ team revealed altered morphology of osteocyte lacunae in OA, where their three-dimensional structure appeared more plate-like rather than rod-like (Figure 3e). Plate-like architecture indicated fragility and reduced sensitivity to stress, while this change was perhaps due to rearrangement of the cytoskeleton in the embedded osteocyte [21]. E11/gp38, a protein expressed during the rearrangement of the cytoskeleton, such in osteoblast-osteocyte differentiation [80], was enriched in OA, especially in the sclerotic lesions [81]. The elongation of osteocytic dendrites in response to mechanical strain was believed to be regulated by E11 [80]. Changes in the canaliculi then followed, which altered the LCN network. Osteocyte lacunae in OA also appeared small, concentrated, and aligned to the principal stress direction [82]. Osteocytes embedded in large lacunae were thought to have higher mechano-sensitivity, as they responded to mechanical loading with a more prominent drop in sclerostin production and significantly higher β-catenin expression [83]. Decreased DMP-1 expression was also associated with large lacunae and a reduced number of canaliculi [84]. As small lacunae again indicated decreased mechano-sensitivity, local bone remodeling in load-bearing regions might favor osteocyte lacunae with lower mechano-sensitivity in adaptation to repetitive or excessive loading [85].
Like the trabecular bones, osteocytes are also well-aligned in OA, likely in response to the cumulative mechanical loading. Osteocyte lacunae align themselves with collagen fibers, which are produced by the ancestral osteoblast [13]. Contractions of actin filaments of the cytoskeleton are believed to introduce intracellular tension in osteocytes. This allows the cell to sense the stress exerted by the extracellular collagen matrix [86]. Collagen orientation is in turn controlled by the dominant loading orientation, either longitudinally with tensile stress or transversely with compression [87,88,89]. Degradation of unloaded collagen fibers by MMPs and preservation of loaded fibers are proposed to mediate collagen alignment (Figure 5b) [90,91]. Meanwhile, fibronectin and integrins are involved in collagen synthesis along with other molecules, which may suggest interactions between cells and ECM to regulate alignment [92]. Sclerostin inhibition was also correlated with high collagen alignment [93], consistent with findings suggesting that periostin was responsible for aligning osteocyte lacunae. However, periostin had no significant effect on lacunae volume [94], which could suggest that it was not the cause of sclerostin-induced modifications of the LCN network. It should also be noted that in contrast to the highly aligned osteocyte lacunae and low BMD observed in end-stage OA, collagen fibers were disorganized, while the subchondral bone was hyper-mineralized in early-stage OA [43]. This could indicate that mineral deposition, rather than the realignment of osteocytes, was the initial response to excessive loading and that there existed dissimilar responses to mechanical loading throughout OA progression.
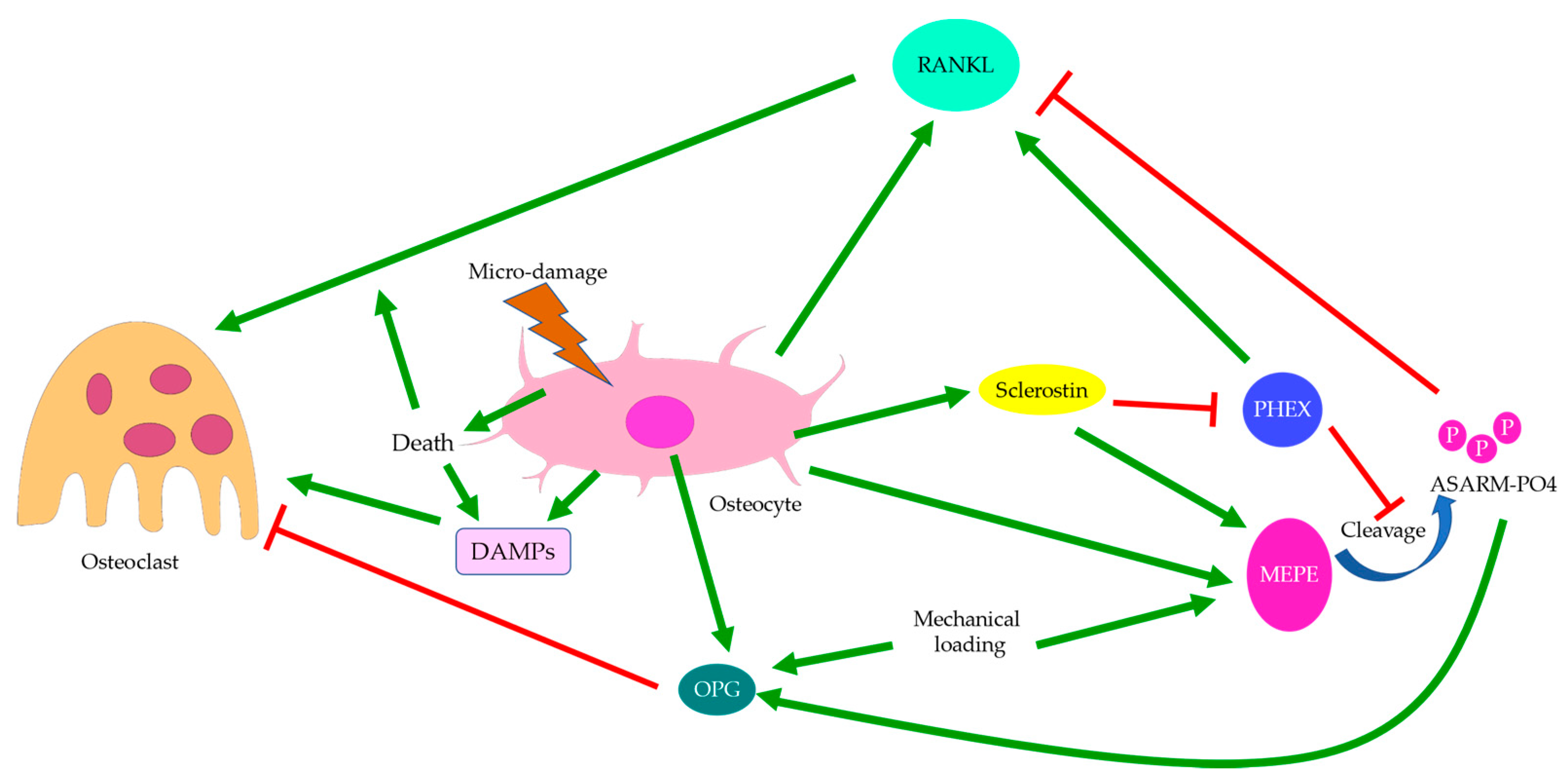
Another cause of sclerostin deficiency and well-aligned osteocyte lacunae may be osteocyte death. Osteocyte lacunae respond to load from perpendicular and parallel directions differently. In the case of tensile load, osteocytes are more susceptible to microdamage when aligned perpendicularly to the loading direction, while the opposite is true for compression [88,89]. Thus, the pre-existing osteocytes not aligned properly with the principal stress direction are subject to microfracture. Microfractures are believed to induce ionic flux that can activate osteocytes [13] or cause osteocyte apoptosis in the damaged region [17]. Selective osteocyte apoptosis may hence indicate that bone remodeling in accordance with the loading orientation will be preferred. Induced apoptotic osteocytes have shown abnormal gene expression resembling that seen in OA [95], which supports the role of osteocyte apoptosis in overload-induced OA progression. Therefore, in addition to stress-induced sclerostin inhibition, osteocyte apoptosis may also contribute to sclerostin deficiency, which in turn affects osteoblast differentiation and consequently collagen production [96,97]. Recent studies have also revealed similarities between OA and osteonecrosis, especially in the spatial distribution of osteocytes [98]. Increased osteocyte apoptosis was detected with TUNEL staining [28], while the proportion of osteocytes stained positive for activated Caspase-3 was similar to that of normal bones [99]. This might suggest that necrosis, in addition to apoptosis, was responsible for osteocyte death in OA [100]. Past findings also supported that microfracture could induce osteocyte necrosis [101] and contribute to bone remodeling. Upon damage, osteocytes might release damage-associated molecular patterns (DAMPs), which then activated osteoclasts through Mincle, a DAMP sensor. In this way, osteoclasts could sense osteocyte necrosis and initiate bone resorption independent of the sclerostin-RANKL pathway [102]. Hence, osteocyte death induced by overload or microfracture might explain the thinning of the subchondral bone in early-stage OA and the uncoupled osteoblastic and osteoclastic activities [8].
3.2. Regulation of Osteoblasts
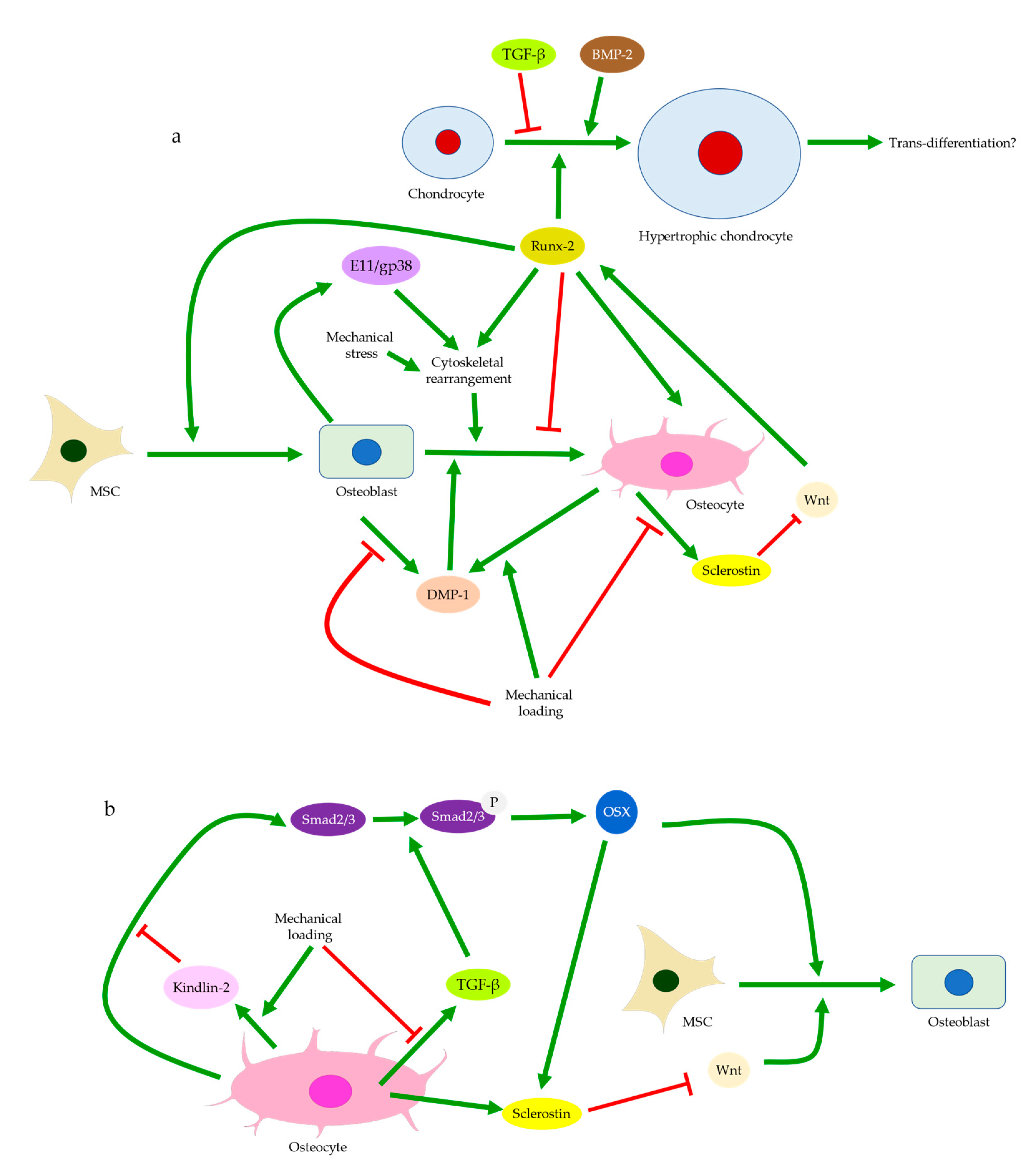
One of the major functions of osteocytes is acting as the key regulator of osteoblast differentiation. Through the secretion of sclerostin, osteocytes modulate osteoblastic activities and maintain bone homeostasis and integrity [96,97]. Sclerostin can inhibit LRP5/6, the activators of Wnt/β-catenin signaling for mechano-transduction (Figure 1c) [103,104]. The percentage of sclerostin-positive osteocytes is significantly lower in OA [105], whereas the fraction of osteocalcin-positive cells is higher, indicating increased osteoblastic activities [106]. While the number of osteocyte lacunae increases upon mechanical loading, the small individual lacuna size and the low level of sclerostin secretion both indicate the status of immature osteocytes [13,99]. Hence, decreased sclerostin expression in early-stage OA may arise from high mechano-sensitivity of osteocytes and osteocyte death, while the erroneous regulation of osteoblast differentiation may contribute to the low level of sclerostin at end-stage OA. Sclerostin deficiency is believed to activate Wnt signaling pathways and thus increase subchondral bone formation, eventually aggravating OA [13,25]. Mature osteocytes secret sclerostin to negatively regulate the differentiation of osteoblasts lineage and then terminate the cycle of bone remodeling [17,22,23,24]. Immature osteocytes with reduced sclerostin expression in OA would inhibit the negative feedback to MSCs, which were recruited and committed to osteoblast differentiation and bone formation [107]. This was supported by the significantly large number of osteoblasts and osteocytes in osteoarthritic subchondral bone, and by the presence of Runx-2-positive or osterix-positive bone cell clusters in marrow space or adjacent to trabeculae. Osteocalcin, Runx-2, or osterix-positive cells from the subchondral bone were also seen to cluster in the load-bearing regions of OA joints. This suggested upregulated activities of both osteoblasts and pre-osteoblasts [108], possibly in response to the increased demand for bone formation due to excessive mechanical loading and osteocyte death. Igf1 is believed to be involved in osteoblast differentiation mediated by miR-29b-3p (Figure 4). Its expression could be reduced by miR-29b-3p, whose production by osteocytes is downregulated upon mechanical loading. Therefore, increased osteoblast differentiation is closely linked with altered secretion of signaling molecules by osteocytes [109].
Osteoblasts and the matrix they deposit can determine the initial size and shape of the descendant osteocyte lacunae [110,111]. Notably, osteocytes derived from dynamic osteogenesis (DO), where migrating osteoblasts differentiate into osteocytes in apposition to pre-existing trabeculae, are smaller and ellipsoidal [13,112]. As DO occurs commonly in response to mechanical deformation and produces osteocytes aligned with the principal loading direction [113], one may assume that the direct recruitment of osteoblasts, in addition to mesenchymal progenitors, is responsible for subchondral bone remodeling in OA. Hence, the abundance of immature osteocytes in OA can also arise from a problematic transformation from the recruited osteoblasts to osteocytes [71]. Cytoskeleton and the distribution of actin filaments and microtubules are believed to affect the differentiation from osteoblasts to osteocytes [114]. Cytoskeletal rearrangement normally allows osteocytes to adopt different shapes and become more sensitive to mechanical stress and strain. Compared with osteoblasts, osteocytes exhibit decreased stiffness and are more dependent on actin filaments instead of microtubules, though both are critical for mechano-transduction [21,75]. Increased expressions of cytoskeletal proteins in osteocytes were induced by shear stress, along with elevated integrin, E11/gp38, and Runx-2 levels [115]. Hence, cytoskeletal arrangement and thus osteoblast differentiation may be disrupted by an abnormal level of stress in OA (Figure 6a). Meanwhile, osteocyte dysfunction induced by advanced glycation end products (AGEs) showed increased production of sclerostin and RANKL [95]. This process was mediated by FOXO1, a regulator of osteoblast differentiation. FOXO1 can enhance Runx-2 activities [116], while Runx-2 can upregulate sclerostin level [117], inhibit osteoblast-osteocyte differentiation, but promote osteoblast differentiation [118]. Hence, osteocyte dysfunction is linked with erroneous osteoblast differentiation in both mechanically and chemically induced OA. DMP-1 is also considered essential in both osteoblast differentiation and the transformation from osteoblasts to osteocytes [84]. Its release by osteoblasts into the ECM is critical to bone mineralization and osteoblast differentiation [51]. Hence, DMP-1 deficiency in osteoblasts upon mechanical loading [53] may explain the arrested differentiation, though it remains unelucidated how the osteocyte-derived DMP-1 may affect this process.
In addition to aberrant protein and RNA expressions, anomalies of the content in the ECM may be equally crucial to the pathophysiology of OA. For instance, morphological changes of bone mineral particles and disorganization of collagen fibers are likely involved in osteoblast and osteocyte dysfunction [119]. Hydroxyapatite (HA) is the major type of bone mineral. HA crystals are formed by phosphate and calcium ions, synthesized by osteoblasts and chondrocytes, and regulated by osteocalcin [120]. Osteocytes are also believed to be responsible for bone mineral control through periosteocytic osteolysis, which can be more prominent in pathological conditions [111]. Osteocyte-derived sclerostin can modulate osteoblast-osteocyte differentiation through enhancing phosphorylated MEPE or suppressing phosphate-regulating neutral endopeptidase (PHEX) expression. Cleaved phosphorylated MEPE can bind to HA crystals and halt mineralization, while PHEX can inhibit this process and promote mineralization [121]. Hence, sclerostin deficiency can also affect HA crystals, leading to increased mineralization. Meanwhile, decreased DMP-1 expression associated with Wnt signaling will likely impact the formation of HA crystals, as DMP-1 is essential for the mineralization of collagen [52]. Examinations of HA crystals by our team suggested that bone mineral particles could directly contribute to OA development. In OA bone, HA crystals were predominantly in a cigarette-like shape, while those extracted from the control group appeared bean-like (Figure 7). Abnormal collagen and mineral production might reflect osteoblastic dysfunction, indicate erroneous osteoblast differentiation, and directly or indirectly correlate with osteocyte dysfunction [6,21,80]. When mixed with MG-63 mature osteoblasts, HA nanoparticles derived from OA bone showed a significant inhibitory effect on proliferation [122] and alkaline phosphatase (ALP) activities. Therefore, malformed HA nanoparticles in OA bone might further impair mineral deposition and disrupt matrix maturation by interfering with osteoblastic and pre-osteoblastic activities [123]. Aberrant mineralization could thus exacerbate osteocyte dysfunction and eventually result in the undermined strength of OA trabecular bone.
3.3. Regulation of Mesenchymal Progenitors
The recruitment of mesenchymal progenitor cells in joint tissues has long been believed to be involved in OA progression. It was reported that mesenchymal progenitor cells were present at the destruction site of the osteoarthritic cartilage and populated in the superficial layer of the articular cartilage [124,125]. They could also be isolated from the synovium and synovial fluids in OA joints [126,127]. Osteocytes regulate bone-marrow-derived cells through sclerostin, as altered sclerostin expression was found to affect adipogenesis and osteogenesis [79]. TGF-β signaling is associated with the increased number of MSCs in the bone marrow. Enhanced osterix expression in induced OA is related to the phosphorylation of Smad2/3 through TGF-β signaling (Figure 6b) [45]. Osterix normally regulates osteoblast differentiation and promotes sclerostin production [117]. However, increased osterix expression, but not sclerostin expression, has been observed in sclerotic bones. This might be explained by the suppression of TGF-β signaling upon mechanical loading [128]. Suppressed TGF-β signaling could have inhibited the phosphorylation of Smad2/3, decreased sclerostin deficiency, and activated Wnt signaling [129]. MSCs recruitment and osteoblast differentiation were hence enhanced, leading to increased osterix expression by osteoblasts and MSCs, but could not rescue sclerostin deficiency. Osteoblastic differentiation of MSCs is also mediated by Igf1 through the activation of the PI3K/AKT/mTOR pathway [130]. Increased Igf1 production in induced OA models [36] might induce osteogenic differentiation of MSCs and eventually facilitate the maturation of osteoblasts. Osteocytes at the bone formation sites expressed immature markers like E11 in OA samples [81], indicating incomplete differentiation. These findings further suggested that MSCs acted as a reservoir of osteoblasts and were regulated by the newly synthesized immature osteocytes. Hence, microfracture of the subchondral bone by mechanical loading could induce osteocyte death, causing compromised sclerostin expression. Abnormal production of signaling molecules then activated and recruited marrow or periosteum-derived progenitor cells to the boundary of subchondral bone and articular cartilage for tissue repair [131].
It has been well documented that the content of the ECM can either induce or inhibit bone formation. For example, the size of bone matrix particles and the structure of collagen fibrils were found to affect the lineage commitment of MSCs [132]. Meanwhile, the shape, crystallinity, surface charge, solubility, and mechanical properties of HA nanoparticles could also influence osteoblastic activities and the osteogenic potential of MSCs [133,134,135,136,137]. It was theorized that MSCs were recruited to osteoclast-mediated bone resorption pits and contacted with the debris after resorption. HA nanoparticles were postulated to be one of the major components of the debris. Differentially shaped HA nanoparticles were suggested to provide dissimilar geometric cues and determine the fate of osteoblasts and MSCs. This was supported by findings suggesting that cigarette-like HA nanoparticles in OA bones could promote cytoskeletal changes of human MSCs and enhance their osteogenic potentials [122]. As there were no significant differences between the HA nanoparticles in healthy and OA bone except for their shape, it is possible that the physical property of HA, rather than the chemical property, is deterministic for controlling the fate of MSCs in bone remodeling. Hence, osteocyte dysfunction may also indirectly promote osteogenic differentiation of MSCs through inducing aberrant mineralization by osteoblasts.
In summary, altered sclerostin expression may cause osteoblast dysfunction and consequently abnormal mineral production, which can directly or indirectly facilitate osteogenic differentiation of MSCs. It was thus conceptualized that the accumulated microdamage to the subchondral bone by mechanical overload would promote tissue repair, and eventually, bone sclerosis, by which bone turnover was enhanced and resulted in disease deterioration [138]. The increased MSCs recruitment and osteoblastic activities appear to suggest an adaptation to compensate for osteocyte dysfunction and death upon mechanical loading, though this may, in fact, exacerbate the anomalies in tissue morphology and protein production as mature and functional osteocytes are depleted.
3.4. Regulation of Cartilage Tissues
Recently, chondrocytes were proposed to be another source of osteoblasts in addition to MSCs, especially since chondrocytes in OA and MSCs appeared to share the fate of hypertrophic differentiation followed by ossification [139,140,141]. Runx-2 and osterix, the regulators of osteoblast differentiation, were assumed to be involved in the possible trans-differentiation from chondrocytes to osteoblasts. Runx-2 could induce chondrocyte hypertrophy, while osterix was expressed by hypertrophic chondrocytes [118,140]. Hypertrophic chondrocytes also express DMP-1, suggesting commitments to the osteogenic lineage and possibly the status of pre-osteoblasts [9,142]. Wnt/β-catenin signaling enhances Runx-2 production and promotes the expression of type X collagen, a hypertrophic marker [143]. Activation of Wnt signaling can be regulated by either TGF-β, BMP-2, with BMP-2 promoting and TGF-β inhibiting chondrocyte hypertrophy (Figure 6a). Previous studies reported that sclerostin was found in chondrocytes of calcified cartilage tissues in normal joints and non-calcified cartilage in OA [25,144]. Sclerostin was hence speculated to be secreted either by chondrocytes themselves or by osteocytes in the subchondral bone and then entered cartilage tissues [145]. Therefore, osteocytes might also function as a regulator of chondrocyte hypertrophy and even trans-differentiation, which could be another possible reason why cell cluster was found at deep and calcified cartilage [28]. It is believed that sclerostin could also inhibit the Wnt/β-catenin signaling pathway and thus downregulate Runx-2, type X collagen, and MMP expressions in cartilage tissue [144,146]. Thus, decreased sclerostin expression might contribute to increased chondrocyte hypertrophy and cartilage degeneration in OA. Notably, sclerostin expression has been observed in cartilage to peak at early-stage chondrogenic differentiation but was decreased at late-stage chondrogenic differentiation [144]. This was echoed by the time-course variation of sclerostin level in both calcified cartilage and subchondral bone seen in induced murine OA models [25]. Differential regulations of sclerostin expressions might explain the increase in chondrocyte sclerostin level observed in mid-stage but not end-stage human OA and could suggest that osteocyte dysfunction preceded or occurred simultaneously with chondrocyte hypertrophy, causing increasingly severe disturbances to the subchondral bone and cartilage as OA progressed [145]. Hence, the disrupted negative feedback to chondrocytes due to a reduction in sclerostin level may contribute to or at least aggravate chondrocyte hypertrophy, though whether trans-differentiation from chondrocytes to osteoblasts and OA progression are mediated by osteocytes and sclerostin requires further investigation.
3.5. Regulation of Osteoclasts
Osterix-positive pre-osteoblasts are often spatially disassociated with TRAP-positive osteoclasts in OA bone remodeling, which might explain why bone formation in OA is not coupled with bone resorption pits [28]. Upregulation of osteoclastic activities in OA is believed to be controlled by osteocytes through sclerostin and the OPG/RANKL/RANK system (Figure 8) [13]. This was supported by the elevation in RANKL expression in induced murine OA models [95]. Osteocytes are the major source of RANKL in bone and are responsible for osteoclastogenesis and bone resorption [147]. Osteocyte-derived sclerostin can promote osteoclastogenesis, which is enhanced by RANKL and suppressed by OPG [148]. The PHEX/MEPE/ASARM-PO4 pathway partially regulated by sclerostin was also found to modulate RANKL/OPG expressions. ASARM-PO4, a phosphorylated peptide derived from MEPE, could inhibit RANKL expression but upregulate OPG, while PHEX mostly promoted RANKL expression [149]. Hence, sclerostin deficiency might cause reduced ASARM-PO4 production and increased PHEX signaling [121], thus upregulating the RANKL/OPG ratio. However, a high RANKL/OPG ratio did not guarantee osteoclastogenesis, which might suggest that PHEX/MEPE/ASARM-PO4 signaling could regulate osteoclastic activities through other pathways. For pathological conditions associated with aberrant bone mineralization and protein production, such as hypophosphatemic rickets, the addition of exogenous PHEX might also contribute to higher OPG levels [149]. This might suggest that osteoblastic and osteoclastic activities were also affected bone mineralization. Inflammatory factors commonly found in arthritis could also increase RANKL and decrease OPG expressions, thus upregulating the RANKL/OPG ratio. This effect could be attenuated by mechanical loading [150], which might suggest that osteoclastic activities were favored by inflammation but suppressed by mechanical stress. Similarly, mechanical loading itself could cause a reduction in osteoclastogenesis through increasing MEPE expression [151]. Interestingly, both RANKL and OPG transcriptions were upregulated by mechanical loading, though the RANKL/OPG ratio was decreased. The release of OPG by osteocytes was upregulated soon after mechanical loading, while the release of RANKL was delayed [152], which could suggest a return to homeostasis or regulation by other pathways. Fatigue loading associated with microdamage and increased osteocyte apoptosis, however, appeared to promote osteoclastogenesis, especially near the site of damage [153]. Hence, damage and osteocyte death are likely critical to induce osteoclastogenesis. In the absence of cell and tissue injury, mechanical loading may preferentially enhance osteoblastic activities.
Past studies reported that MSCs and immature osteocytes also expressed OPG, the RANKL inhibitor, while RANKL expression was not affected [81]. As RANKL expression is strongly correlated with osteocyte death, one may assume that osteocyte dysfunction or apoptosis is required to trigger RANKL expression [154], while OPG expression is predominant in osteoblasts and early osteocytes. Thus, temporally and spatially differential regulation of osteoclastogenesis can be achieved depending on the status of osteocytes [17]. Meanwhile, osteoclastic activities can also be mediated by RANKL-independent pathways, such as by DAMPs released during osteocyte necrosis [102], which promotes osteoclastic activities; or by cysteine-rich protein 61 (CYR-61), which inhibits osteoclastogenesis [155]. Localization of living osteocytes in OA suggested that cells were absent from the cores of the enlarged trabeculae but present in the newly formed bone structure in apposition to pre-existing dead trabeculae [98]. Thus, osteocyte death in trabecular cores and osteocyte activities in the surroundings might be correlated with the disassociation between osteoclastic and osteoblastic activities. Likewise, RANKL expressions were generally closer to the site of damage and apoptotic osteocytes, while OPG-positive cells were farther away [153]. These might also partially explain our findings regarding bone remodeling in hip OA. Osteoid formation in apposition to pre-existing trabeculae was not accompanied by bone resorption, while osteoid formation in the control group was closely associated with bone resorption pits spatially (Figure 3c). Meanwhile, in induced murine OA models, bone loss was more prominent at early-stage OA, while bone formation became significant at end-stage OA [156]. Therefore, there appeared to be both spatial and temporal differences in osteoclastic and osteoblastic activities, with osteoclastic activities seemingly preceding osteoblastic activities and showing different site preferences. Hence, OA development may be the result of temporally and spatially differential regulations of osteoblastic and osteoclastic activities by osteocytes in response to mechanical loading and other stimuli.
4. Allometry
Allometry, or biological scaling, refers to the process where one organismal trait varies with another, typically with an emphasis on the correlation between cell size and mass, shape, growth rate, or metabolic rate [157]. In most mammalian cells, cell size and growth rates are believed to be controlled by metabolism, where mitochondrial activities are postulated as the potential sensing mechanism of cell size. Increased cell size is often accompanied by compromised mitochondrial metabolism and upregulation of cytoskeletal genes [158,159], while the optimal cell size is normally of intermediate volume and yields maximal functionality [160]. Chondrocyte hypertrophy, for instance, is known to be associated with OA development. Hypertrophic chondrocytes secreted MMP-13 but expressed fewer cartilage makers, which was linked with increased catabolic activities and reduced anabolic signaling [63,161]. As free energy was required for protein synthesis, a preference for catabolism over anabolism might imply compromised metabolic activities, which was consistent with the observed metabolic inefficiency in other oversized cells [159,160]. Hence, changes in the protein expression profile of an enlarged chondrocyte might correlate with its inability to maintain the metabolic efficiency of a healthy cell. It should also be noted that the variation in metabolism and thus protein synthesis could be phase-dependent, as three distinct phases were previously identified in chondrocyte hypertrophy. Only the second phase was characterized by a drop in dry mass density and presumably low metabolic rates [58]. Meanwhile, the third phase of chondrocyte enlargement was found to be regulated by Igf1 (Figure 4), which was usually secreted by hepatic cells and bone cells, including osteocytes. Upregulation of Igf1 production by osteocytes was observed in response to mechanical loading, especially in the loaded region [58,59]. As Igf1 expression by osteoarthritic chondrocytes themselves were reduced [61], this might suggest that chondrocyte hypertrophy was regulated through osteocyte-derived Igf1 and that Igf1 overexpression by osteocytes could link cartilage degeneration by MMP-13 with excessive loading.
In addition to chondrocytes, osteocytes could also change the size of their lacunae even after being embedded into the bone matrix. This might at least partially explain the morphological and functional changes in osteocytes in response to stimuli [88]. For example, in avian bones, mechanical loading appeared to induce osteocytes responsible for sensing compressive stress to grow along the longitudinal axis, though this effect was modest and did not affect the overall lacuna size. The volume of osteocytes was found to scale with mass-specific basal metabolic rate, though their correlation was relatively weak [110]. An abnormally small lacuna size and an increased number of osteocytes were observed both under excessive mechanical loading and in OA bones [21]. The altered protein expression patterns, such as sclerostin deficiency, might be associated with aberrant metabolism, which then disrupted the regulation of bone cells and joint homeostasis. Meanwhile, the ellipsoidal osteocytes predominantly produced under mechanical loading might suggest elevated metabolism. Elongated cells are often linked with a higher surface-to-volume ratio and metabolic rate compared with spherical cells of the same volume, as the intracellular distances traveled by biomolecules are reduced [160]. Hence, future studies should investigate whether there exists a link between signaling molecule production, cellular metabolic rate, and cell size and whether mechanical loading can alter osteocyte function by changing osteocyte morphology and impacting cell metabolism.
Overall, in addition to increased osteocyte apoptosis upon damage and osteoblast differentiation for bone repair, mechanical loading may also contribute to osteocyte dysfunction, chondrocyte hypertrophy, and thus OA development through acting on cell volume and metabolism directly or indirectly. Future research should also elucidate whether osteocytes display abnormal metabolic rates under mechanical loading and whether the observed morphological and functional changes can contribute to OA before or independent of osteocyte death.
5. Conclusions
In a healthy individual, bone remodeling occurs continuously and actively to maintain homeostasis, notably of the articular joint [17]. This process is believed to be regulated by osteocytes, which recruit and activate osteoblasts, osteoclasts, and progenitors with an osteogenic potential to designated sites for bone formation or absorption. Sclerostin expressed by osteocytes, bone minerals deposited by osteoblasts, and MMPs are among the molecules proposed to affect cellular activities and joint integrity [96,97]. A balance of osteoblastic and osteoclastic activities ensures the mechanical strength and uniform distribution of trabeculae at the tissue level, which enables the joint to withstand compressive forces. If disrupted, bone pathologies such as OA or OP may manifest. The pathomechanism of OA is involuted and likely involves exogenous and indigenous factors, such as trauma and age-related oxidative stress [63,162]. In this review, we attempt to provide a possible mechanism of OA development, with an emphasis on the role of mechanical loading in pathogenesis. At the molecular level, excessive loading can affect protein synthesis, notably inhibiting sclerostin expression, which may partially explain the observed drop in sclerostin level in OA [71]. This process interacts with various signaling pathways, possibly including periostin, DMP-1, and TGF-β signaling, and may be associated with aberrant metabolic activities [29,74,128]. Meanwhile, the low level of sclerostin expression may also arise from the deaths of pre-existing osteocytes due to microdamage and their replacement by the newly synthesized immature or dysfunctional osteocytes. Sclerostin deficiency then activates the Wnt signaling pathways and regulates the OPG/RANKL/RANK system, which recruits osteoclasts and osteoblasts in a time-and-space-dependent manner [13]. It is hypothesized that osteoclasts will first absorb the existing bones in early-stage OA, while osteoblasts will deposit collagens and minerals to align the osteocytes along the principal loading direction and enhance the osteogenic potential of mesenchymal progenitors in end-stage OA [98]. Thus, the spatially and temporally uncoupled bone remodeling renders the trabecular bone mechanically inferior and results in structural disturbances of the subchondral bone. Abnormal bone microarchitecture may undermine the ability of the joint to sustain compression and gradually contribute to cartilage degeneration.
In summary, as mechano-sensors, osteocytes display aberrant and altered protein secretion under excessive mechanical loading, which is believed to link osteocytes with the pathophysiology of arthritis, including ankylosing spondylitis and osteoarthritis [82,163]. Thus, osteocyte dysfunction and the relevant signaling pathways may serve as a potential therapeutic target for OA treatment, especially if induced by cumulative or repetitive loading [18]. However, whether osteocytes and their secretions remain to be the central regulator and an essential signaling molecule in other means of pathogeneses of OA is not fully elucidated and should be further investigated in the future.
Author Contributions
C.W. proposed the focus and framework of the study. L.Z. prepared the draft of the manuscript, which was revised by C.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work received support from the Research Grants Council of Hong Kong’s Early Career Scheme (PolyU 251008/18M), PROCORE-France/Hong Kong Joint Research Scheme (F-PolyU504/18), Health and Medical Research Fund Scheme (01150087, 15161391, 16172691), PolyU project of strategic importance (ZE2C), Hong Kong Polytechnic.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were generated in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- Kendzerska, T.; Juni, P.; King, L.K.; Croxford, R.; Stanaitis, I.; Hawker, G.A. The longitudinal relationship between hand, hip and knee osteoarthritis and cardiovascular events: A population-based cohort study. Osteoarthr. Cartil. 2017, 25, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Brandt, K.D.; Dieppe, P.; Radin, E. Etiopathogenesis of osteoarthritis. Med. Clin. N. Am. 2009, 93, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; Gerstenfeld, L.; Bishop, G.; Davis, A.D.; Mason, Z.D.; Einhorn, T.A.; Maciewicz, R.A.; Newham, P.; Foster, M.; Jackson, S.; et al. Bone marrow lesions from osteoarthritis knees are characterized by sclerotic bone that is less well mineralized. Arthritis Res. Ther. 2009, 11, R11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, K.; Nango, N.; Kubota, S.; Okazaki, N.; Taguchi, K.; Osaki, M.; Ito, M. Relationship between microstructure and degree of mineralization in subchondral bone of osteoarthritis: A synchrotron radiation µCT study. J. Bone Miner. Res. 2012, 27, 1511–1517. [Google Scholar] [CrossRef]
- Coats, A.M.; Zioupos, P.; Aspden, R.M. Material properties of subchondral bone from patients with osteoporosis or osteoarthritis by microindentation testing and electron probe microanalysis. Calcif. Tissue Int. 2003, 73, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Couchourel, D.; Aubry, I.; Delalandre, A.; Lavigne, M.; Martel-Pelletier, J.; Pelletier, J.-P.; Lajeunesse, D. Altered mineralization of human osteoarthritic osteoblasts is attributable to abnormal type I collagen production. Arthritis Rheumatol. 2009, 60, 1438–1450. [Google Scholar] [CrossRef] [PubMed]
- Bettica, P.; Cline, G.; Hart, D.J.; Meyer, J.; Spector, T.D. Evidence for increased bone resorption in patients with progressive knee osteoarthritis: Longitudinal results from the Chingford study. Arthritis Rheumatol. 2002, 46, 3178–3184. [Google Scholar] [CrossRef] [PubMed]
- Dai, G.; Xiao, H.; Liao, J.; Zhou, N.; Zhao, C.; Xu, W.; Xu, W.; Liang, X.; Huang, W. Osteocyte TGFβ1-Smad2/3 is positively associated with bone turnover parameters in subchondral bone of advanced osteoarthritis. Int. J. Mol. Med. 2020, 46, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Lin, S.; Liu, Y.; Yuan, B.; Harris, S.E.; Feng, J.Q. Dmp1 null mice develop a unique osteoarthritis-like phenotype. Int. J. Biol. Sci. 2016, 12, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, N.; Standley, K.N.; Bianchi, E.N.; Stadelmann, V.; Foti, M.; Conway, S.J.; Ferrari, S.L. The matricellular protein periostin is required for sost inhibition and the anabolic response to mechanical loading and physical activity. J. Biol. Chem. 2009, 284, 35939–35950. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Sun, X.; Ma, J.; Ma, X.; Xing, G.; Wang, Y.; Sun, L.; Wang, J.; Li, F.; Li, Y. Involvement of periostin-sclerostin-Wnt/β-catenin signaling pathway in the prevention of neurectomy-induced bone loss by naringin. Biochem. Biophys. Res. Commun. 2015, 468, 587–593. [Google Scholar] [CrossRef]
- Funck-Brentano, T.; Bouaziz, W.; Marty, C.; Geoffroy, V.; Hay, E.; Cohen-Solal, M. Dkk-1-Mediated inhibition of Wnt signaling in bone ameliorates osteoarthritis in mice. Arthritis Rheumatol. 2014, 66, 3028–3039. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, C.; Ferretti, M. The osteocyte: From “prisoner” to “orchestrator”. J. Funct. Morphol. Kinesiol. 2021, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Carbone, L.D.; Nevitt, M.C.; Wildy, K.; Barrow, K.D.; Harris, F.; Felson, D.; Peterfy, C.; Visser, M.; Harris, T.B.; Wang, B.W.E.; et al. The relationship of antiresorptive drug use to structural findings and symptoms of knee osteoarthritis. Arthritis Rheumatol. 2004, 50, 3516–3525. [Google Scholar] [CrossRef]
- Ding, M.; Danielsen, C.C.; Hvid, I. The Effects of bone remodeling inhibition by alendronate on three-dimensional microarchitecture of subchondral bone tissues in guinea pig primary osteoarthrosis. Calcif. Tissue Int. 2008, 82, 77–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayami, T.; Pickarski, M.; Wesolowski, G.A.; Mclane, J.; Bone, A.; Destefano, J.; Rodan, G.A.; Duong, L.T. The role of subchondral bone remodeling in osteoarthritis: Reduction of cartilage degeneration and prevention of osteophyte formation by alendronate in the rat anterior cruciate ligament transection model. Arthritis Rheumatol. 2004, 50, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, K.; Neutzsky-Wulff, A.V.; Bonewald, L.F.; Karsdal, M.A. Local communication on and within bone controls bone remodeling. Bone 2009, 44, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Lajeunesse, D.; Reboul, P. Subchondral bone in osteoarthritis: A biologic link with articular cartilage leading to abnormal remodeling. Curr. Opin. Rheumatol. 2003, 15, 628–633. [Google Scholar] [CrossRef]
- Massicotte, F.; Aubry, I.; Martel-Pelletier, J.; Pelletier, J.-P.; Fernandes, J.; Lajeunesse, D. Abnormal insulin-like growth factor 1 signaling in human osteoarthritic subchondral bone osteoblasts. Arthritis Res. Ther. 2006, 8, R177. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, C.; Deberg, M.A.; Bellahcène, A.; Castronovo, V.; Msika, P.; Delcour, J.P.; Crielaard, J.M.; Henrotin, Y.E. Phenotypic characterization of osteoblasts from the sclerotic zones of osteoarthritic subchondral bone. Arthritis Rheumatol. 2008, 58, 442–455. [Google Scholar] [CrossRef]
- Hemmatian, H.; Bakker, A.D.; Klein-Nulend, J.; van Lenthe, G.H. Aging, osteocytes, and mechanotransduction. Curr. Osteoporos. Rep. 2017, 15, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Ten Dijke, P.; Krause, C.; de Gorter, D.J.J.; Löwik, C.W.G.M.; van Bezooijen, R.L. Osteocyte-Derived sclerostin inhibits bone formation: Its role in bone morphogenetic protein and Wnt signaling. J. Bone Jt. Surg. 2008, 90, 31–35. [Google Scholar] [CrossRef]
- Van Bezooijen, R.L.; Roelen, B.A.J.; Visser, A.; van der Wee-Pals, L.; de Wilt, E.; Karperien, M.; Hamersma, H.; Papapoulos, S.E.; ten Dijke, P.; Löwik, C.W.G.M. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J. Exp. Med. 2004, 199, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Bonewald, L.F. Osteocytes: A proposed multifunctional bone cell. J. Musculoskelet. Neuronal Interact. 2002, 2, 239–241. [Google Scholar]
- Li, J.; Xue, J.; Jing, Y.; Wang, M.; Shu, R.; Xu, H.; Xue, C.; Feng, J.; Wang, P.; Bai, D. SOST deficiency aggravates osteoarthritis in mice by promoting sclerosis of subchondral bone. Biomed. Res. Int. 2019, 2019, 7623562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chappard, C.; Peyrin, F.; Bonnassie, A.; Lemineur, G.; Brunet-Imbault, B.; Lespessailles, E.; Benhamou, C.L. Subchondral bone micro-architectural alterations in osteoarthritis: A synchrotron micro-computed tomography study. Osteoarthr. Cartil. 2006, 14, 215–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, K.; Ito, M.; Osaki, M.; Uetani, M.; Shindo, H. In vivo structural analysis of subchondral trabecular bone in osteoarthritis of the hip using multi-detector row CT. Osteoarthr. Cartil. 2011, 19, 180–185. [Google Scholar] [CrossRef] [Green Version]
- Jaiprakash, A.; Prasadam, I.; Feng, J.Q.; Liu, Y.; Crawford, R.; Xiao, Y. Phenotypic characterization of osteoarthritic osteocytes from the sclerotic zones: A possible pathological role in subchondral bone sclerosis. Int. J. Biol. Sci. 2012, 8, 406–417. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Jiang, X.; Dai, Z.; Guo, X.; Weng, T.; Wang, J.; Li, Y.; Feng, G.; Gao, X.; He, L. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/β-Catenin signaling. J. Bone Miner. Res. 2009, 24, 1651–1661. [Google Scholar] [CrossRef]
- Tu, X.; Rhee, Y.; Condon, K.W.; Bivi, N.; Allen, M.R.; Dwyer, D.; Stolina, M.; Turner, C.H.; Robling, A.G.; Plotkin, L.I.; et al. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone 2012, 50, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.; Ma, X.; Wei, Y.; Tong, W.; Tower, R.J.; Chandra, A.; Wang, L.; Sun, Z.; Yang, Z.; Badar, F.; et al. Loading-Induced reduction in sclerostin as a mechanism of subchondral bone plate sclerosis in mouse knee joints during late-stage osteoarthritis. Arthritis Rheumatol. 2018, 70, 230–241. [Google Scholar] [CrossRef] [Green Version]
- Ko, F.C.; Dragomir, C.; Plumb, D.A.; Goldring, S.R.; Wright, T.M.; Goldring, M.B.; van der Meulen, M.C.H. In vivo cyclic compression causes cartilage degeneration and subchondral bone changes in mouse tibiae. Arthritis Rheumatol. 2013, 65, 1569–1578. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, N.; Garnero, P.; Ferrari, S. Periostin action in bone. Mol. Cell. Endocrinol. 2016, 432, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Tajika, Y.; Moue, T.; Ishikawa, S.; Asano, K.; Okumo, T.; Takagi, H.; Hisamitsu, T. Influence of periostin on synoviocytes in knee osteoarthritis. In Vivo 2017, 31, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Chijimatsu, R.; Kunugiza, Y.; Taniyama, Y.; Nakamura, N.; Tomita, T.; Yoshikawa, H. Expression and pathological effects of periostin in human osteoarthritis cartilage. BMC Musculoskelet. Disord. 2015, 16, 215. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Fang, H.; Chen, Y.; Shen, J.; Lu, H.; Zeng, C.; Ren, J.; Zeng, H.; Li, Z.; Chen, S.; et al. Gene expression analyses of subchondral bone in early experimental osteoarthritis by microarray. PLoS ONE 2012, 7, e32356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnilä, M.A.J.; Thevenot, J.; Aho, O.-M.; Tiitu, V.; Rautiainen, J.; Kauppinen, S.; Nieminen, M.T.; Pritzker, K.; Valkealahti, M.; Lehenkari, P.; et al. Association between subchondral bone structure and osteoarthritis histopathological grade. J. Orthop. Res. 2017, 35, 785–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, M.; Odgaard, A.; Danielsen, C.C.; Hvid, I. Mutual associations among microstructural, physical and mechanical properties of human cancellous bone. J. Bone Jt. Surg. Br. 2002, 84, 900–907. [Google Scholar] [CrossRef]
- Wang, T.; Wen, C.Y.; Yan, C.H.; Lu, W.W.; Chiu, K.Y. Spatial and temporal changes of subchondral bone proceed to microscopic articular cartilage degeneration in guinea pigs with spontaneous osteoarthritis. Osteoarthr. Cartil. 2013, 21, 574–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, S.J.; Izuhara, K.; Kudo, Y.; Litvin, J.; Markwald, R.; Ouyang, G.; Arron, J.R.; Holweg, C.T.; Kudo, A. The role of periostin in tissue remodeling across health and disease. Cell. Mol. Life. Sci. 2014, 71, 1279–1288. [Google Scholar] [CrossRef] [Green Version]
- Spatz, J.M.; Ellman, R.; Cloutier, A.M.; Louis, L.; van Vliet, M.; Suva, L.J.; Dwyer, D.; Stolina, M.; Ke, H.Z.; Bouxsein, M.L. Sclerostin antibody inhibits skeletal deterioration due to reduced mechanical loading. J. Bone Miner. Res. 2013, 28, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Gingery, A.; Subramaniam, M.; Pitel, K.S.; Li, X.; Ke, H.Z.; Turner, R.T.; Iwaniec, U.T.; Hawse, J.R. Sclerostin antibody treatment rescues the osteopenic bone phenotype of TGFβ inducible early gene-1 knockout female mice. J. Cell Physiol. 2020, 235, 5679–5688. [Google Scholar] [CrossRef]
- Mazur, C.M.; Woo, J.J.; Yee, C.S.; Fields, A.J.; Acevedo, C.; Bailey, K.N.; Kaya, S.; Fowler, T.W.; Lotz, J.C.; Dang, A.; et al. Osteocyte dysfunction promotes osteoarthritis through MMP13-dependent suppression of subchondral bone homeostasis. Bone Res. 2019, 7, 34. [Google Scholar] [CrossRef] [Green Version]
- Rowe, P.S.N. Regulation of bone-renal mineral and energy metabolism: The PHEX, FGF23, DMP1, MEPE ASARM pathway. Crit. Rev. Eukaryot. Gene Expr. 2012, 22, 61–86. [Google Scholar] [CrossRef] [PubMed]
- Zhen, G.; Wen, C.; Jia, X.; Li, Y.; Crane, J.L.; Mears, S.C.; Askin, F.B.; Frassica, F.J.; Chang, W.; Yao, J.; et al. Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med. 2013, 19, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Rios, H.F.; Myers, E.R.; Lu, Y.; Feng, J.Q.; Boskey, A.L. DMP1 depletion decreases bone mineralization in vivo: An FTIR imaging analysis. J. Bone Miner. Res. 2005, 20, 2169–2177. [Google Scholar] [CrossRef]
- Baranyay, F.J.; Wang, Y.; Wluka, A.E.; English, D.R.; Giles, G.G.; Sullivan, R.O.; Cicuttini, F.M. Association of bone marrow lesions with knee structures and risk factors for bone marrow lesions in the knees of clinically healthy, community-based adults. Semin. Arthritis Rheum. 2007, 37, 112–118. [Google Scholar] [CrossRef]
- Davies-Tuck, M.L.; Wluka, A.E.; Forbes, A.; Wang, Y.; English, D.R.; Giles, G.G.; O’Sullivan, R.; Cicuttini, F.M. Development of bone marrow lesions is associated with adverse effects on knee cartilage while resolution is associated with improvement--a potential target for prevention of knee osteoarthritis: A longitudinal study. Arthritis Res. Ther. 2010, 12, R10. [Google Scholar] [CrossRef] [Green Version]
- Dore, D.; Martens, A.; Quinn, S.; Ding, C.; Winzenberg, T.; Zhai, G.; Pelletier, J.-P.; Martel-Pelletier, J.; Abram, F.; Cicuttini, F.; et al. Bone marrow lesions predict site-specific cartilage defect development and volume loss: A prospective study in older adults. Arthritis Res. Ther. 2010, 12, R222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornaat, P.R.; Kloppenburg, M.; Sharma, R.; Botha-Scheepers, S.A.; Le Graverand, M.-P.H.; Coene, L.N.J.E.M.; Bloem, J.L.; Watt, I. Bone marrow edema-like lesions change in volume in the majority of patients with osteoarthritis; associations with clinical features. Eur. Radiol. 2007, 17, 3073–3078. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, K.; Ramachandran, A.; Hao, J.; He, G.; Park, K.W.; Cho, M.; George, A. Dual functional roles of dentin matrix protein 1. Implications in biomineralization and gene transcription by activation of intracellular Ca2+ store. J. Biol. Chem. 2003, 278, 17500–17508. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Lin, J.; Shao, J.; Zuo, Q.; Wang, S.; Wolff, A.; Nguyen, D.T.; Rintoul, L.; Du, Z.; Gu, Y.; et al. Aberrant activation of Wnt signaling pathway altered osteocyte mineralization. Bone 2019, 127, 324–333. [Google Scholar] [CrossRef]
- Gluhak-Heinrich, J.; Ye, L.; Bonewald, L.F.; Feng, J.Q.; MacDougall, M.; Harris, S.E.; Pavlin, D. Mechanical loading stimulates dentin matrix protein 1 (DMP1) expression in osteocytes in vivo. J. Bone Miner. Res. 2003, 18, 807–817. [Google Scholar] [CrossRef]
- Ding, M.; Odgaard, A.; Hvid, I. Changes in the three-dimensional microstructure of human tibial cancellous bone in early osteoarthritis. J. Bone Jt. Surg. Br. 2003, 85, 906–912. [Google Scholar] [CrossRef]
- Brown, S.J.; Pollintine, P.; Powell, D.E.; Davie, M.W.J.; Sharp, C.A. Regional differences in mechanical and material properties of femoral head cancellous bone in health and osteoarthritis. Calcif. Tissue Int. 2002, 71, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Cicuttini, F.; Jones, G. Tibial subchondral bone size and knee cartilage defects: Relevance to knee osteoarthritis. Osteoarthr. Cartil. 2007, 15, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Xuan, S.; Bouxsein, M.L.; von Stechow, D.; Akeno, N.; Faugere, M.C.; Malluche, H.; Zhao, G.; Rosen, C.J.; Efstratiadis, A.; et al. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J. Biol. Chem. 2002, 277, 44005–44012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, K.L.; Oh, S.; Sung, Y.; Dasari, R.R.; Kirschner, M.W.; Tabin, C.J. Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature 2013, 495, 375–378. [Google Scholar] [CrossRef]
- Sheng, M.H.; Lau, K.H.; Baylink, D.J. Role of osteocyte-derived insulin-like growth factor I in developmental growth, modeling, remodeling, and regeneration of the bone. J. Bone Metab. 2014, 21, 41–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, F.; Wang, Y.; Bikle, D.D. IGF-1 signaling mediated cell-specific skeletal mechano-transduction. J. Orthop. Res. 2018, 36, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Chubinskaya, S.; Hakimiyan, A.; Pacione, C.; Yanke, A.; Rappoport, L.; Aigner, T.; Rueger, D.C.; Loeser, R.F. Synergistic effect of IGF-1 and OP-1 on matrix formation by normal and OA chondrocytes cultured in alginate beads. Osteoarthr. Cartil. 2007, 15, 421–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeser, R.F. Molecular mechanisms of cartilage destruction: Mechanics, inflammatory mediators, and aging collide. Arthritis Rheumatol. 2006, 54, 1357–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rim, Y.A.; Nam, Y.; Ju, J.H. The role of chondrocyte hypertrophy and senescence in osteoarthritis initiation and progression. Int. J. Mol. Sci. 2020, 21, 2358. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Adams, J.; Leddy, H.A.; McNulty, A.L.; O’Conor, C.J.; Guilak, F. The mechanobiology of articular cartilage: Bearing the burden of osteoarthritis. Curr. Rheumatol. Rep. 2014, 16, 451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plumb, M.S.; Treon, K.; Aspden, R.M. Competing regulation of matrix biosynthesis by mechanical and IGF-1 signalling in elderly human articular cartilage in vitro. Biochim. Biophys. Acta 2006, 1760, 762–767. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, Y.; Yu, Y.E.; Zhang, X.; Watts, T.; Zhou, B.; Wang, J.; Wang, T.; Zhao, W.; Chiu, K.Y.; et al. Subchondral trabecular rod loss and plate thickening in the development of osteoarthritis. J. Bone Miner. Res. 2018, 33, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Muraoka, T.; Hagino, H.; Okano, T.; Enokida, M.; Teshima, R. Role of subchondral bone in osteoarthritis development: A comparative study of two strains of guinea pigs with and without spontaneously occurring osteoarthritis. Arthritis Rheumatol. 2007, 56, 3366–3374. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Fukuda, K.; Yamazaki, K.; Hashimoto, K.; Ueda, H.; Mori, S.; Hamanishi, C. Cyclic compression loaded on cartilage explants enhances the production of reactive oxygen species. J. Rheumatol. 2007, 34, 556–562. [Google Scholar]
- Wolff, K.J.; Ramakrishnan, P.S.; Brouillette, M.J.; Journot, B.J.; McKinley, T.O.; Buckwalter, J.A.; Martin, J.A. Mechanical stress and ATP synthesis are coupled by mitochondrial oxidants in articular cartilage. J. Orthop. Res. 2013, 31, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youlten, S.E.; Kemp, J.P.; Logan, J.G.; Ghirardello, E.J.; Sergio, C.M.; Dack, M.R.G.; Guilfoyle, S.E.; Leitch, V.D.; Butterfield, N.C.; Komla-Ebri, D.; et al. Osteocyte transcriptome mapping identifies a molecular landscape controlling skeletal homeostasis and susceptibility to skeletal disease. Nat. Commun. 2021, 12, 2444. [Google Scholar] [CrossRef]
- Robling, A.G.; Niziolek, P.J.; Baldridge, L.A.; Condon, K.W.; Allen, M.R.; Alam, I.; Mantila, S.M.; Gluhak-Heinrich, J.; Bellido, T.M.; Harris, S.E.; et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of sost/sclerostin. J. Biol. Chem. 2008, 283, 5866–5875. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.F.; Saunders, M.M.; Shingle, D.L.; Cimbala, J.M.; Zhou, Z.; Donahue, H.J. Mechanically stimulated osteocytes regulate osteoblastic activity via gap junctions. Am. J. Physiol. Cell Physiol. 2007, 292, C545–C552. [Google Scholar] [CrossRef] [Green Version]
- Staines, K.A.; Poulet, B.; Farquharson, C.; Pitsillides, A.A. The sclerostin and MEPE axis in the development of osteoarthritis. Osteoarthr. Cartil. 2013, 21. [Google Scholar] [CrossRef] [Green Version]
- Morse, A.; Schindeler, A.; McDonald, M.M.; Kneissel, M.; Kramer, I.; Little, D.G. Sclerostin antibody augments the anabolic bone formation response in a mouse model of mechanical tibial loading. J. Bone Miner. Res. 2018, 33, 486–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein-Nulend, J.; Bacabac, R.G.; Bakker, A.D. Mechanical loading and how it affects bone cells: The role of the osteocyte cytoskeleton in maintaining our skeleton. Eur. Cells Mater. 2012, 24, 278–291. [Google Scholar] [CrossRef]
- Lyons, J.S.; Joca, H.C.; Law, R.A.; Williams, K.M.; Kerr, J.P.; Shi, G.; Khairallah, R.J.; Martin, S.S.; Konstantopoulos, K.; Ward, C.W.; et al. Microtubules tune mechanotransduction through NOX2 and TRPV4 to decrease sclerostin abundance in osteocytes. Sci. Signal. 2017, 10, eaan5748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, L.; Fu, X.; Ma, J.; Lin, M.; Zhang, P.; Wang, Y.; Yan, Q.; Tao, C.; Liu, W.; Tang, B.; et al. Kindlin-2 mediates mechanotransduction in bone by regulating expression of Sclerostin in osteocytes. Commun. Biol. 2021, 4, 402. [Google Scholar] [CrossRef] [PubMed]
- Rabelo, G.D.; vom Scheidt, A.; Klebig, F.; Hemmatian, H.; Citak, M.; Amling, M.; Busse, B.; Jähn, K. Multiscale bone quality analysis in osteoarthritic knee joints reveal a role of the mechanosensory osteocyte network in osteophytes. Sci. Rep. 2020, 10, 673. [Google Scholar] [CrossRef] [Green Version]
- Intemann, J.; De Gorter, D.J.J.; Naylor, A.J.; Dankbar, B.; Wehmeyer, C. Importance of osteocyte-mediated regulation of bone remodelling in inflammatory bone disease. Swiss Med. Wkly. 2020, 150, w20187. [Google Scholar] [CrossRef]
- Prideaux, M.; Loveridge, N.; Pitsillides, A.A.; Farquharson, C. Extracellular matrix mineralization promotes E11/gp38 glycoprotein expression and drives osteocytic differentiation. PLoS ONE 2012, 7, e36786. [Google Scholar] [CrossRef] [Green Version]
- Ilas, D.C.; Churchman, S.M.; Baboolal, T.; Giannoudis, P.V.; Aderinto, J.; McGonagle, D.; Jones, E. The simultaneous analysis of mesenchymal stem cells and early osteocytes accumulation in osteoarthritic femoral head sclerotic bone. Rheumatology 2019, 58, 1777–1783. [Google Scholar] [CrossRef] [Green Version]
- Van Hove, R.P.; Nolte, P.A.; Vatsa, A.; Semeins, C.M.; Salmon, P.L.; Smit, T.H.; Klein-Nulend, J. Osteocyte morphology in human tibiae of different bone pathologies with different bone mineral density—Is there a role for mechanosensing? Bone 2009, 45, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Hemmatian, H.; Jalali, R.; Semeins, C.M.; Hogervorst, J.M.A.; van Lenthe, G.H.; Klein-Nulend, J.; Bakker, A.D. Mechanical loading differentially affects osteocytes in fibulae from lactating mice compared to osteocytes in virgin mice: Possible role for lacuna size. Calcif. Tissue Int. 2018, 103, 675–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Yuan, B.; Qin, C.; Cao, Z.; Xie, Y.; Dallas, S.L.; McKee, M.D.; Drezner, M.K.; Bonewald, L.F.; Feng, J.Q. The biological function of DMP-1 in osteocyte maturation is mediated by its 57-kDa C-terminal fragment. J. Bone Miner. Res. 2011, 26, 331–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacabac, R.G.; Mizuno, D.; Schmidt, C.F.; MacKintosh, F.C.; Van Loon, J.J.W.A.; Klein-Nulend, J.; Smit, T.H. Round versus flat: Bone cell morphology, elasticity, and mechanosensing. J. Biomech. 2008, 41, 1590–1598. [Google Scholar] [CrossRef]
- Kim, J.; Ishikawa, K.; Sunaga, J.; Adachi, T. Uniaxially fixed mechanical boundary condition elicits cellular alignment in collagen matrix with induction of osteogenesis. Sci. Rep. 2021, 11, 9009. [Google Scholar] [CrossRef]
- Kerschnitzki, M.; Wagermaier, W.; Roschger, P.; Seto, J.; Shahar, R.; Duda, G.N.; Mundlos, S.; Fratzl, P. The organization of the osteocyte network mirrors the extracellular matrix orientation in bone. J. Struct. Biol. 2011, 173, 303–311. [Google Scholar] [CrossRef]
- Van Oers, R.F.M.; Wang, H.; Bacabac, R.G. Osteocyte shape and mechanical loading. Curr. Osteoporos. Rep. 2015, 13, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Turner, C.H. Skeletal adaptation to mechanical loading. Clin. Rev. Bone Miner. Metab. 2008, 5, 181–194. [Google Scholar] [CrossRef]
- Wittkowske, C.; Reilly, G.C.; Lacroix, D.; Perrault, C.M. In vitro bone cell models: Impact of fluid shear stress on bone formation. Front Bioeng. Biotechnol. 2016, 4, 87. [Google Scholar] [CrossRef] [Green Version]
- Flynn, B.P.; Bhole, A.P.; Saeidi, N.; Liles, M.; Dimarzio, C.A.; Ruberti, J.W. Mechanical strain stabilizes reconstituted collagen fibrils against enzymatic degradation by mammalian collagenase matrix metalloproteinase 8 (MMP-8). PLoS ONE 2010, 5, e12337. [Google Scholar] [CrossRef] [Green Version]
- Kadler, K.E.; Hill, A.; Canty-Laird, E.G. Collagen fibrillogenesis: Fibronectin, integrins, and minor collagens as organizers and nucleators. Curr. Opin. Cell Biol. 2008, 20, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Kormpakis, I.; Havlioglu, N.; Ominsky, M.S.; Galatz, L.M.; Thomopoulos, S. Sclerostin antibody treatment enhances rotator cuff tendon-to-bone healing in an animal model. J. Bone Jt. Surg. Am. 2017, 99, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, N.; Gineyts, E.; Ammann, P.; Conway, S.J.; Garnero, P.; Ferrari, S. Periostin deficiency increases bone damage and impairs injury response to fatigue loading in adult mice. PLoS ONE 2013, 8, e78347. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wei, W.; Chi, M.; Wan, Y.; Li, X.; Qi, M.; Zhou, Y. FOXO1 Mediates advanced glycation end products induced mouse osteocyte-like MLO-Y4 cell apoptosis and dysfunctions. J. Diabetes Res. 2019, 2019, 6757428. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Hulley, P. The osteocyte—A novel endocrine regulator of body phosphate homeostasis. Maturitas 2010, 67, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Goldring, S.R. The osteocyte: Key player in regulating bone turnover. RMD Open 2015, 1, e000049. [Google Scholar] [CrossRef]
- Rony, L.; Perrot, R.; Hubert, L.; Chappard, D. Osteocyte staining with rhodamine in osteonecrosis and osteoarthritis of the femoral head. Microsc. Res. Tech. 2019, 82, 2072–2078. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Arozamena, J.; García-Renedo, R.; García-Ibarbia, C.; Pascual-Carra, M.A.; González-Macías, J.; Riancho, J.A. Osteocyte deficiency in hip fractures. Calcif. Tissue Int. 2011, 89, 327. [Google Scholar] [CrossRef]
- Duan, W.R.; Garner, D.S.; Williams, S.D.; Funckes-Shippy, C.L.; Spath, I.S.; Blomme, E.A. Comparison of immunohistochemistry for activated caspase-3 and cleaved cytokeratin 18 with the TUNEL method for quantification of apoptosis in histological sections of PC-3 subcutaneous xenografts. J. Pathol. 2003, 199, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Sun, J.; Hoemann, C.D.; Lascau-Coman, V.; Ouyang, W.; McKee, M.D.; Shive, M.S.; Buschmann, M.D. Drilling and microfracture lead to different bone structure and necrosis during bone-marrow stimulation for cartilage repair. J. Orthop. Res. 2009, 27, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Andreev, D.; Liu, M.; Weidner, D.; Kachler, K.; Faas, M.; Grüneboom, A.; Schlötzer-Schrehardt, U.; Muñoz, L.E.; Steffen, U.; Grötsch, B.; et al. Osteocyte necrosis triggers osteoclast-mediated bone loss through macrophage-inducible C-type lectin. J. Clin. Investig. 2020, 130, 4811–4830. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Yang, S.; Shao, J.; Li, Y.-P. Signaling and transcriptional regulation in osteoblast commitment and differentiation. Front. Biosci. 2007, 12, 3068–3092. [Google Scholar] [CrossRef] [Green Version]
- Sawakami, K.; Robling, A.G.; Ai, M.; Pitner, N.D.; Liu, D.; Warden, S.J.; Li, J.; Maye, P.; Rowe, D.W.; Duncan, R.L.; et al. The Wnt co-receptor LRP5 is essential for skeletal mechanotransduction but not for the anabolic bone response to parathyroid hormone treatment. J. Biol. Chem. 2006, 281, 23698–23711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruotti, N.; Corrado, A.; Cantatore, F.P. Osteoblast role in osteoarthritis pathogenesis. J. Cell. Physiol. 2017, 232, 2957–2963. [Google Scholar] [CrossRef] [Green Version]
- Zoch, M.L.; Clemens, T.L.; Riddle, R.C. New insights into the biology of osteocalcin. Bone 2016, 82, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-β1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Kim, D.J.; Shen, J.; Zou, Z.; O’Keefe, R.J. Runx2 plays a central role in osteoarthritis development. J. Orthop. Translat. 2020, 23, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Wang, Y.; Gao, J.; Yan, Z.; Li, Z.; Zou, X.; Li, Y.; Wang, J.; Guo, Y. miR-29b-3p regulated osteoblast differentiation via regulating IGF-1 secretion of mechanically stimulated osteocytes. Cell. Mol. Biol. Lett. 2019, 24, 11. [Google Scholar] [CrossRef] [Green Version]
- D’Emic, M.D.; Benson, R.B.J. Measurement, variation, and scaling of osteocyte lacunae: A case study in birds. Bone 2013, 57, 300–310. [Google Scholar] [CrossRef]
- Teti, A.; Zallone, A. Do osteocytes contribute to bone mineral homeostasis? Osteocytic osteolysis revisited. Bone 2009, 44, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Grunmeier, O., 3rd; D’Emic, M.D. Scaling of statically derived osteocyte lacunae in extant birds: Implications for palaeophysiological reconstruction. Biol. Lett. 2019, 15, 20180837. [Google Scholar] [CrossRef] [Green Version]
- Ferretti, M.; Palumbo, C. Static osteogenesis versus dynamic osteogenesis: A comparison between two different types of bone formation. Appl. Sci. 2021, 11, 2025. [Google Scholar] [CrossRef]
- Dallas, S.L.; Bonewald, L.F. Dynamics of the transition from osteoblast to osteocyte. Ann. N. Y. Acad. Sci. 2010, 1192, 437–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Zhang, J.; Wu, J.; Guan, Y.; Weng, Y.; Shang, P. Oscillatory fluid flow elicits changes in morphology, cytoskeleton and integrin-associated molecules in MLO-Y4 cells, but not in MC3T3-E1 cells. Biol. Res. 2012, 45, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, C.C.; Liu, Y.; Thant, L.M.; Pang, J.; Palmer, G.; Alikhani, M. Foxo1, a novel regulator of osteoblast differentiation and skeletogenesis. J. Biol. Chem. 2010, 285, 31055–31065. [Google Scholar] [CrossRef] [Green Version]
- Perez-Campo, F.M.; Santurtun, A.; Garcia-Ibarbia, C.; Pascual, M.A.; Valero, C.; Garces, C.; Sanudo, C.; Zarrabeitia, M.T.; Riancho, J.A. Osterix and RUNX2 are transcriptional regulators of sclerostin in human bone. Calcif. Tissue Int. 2016, 99, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Komori, T. Runx2, an inducer of osteoblast and chondrocyte differentiation. Histochem. Cell Biol. 2018, 149, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Weiner, S.; Raguin, E.; Shahar, R. High resolution 3D structures of mineralized tissues in health and disease. Nat. Rev. Endocrinol. 2021, 17, 307–316. [Google Scholar] [CrossRef]
- Arnold, A.; Dennison, E.; Kovacs, C.S.; Mannstadt, M.; Rizzoli, R.; Brandi, M.L.; Clarke, B.; Thakker, R.V. Hormonal regulation of biomineralization. Nat. Rev. Endocrinol. 2021, 17, 261–275. [Google Scholar] [CrossRef]
- Atkins, G.J.; Rowe, P.S.; Lim, H.P.; Welldon, K.J.; Ormsby, R.; Wijenayaka, A.R.; Zelenchuk, L.; Evdokiou, A.; Findlay, D.M. Sclerostin is a locally acting regulator of late-osteoblast/preosteocyte differentiation and regulates mineralization through a MEPE-ASARM-dependent mechanism. J. Bone Miner. Res. 2011, 26, 1425–1436. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.L.; Khor, K.A.; Sui, J.J.; Zhang, J.H.; Chen, W.N. Protein expression profiles in osteoblasts in response to differentially shaped hydroxyapatite nanoparticles. Biomaterials 2009, 30, 5385–5391. [Google Scholar] [CrossRef]
- Karperien, M.; Roelen, B.A.J.; Poelmann, R.E.; Gittenberger-de Groot, A.C.; Hierck, B.P.; DeRuiter, M.C.; Meijer, D.; Gibbs, S. Tissue formation during embryogenesis. In Tissue Engineering, 2nd ed.; Blitterswijk, C.A.V., De Boer, J., Eds.; Academic Press: Oxford, UK, 2015; pp. 67–109. [Google Scholar]
- Koelling, S.; Kruegel, J.; Irmer, M.; Path, J.R.; Sadowski, B.; Miro, X.; Miosge, N. Migratory chondrogenic progenitor cells from repair tissue during the later stages of human osteoarthritis. Cell Stem Cell 2009, 4, 324–335. [Google Scholar] [CrossRef] [Green Version]
- Grogan, S.P.; Miyaki, S.; Asahara, H.; D’Lima, D.D.; Lotz, M.K. Mesenchymal progenitor cell markers in human articular cartilage: Normal distribution and changes in osteoarthritis. Arthritis Res. Ther. 2009, 11, R85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannoudis, P.V.; Goff, T.; Roshdy, T.; Jones, E.; McGonagle, D. Does mobilisation and transmigration of mesenchymal stem cells occur after trauma? Injury 2010, 41, 1099–1102. [Google Scholar] [CrossRef]
- Jones, E.A.; Crawford, A.; English, A.; Henshaw, K.; Mundy, J.; Corscadden, D.; Chapman, T.; Emery, P.; Hatton, P.; McGonagle, D. Synovial fluid mesenchymal stem cells in health and early osteoarthritis: Detection and functional evaluation at the single-cell level. Arthritis Rheumatol. 2008, 58, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.; Tang, S.Y.; Nguyen, D.; Alliston, T. Load regulates bone formation and sclerostin expression through a TGFβ-dependent mechanism. PLoS ONE 2013, 8, e53813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherifi, C.; Monteagudo, S.; Lories, R.J. Promising targets for therapy of osteoarthritis: A review on the Wnt and TGF-β signalling pathways. Ther. Adv. Musculoskelet. Dis. 2021, 13. [Google Scholar] [CrossRef]
- Xian, L.; Wu, X.; Pang, L.; Lou, M.; Rosen, C.J.; Qiu, T.; Crane, J.; Frassica, F.; Zhang, L.; Rodriguez, J.P.; et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat. Med. 2012, 18, 1095–1101. [Google Scholar] [CrossRef] [Green Version]
- Lories, R.J.; Luyten, F.P. The bone-cartilage unit in osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 43–49. [Google Scholar] [CrossRef]
- Dozza, B.; Lesci, I.G.; Duchi, S.; Della Bella, E.; Martini, L.; Salamanna, F.; Falconi, M.; Cinotti, S.; Fini, M.; Lucarelli, E.; et al. When size matters: Differences in demineralized bone matrix particles affect collagen structure, mesenchymal stem cell behavior, and osteogenic potential. J. Biomed. Mater. Res. A 2017, 105, 1019–1033. [Google Scholar] [CrossRef]
- Roohani-Esfahani, S.-I.; Nouri-Khorasani, S.; Lu, Z.; Appleyard, R.; Zreiqat, H. The influence hydroxyapatite nanoparticle shape and size on the properties of biphasic calcium phosphate scaffolds coated with hydroxyapatite–PCL composites. Biomaterials 2010, 31, 5498–5509. [Google Scholar] [CrossRef] [PubMed]
- Zandi, M.; Mirzadeh, H.; Mayer, C.; Urch, H.; Eslaminejad, M.B.; Bagheri, F.; Mivehchi, H. Biocompatibility evaluation of nano-rod hydroxyapatite/gelatin coated with nano-HAp as a novel scaffold using mesenchymal stem cells. J. Biomed. Mater. Res. A 2010, 92A, 1244–1255. [Google Scholar] [CrossRef] [PubMed]
- Bodhak, S.; Bose, S.; Bandyopadhyay, A. Role of surface charge and wettability on early stage mineralization and bone cell–materials interactions of polarized hydroxyapatite. Acta Biomater. 2009, 5, 2178–2188. [Google Scholar] [CrossRef] [PubMed]
- Bertazzo, S.; Zambuzzi, W.F.; Campos, D.D.P.; Ogeda, T.L.; Ferreira, C.V.; Bertran, C.A. Hydroxyapatite surface solubility and effect on cell adhesion. Colloids Surf. B Biointerfaces 2010, 78, 177–184. [Google Scholar] [CrossRef]
- Lin, L.; Chow, K.L.; Leng, Y. Study of hydroxyapatite osteoinductivity with an osteogenic differentiation of mesenchymal stem cells. J. Biomed. Mater. Res. A 2009, 89A, 326–335. [Google Scholar] [CrossRef]
- Burr, D.B.; Radin, E.L. Microfractures and microcracks in subchondral bone: Are they relevant to osteoarthrosis? Rheum. Dis. Clin. N. Am. 2003, 29, 675–685. [Google Scholar] [CrossRef]
- Ruscitto, A.; Morel, M.M.; Shawber, C.J.; Reeve, G.; Lecholop, M.K.; Bonthius, D.; Yao, H.; Embree, M.C. Evidence of vasculature and chondrocyte to osteoblast transdifferentiation in craniofacial synovial joints: Implications for osteoarthritis diagnosis and therapy. FASEB J. 2020, 34, 4445–4461. [Google Scholar] [CrossRef] [Green Version]
- Aghajanian, P.; Mohan, S. The art of building bone: Emerging role of chondrocyte-to-osteoblast transdifferentiation in endochondral ossification. Bone Res. 2018, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedl, M.; Witzmann, C.; Koch, M.; Lang, S.; Kerschbaum, M.; Baumann, F.; Krutsch, W.; Docheva, D.; Alt, V.; Pfeifer, C. Attenuation of hypertrophy in human MSCs via treatment with a retinoic acid receptor inverse agonist. Int. J. Mol. Sci. 2020, 21, 1444. [Google Scholar] [CrossRef] [Green Version]
- Aghajanian, P.; Xing, W.; Cheng, S.; Mohan, S. Epiphyseal bone formation occurs via thyroid hormone regulation of chondrocyte to osteoblast transdifferentiation. Sci. Rep. 2017, 7, 10432. [Google Scholar] [CrossRef]
- Dong, Y.-F.; Soung, D.Y.; Schwarz, E.M.; O’Keefe, R.J.; Drissi, H. Wnt induction of chondrocyte hypertrophy through the Runx2 transcription factor. J. Cell Physiol. 2006, 208, 77–86. [Google Scholar] [CrossRef]
- Chan, B.Y.; Fuller, E.S.; Russell, A.K.; Smith, S.M.; Smith, M.M.; Jackson, M.T.; Cake, M.A.; Read, R.A.; Bateman, J.F.; Sambrook, P.N.; et al. Increased chondrocyte sclerostin may protect against cartilage degradation in osteoarthritis. Osteoarthr. Cartil. 2011, 19, 874–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Ma, L.; Wu, L.; Jin, Q. Wnt-β-Catenin signaling pathway inhibition by sclerostin may protect against degradation in healthy but not osteoarthritic cartilage. Mol. Med. Rep. 2017, 15, 2423–2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, Y.; Kumagai, K.; Imai, S.; Miyatake, K.; Saito, T. Sclerostin is upregulated in the early stage of chondrogenic differentiation, but not required in endochondral ossification in vitro. PLoS ONE 2018, 13, e0201839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.; Piemontese, M.; Onal, M.; Campbell, J.; Goellner, J.J.; Dusevich, V.; Bonewald, L.; Manolagas, S.C.; O’Brien, C.A. Osteocytes, not osteoblasts or lining cells, are the main source of the RANKL required for osteoclast formation in remodeling bone. PLoS ONE 2015, 10, e0138189. [Google Scholar] [CrossRef]
- Wijenayaka, A.R.; Kogawa, M.; Lim, H.P.; Bonewald, L.F.; Findlay, D.M.; Atkins, G.J. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS ONE 2011, 6, e25900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, A.; David, V.; Laurence, J.S.; Schwarz, P.M.; Lafer, E.M.; Hedge, A.M.; Rowe, P.S. Degradation of MEPE, DMP1, and release of SIBLING ASARM-peptides (minhibins): ASARM-peptide(s) are directly responsible for defective mineralization in HYP. Endocrinology 2008, 149, 1757–1772. [Google Scholar] [CrossRef] [Green Version]
- Pathak, J.L.; Bravenboer, N.; Luyten, F.P.; Verschueren, P.; Lems, W.F.; Klein-Nulend, J.; Bakker, A.D. Mechanical loading reduces inflammation-induced human osteocyte-to-osteoclast communication. Calcif. Tissue Int. 2015, 97, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, R.N.; Bakker, A.D.; Everts, V.; Klein-Nulend, J. Inhibition of osteoclastogenesis by mechanically loaded osteocytes: Involvement of MEPE. Calcif. Tissue Int. 2010, 87, 461–468. [Google Scholar] [CrossRef] [Green Version]
- You, L.; Temiyasathit, S.; Lee, P.; Kim, C.H.; Tummala, P.; Yao, W.; Kingery, W.; Malone, A.M.; Kwon, R.Y.; Jacobs, C.R. Osteocytes as mechanosensors in the inhibition of bone resorption due to mechanical loading. Bone 2008, 42, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, O.D.; Herman, B.C.; Laudier, D.M.; Majeska, R.J.; Sun, H.B.; Schaffler, M.B. Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone 2012, 50, 1115–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, L.I.; Gortazar, A.R.; Davis, H.M.; Condon, K.W.; Gabilondo, H.; Maycas, M.; Allen, M.R.; Bellido, T. Inhibition of osteocyte apoptosis prevents the increase in osteocytic receptor activator of nuclear factor κB ligand (RANKL) but does not stop bone resorption or the loss of bone induced by unloading. J. Biol. Chem. 2015, 290, 18934–18942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crockett, J.C.; Schutze, N.; Tosh, D.; Jatzke, S.; Duthie, A.; Jakob, F.; Rogers, M.J. The matricellular protein CYR61 inhibits osteoclastogenesis by a mechanism independent of αvβ3 and αvβ5. Endocrinology 2007, 148, 5761–5768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botter, S.M.; van Osch, G.J.V.M.; Clockaerts, S.; Waarsing, J.H.; Weinans, H.; van Leeuwen, J.P.T.M. Osteoarthritis induction leads to early and temporal subchondral plate porosity in the tibial plateau of mice: An in vivo microfocal computed tomography study. Arthritis Rheumatol. 2011, 63, 2690–2699. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, T.P.; Bjorklund, M. Cellular allometry of mitochondrial functionality establishes the optimal cell size. Dev. Cell 2016, 39, 370–382. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, T.P.; Pessa, H.K.; Caldez, M.J.; Fuhrer, T.; Diril, M.K.; Sauer, U.; Kaldis, P.; Bjorklund, M. Identification of transcriptional and metabolic programs related to mammalian cell size. Curr. Biol. 2014, 24, 598–608. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, T.P.; Bjorklund, M. Mitochondrial function and cell size: An allometric relationship. Trends Cell Biol. 2017, 27, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, T.P.; Caldez, M.J.; Kaldis, P.; Björklund, M. Cell size control—A mechanism for maintaining fitness and function. BioEssays 2017, 39, 1700058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsyth, C.B.; Cole, A.; Murphy, G.; Bienias, J.L.; Im, H.-J.; Loeser, R.F., Jr. Increased matrix metalloproteinase-13 production with aging by human articular chondrocytes in response to catabolic stimuli. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 1118–1124. [Google Scholar] [CrossRef] [Green Version]
- Loeser, R.F. Aging and osteoarthritis: The role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr. Cartil. 2009, 17, 971–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appel, H.; Ruiz-Heiland, G.; Listing, J.; Zwerina, J.; Herrmann, M.; Mueller, R.; Haibel, H.; Baraliakos, X.; Hempfing, A.; Rudwaleit, M.; et al. Altered skeletal expression of sclerostin and its link to radiographic progression in ankylosing spondylitis. Arthritis Rheumatol. 2009, 60, 3257–3262. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The representative uCT images and data show the comparisons of hip osteoarthritis (OA) with a fracture in two- (a) and three-dimensional architecture (b). Osteoarthritic bone was characterized by high bone mass disproportionately concentrated in the load-bearing regions in comparison with hip fracture bone. This is believed to be regulated by periostin and sclerostin (c). Enhanced periostin and decreased sclerostin expressions upon mechanical loading can activate Wnt/β-catenin signaling through LRP5/6. β-catenin then promotes gene transcription in the targeted cell, such as osteoblasts, which results in bone formation.
Figure 1.
The representative uCT images and data show the comparisons of hip osteoarthritis (OA) with a fracture in two- (a) and three-dimensional architecture (b). Osteoarthritic bone was characterized by high bone mass disproportionately concentrated in the load-bearing regions in comparison with hip fracture bone. This is believed to be regulated by periostin and sclerostin (c). Enhanced periostin and decreased sclerostin expressions upon mechanical loading can activate Wnt/β-catenin signaling through LRP5/6. β-catenin then promotes gene transcription in the targeted cell, such as osteoblasts, which results in bone formation.

Figure 2.
The representative micro-CT images show the microstructure of trabecular bone in the medial plateau of healthy (a) and osteoarthritic (OA) knees (b). The color-coded images of trabecular bone show the distribution of bone mineral density (BMD). The red-labeled trabecular bone represented high BMD. Bone mineralization is regulated by sclerostin, dentin matrix protein 1 (DMP-1), matrix extracellular phosphoglycoprotein (MEPE), and phosphate-regulating neutral endopeptidase (PHEX) (c). Osteoblast-derived DMP-1 is critical for the mineralization of collagen fibers, while sclerostin can regulate MEPE and PHEX to control further mineralization of hydroxyapatite (HA) crystals. Mechanical loading regulates DMP-1 expressions by osteoblasts and osteocytes differently. Abnormally shaped HA crystals can induce osteogenic differentiation of mesenchymal stem cells (MSCs) or trigger osteoblast dysfunction.
Figure 2.
The representative micro-CT images show the microstructure of trabecular bone in the medial plateau of healthy (a) and osteoarthritic (OA) knees (b). The color-coded images of trabecular bone show the distribution of bone mineral density (BMD). The red-labeled trabecular bone represented high BMD. Bone mineralization is regulated by sclerostin, dentin matrix protein 1 (DMP-1), matrix extracellular phosphoglycoprotein (MEPE), and phosphate-regulating neutral endopeptidase (PHEX) (c). Osteoblast-derived DMP-1 is critical for the mineralization of collagen fibers, while sclerostin can regulate MEPE and PHEX to control further mineralization of hydroxyapatite (HA) crystals. Mechanical loading regulates DMP-1 expressions by osteoblasts and osteocytes differently. Abnormally shaped HA crystals can induce osteogenic differentiation of mesenchymal stem cells (MSCs) or trigger osteoblast dysfunction.
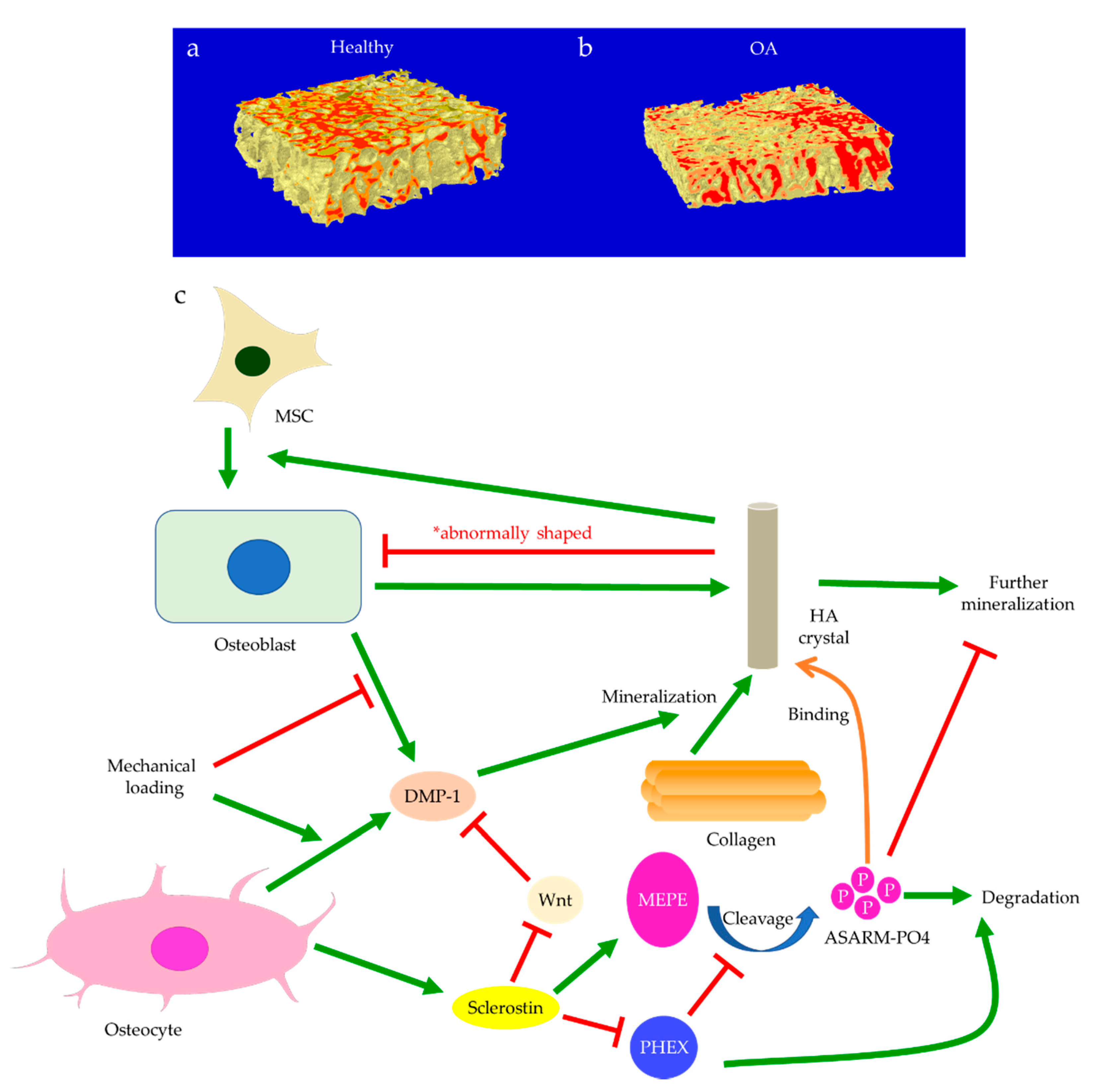
Figure 3.
The representative uCT (a–f) and histological images (g–l) show the comparisons between hip osteoarthritis (OA) (a,c,e,g,i,k) and fracture (b,d,f,h,j,l). As shown by uCT images, osteoid formation (blue arrow) was found in apposition to pre-existing trabeculae for both hip OA (c) and fracture (d). The newly formed bone was associated spatially with bone resorption pits (red arrow) in hip fracture, yet it was not the case for hip OA. It was revealed by histology that bone cell clusters (black arrow) were present close to osteoid formation (block arrow) either in trabecular bone (g) or marrow space (i). In contrast, bone cell clusters were not found in hip fractures (h,j). The two (k,l) and three (e,f) dimensional morphology of osteocytes lacunae both revealed that the number of lacunae from hip OA bone was higher than that of hip fracture. In addition, OA osteocytes lacunae were well aligned and more plate-like as compared with hip fracture ones. (g–j: Hematoxylin and eosin staining with a magnification of 20×).
Figure 3.
The representative uCT (a–f) and histological images (g–l) show the comparisons between hip osteoarthritis (OA) (a,c,e,g,i,k) and fracture (b,d,f,h,j,l). As shown by uCT images, osteoid formation (blue arrow) was found in apposition to pre-existing trabeculae for both hip OA (c) and fracture (d). The newly formed bone was associated spatially with bone resorption pits (red arrow) in hip fracture, yet it was not the case for hip OA. It was revealed by histology that bone cell clusters (black arrow) were present close to osteoid formation (block arrow) either in trabecular bone (g) or marrow space (i). In contrast, bone cell clusters were not found in hip fractures (h,j). The two (k,l) and three (e,f) dimensional morphology of osteocytes lacunae both revealed that the number of lacunae from hip OA bone was higher than that of hip fracture. In addition, OA osteocytes lacunae were well aligned and more plate-like as compared with hip fracture ones. (g–j: Hematoxylin and eosin staining with a magnification of 20×).

Figure 4.
Insulin-like growth factor 1 (Igf1) signaling in the crosstalk between osteocytes and chondrocytes. Mechanical loading regulates Igf1 expressions by osteocytes and chondrocytes differently. Inhibition of miR-29b-3p production by osteocytes can increase Igf1 expression. Meanwhile, elevated reactive oxygen species (ROS) production by chondrocytes can suppress Igf1 expression. Igf1 also regulates sclerostin expression, chondrocyte hypertrophy, and osteogenic differentiation of MSCs through the PI3K/AKT/mTOR pathway.
Figure 4.
Insulin-like growth factor 1 (Igf1) signaling in the crosstalk between osteocytes and chondrocytes. Mechanical loading regulates Igf1 expressions by osteocytes and chondrocytes differently. Inhibition of miR-29b-3p production by osteocytes can increase Igf1 expression. Meanwhile, elevated reactive oxygen species (ROS) production by chondrocytes can suppress Igf1 expression. Igf1 also regulates sclerostin expression, chondrocyte hypertrophy, and osteogenic differentiation of MSCs through the PI3K/AKT/mTOR pathway.

Figure 5.
Regulation of osteocyte morphology (a) and alignment (b). Osteocyte-derived DMP-1 and E11/gp38 can alter the lacuno-canalicular networks (LCN) and hence mechano-transduction. In OA, matrix metalloproteinase (MMP)-13 production by osteocytes is decreased. This may contribute to increased bone formation in subchondral bone and elevated MMP-13 expression by chondrocytes (a). Mechanical loading can determine the alignment of collagen fibers, possibly by degrading the unstrained fibers. Collagen alignment, in turn, affects the alignment of osteocyte lacunae through cytoskeleton and integrins. Sclerostin expression is more prominent in unaligned osteocytes. Micro-damage may induce dysfunction, apoptosis, or necrosis of the unaligned osteocytes (b).
Figure 5.
Regulation of osteocyte morphology (a) and alignment (b). Osteocyte-derived DMP-1 and E11/gp38 can alter the lacuno-canalicular networks (LCN) and hence mechano-transduction. In OA, matrix metalloproteinase (MMP)-13 production by osteocytes is decreased. This may contribute to increased bone formation in subchondral bone and elevated MMP-13 expression by chondrocytes (a). Mechanical loading can determine the alignment of collagen fibers, possibly by degrading the unstrained fibers. Collagen alignment, in turn, affects the alignment of osteocyte lacunae through cytoskeleton and integrins. Sclerostin expression is more prominent in unaligned osteocytes. Micro-damage may induce dysfunction, apoptosis, or necrosis of the unaligned osteocytes (b).

Figure 6.
Osteoblast differentiation and osteoblast-osteocyte transformation (a) and the regulation by TGF-β signaling (b). Mechanical loading can promote osteogenic differentiation of MSCs. This is regulated by sclerostin and Wnt signaling and likely involves Runx-2 expression. Rearrangement of the cytoskeleton and DMP-1 are both believed to regulate osteoblast to osteocyte transformation (a). Osteoblast differentiation can also be regulated by TGF-β signaling and phosphorylation of Smad2/3. Mechanical loading can suppress Smad2/3 expression or their phosphorylation, which can lead to sclerostin deficiency and impact osteoblast differentiation (b). Chondrocyte hypertrophy is believed to be regulated by factors including Runx-2, BMP-2, TGF-β, and sclerostin, which is possibly enhanced by mechanical loading. Hypertrophic chondrocytes are speculated to possess the potential to trans-differentiate into osteoblasts.
Figure 6.
Osteoblast differentiation and osteoblast-osteocyte transformation (a) and the regulation by TGF-β signaling (b). Mechanical loading can promote osteogenic differentiation of MSCs. This is regulated by sclerostin and Wnt signaling and likely involves Runx-2 expression. Rearrangement of the cytoskeleton and DMP-1 are both believed to regulate osteoblast to osteocyte transformation (a). Osteoblast differentiation can also be regulated by TGF-β signaling and phosphorylation of Smad2/3. Mechanical loading can suppress Smad2/3 expression or their phosphorylation, which can lead to sclerostin deficiency and impact osteoblast differentiation (b). Chondrocyte hypertrophy is believed to be regulated by factors including Runx-2, BMP-2, TGF-β, and sclerostin, which is possibly enhanced by mechanical loading. Hypertrophic chondrocytes are speculated to possess the potential to trans-differentiate into osteoblasts.

Figure 7.
The representative SEM images show the differentially shaped HA nanoparticles of the trabecular bone from donors with (a) knee OA or (b) osteoporosis at different magnifications. In osteoporosis patients, HA crystals present with a bean-like shape. In contrast, HA nanoparticles from OA patients presented with a cigarette-like shape.
Figure 7.
The representative SEM images show the differentially shaped HA nanoparticles of the trabecular bone from donors with (a) knee OA or (b) osteoporosis at different magnifications. In osteoporosis patients, HA crystals present with a bean-like shape. In contrast, HA nanoparticles from OA patients presented with a cigarette-like shape.

Figure 8.
Regulation of osteoclasts. Osteoclastogenesis is regulated through the OPG/RANKL/RANK system and sometimes by damage-associated molecular patterns (DAMPs). The RANKL/OPG ratio can be regulated by PHEX, MEPE, and sclerostin. Sclerostin deficiency may suppress MEPE but enhance PHEX expressions and increase the RANKL/OPG ratio. Osteoclastic activities are associated with mechanical loading and osteocyte death, with osteoclasts more likely to be recruited to the site of micro-damage.
Figure 8.
Regulation of osteoclasts. Osteoclastogenesis is regulated through the OPG/RANKL/RANK system and sometimes by damage-associated molecular patterns (DAMPs). The RANKL/OPG ratio can be regulated by PHEX, MEPE, and sclerostin. Sclerostin deficiency may suppress MEPE but enhance PHEX expressions and increase the RANKL/OPG ratio. Osteoclastic activities are associated with mechanical loading and osteocyte death, with osteoclasts more likely to be recruited to the site of micro-damage.
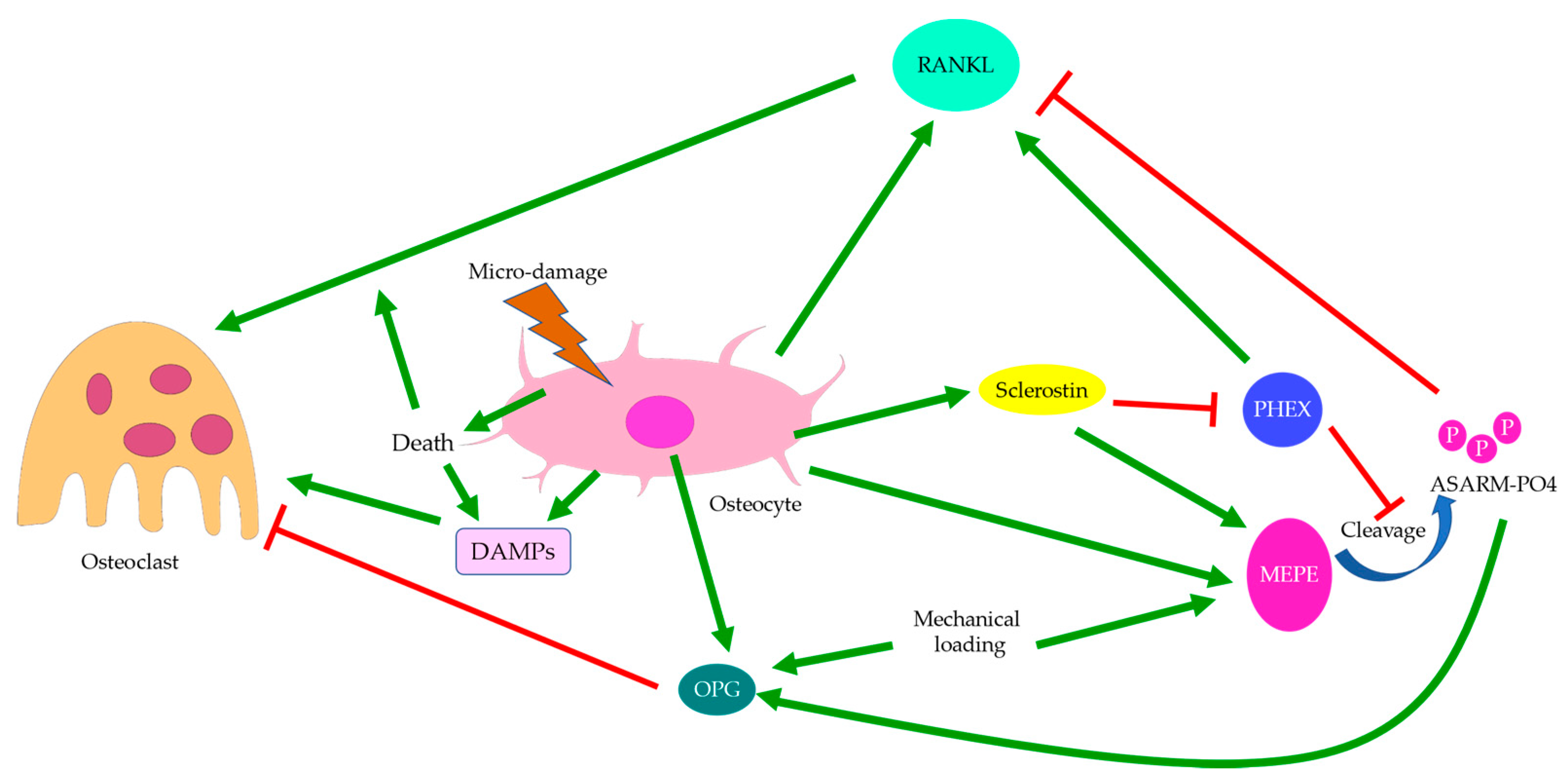
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, L.; Wen, C. Osteocyte Dysfunction in Joint Homeostasis and Osteoarthritis. Int. J. Mol. Sci. 2021, 22, 6522. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126522
AMA Style
Zhang L, Wen C. Osteocyte Dysfunction in Joint Homeostasis and Osteoarthritis. International Journal of Molecular Sciences. 2021; 22(12):6522. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126522
Chicago/Turabian StyleZhang, Lanlan, and Chunyi Wen. 2021. "Osteocyte Dysfunction in Joint Homeostasis and Osteoarthritis" International Journal of Molecular Sciences 22, no. 12: 6522. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126522
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.