
Epitranscriptomics of Ischemic Heart Disease—The IHD-EPITRAN Study Design and Objectives

 , , , add
Show full author list
, , , add
Show full author list
Abstract
:1. Introduction
1.1. Ischemic Heart Disease
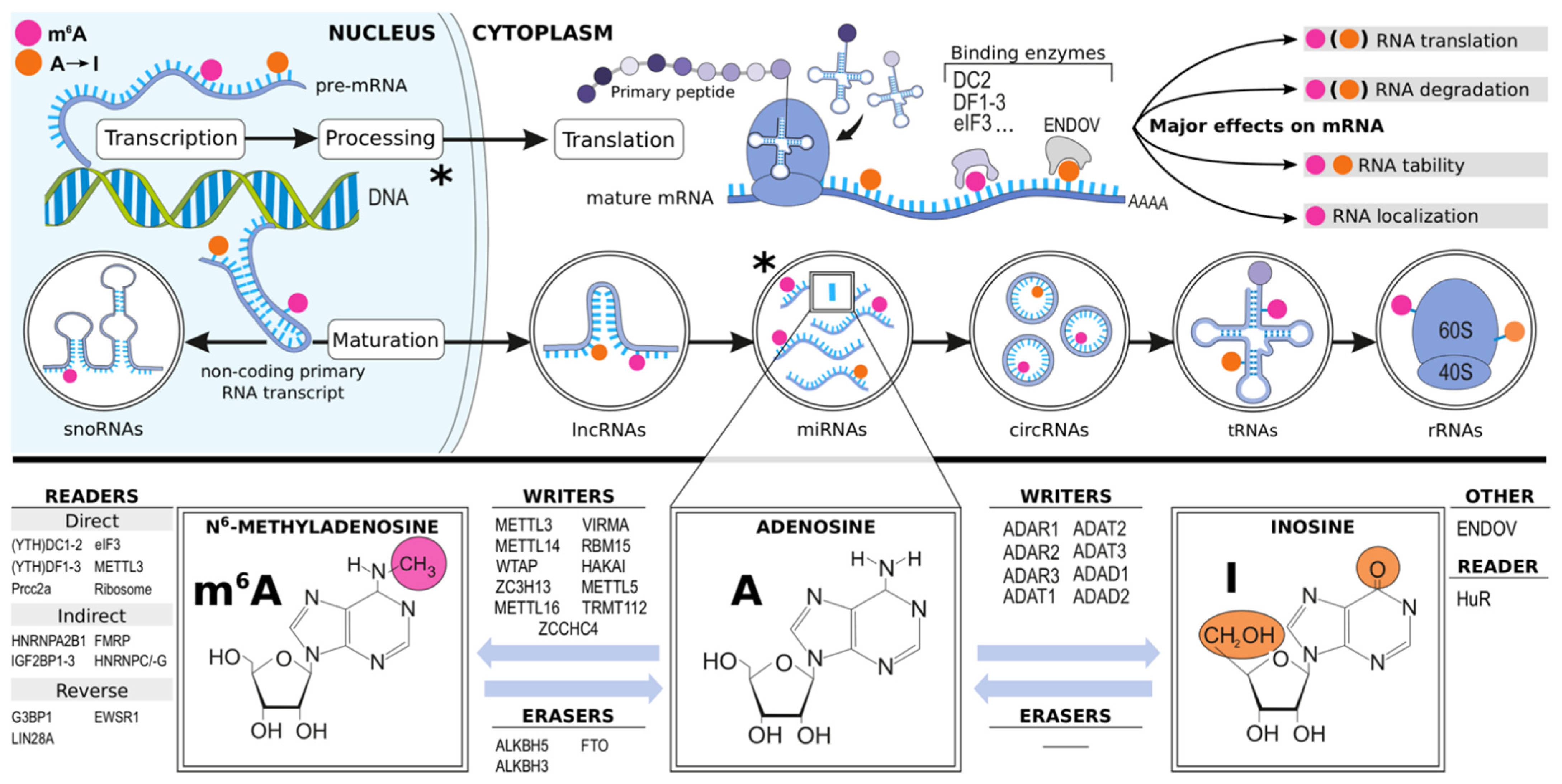
1.2. Epitranscriptomics
1.3. Rationale and Goals
2. Materials and Methods
2.1. Overview
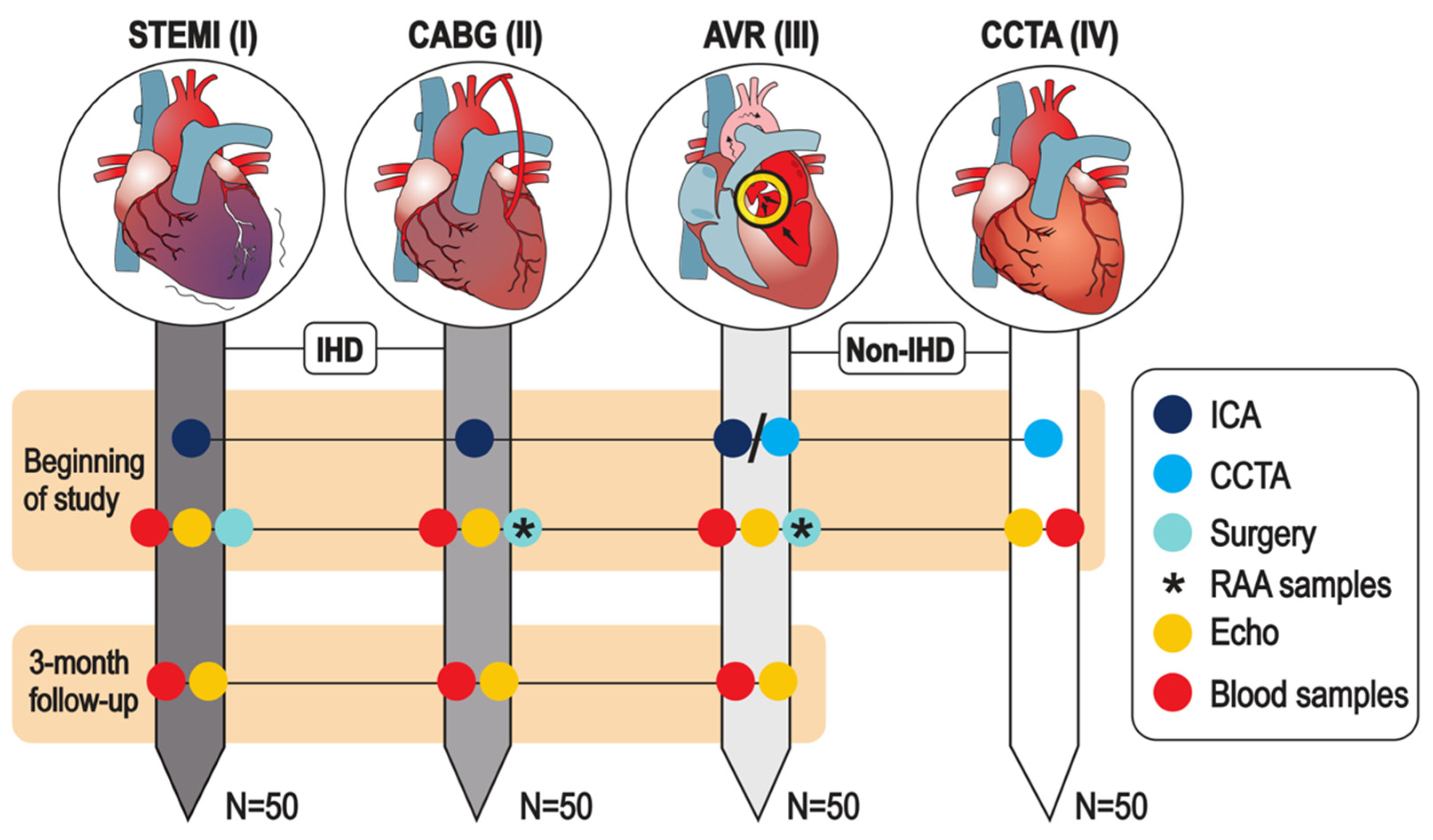
2.2. Study Groups, Recruitment and Exclusion Criteria
2.3. Study Measures
2.3.1. Baseline Morbidity
2.3.2. NYHA, CCS and SF-36
2.3.3. Coronary Angiography, Computed Tomography Angiogram, SYNTAX Score
2.3.4. Echocardiography, 3-Month Control Visit, and Follow-Up Period
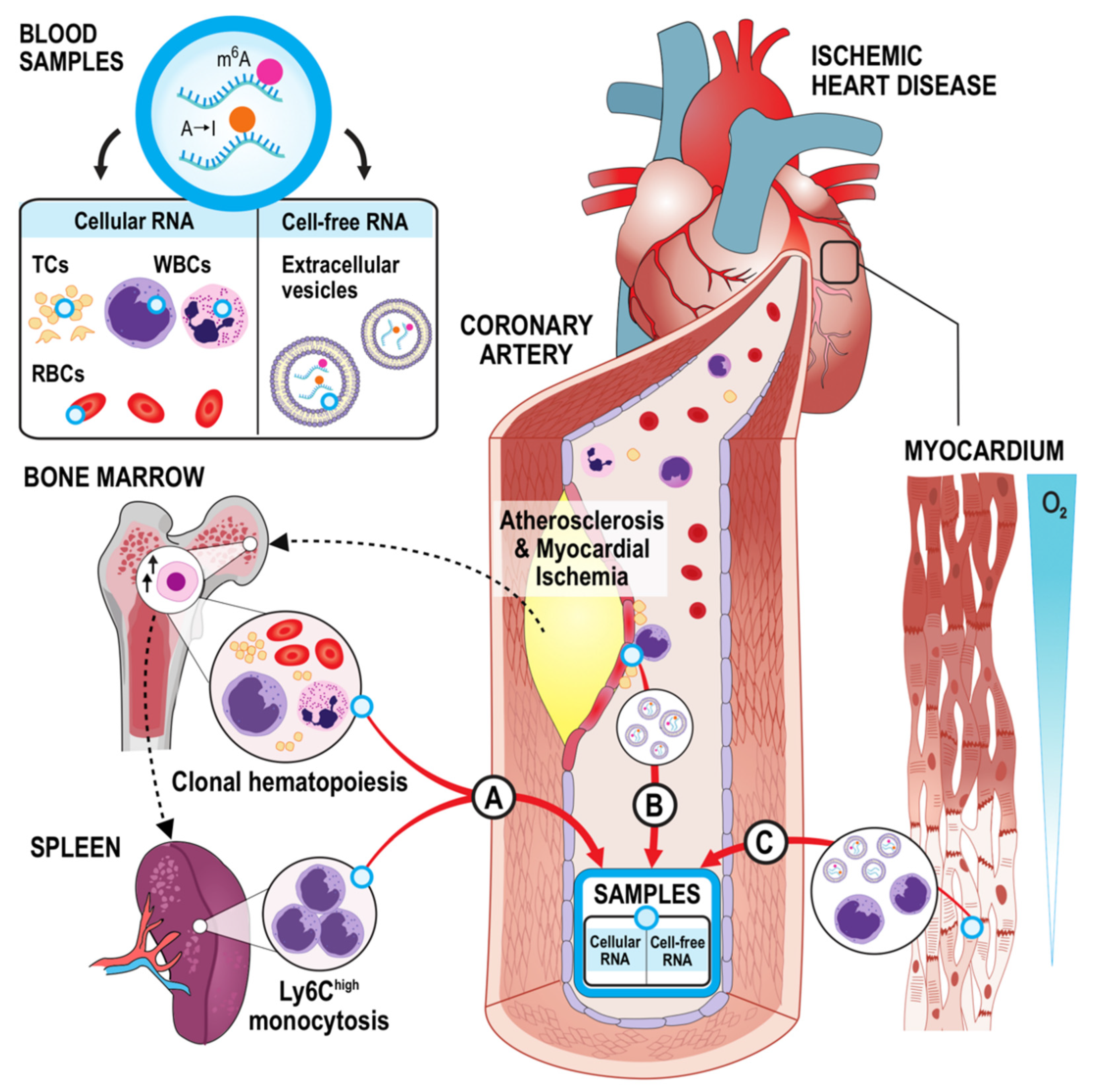
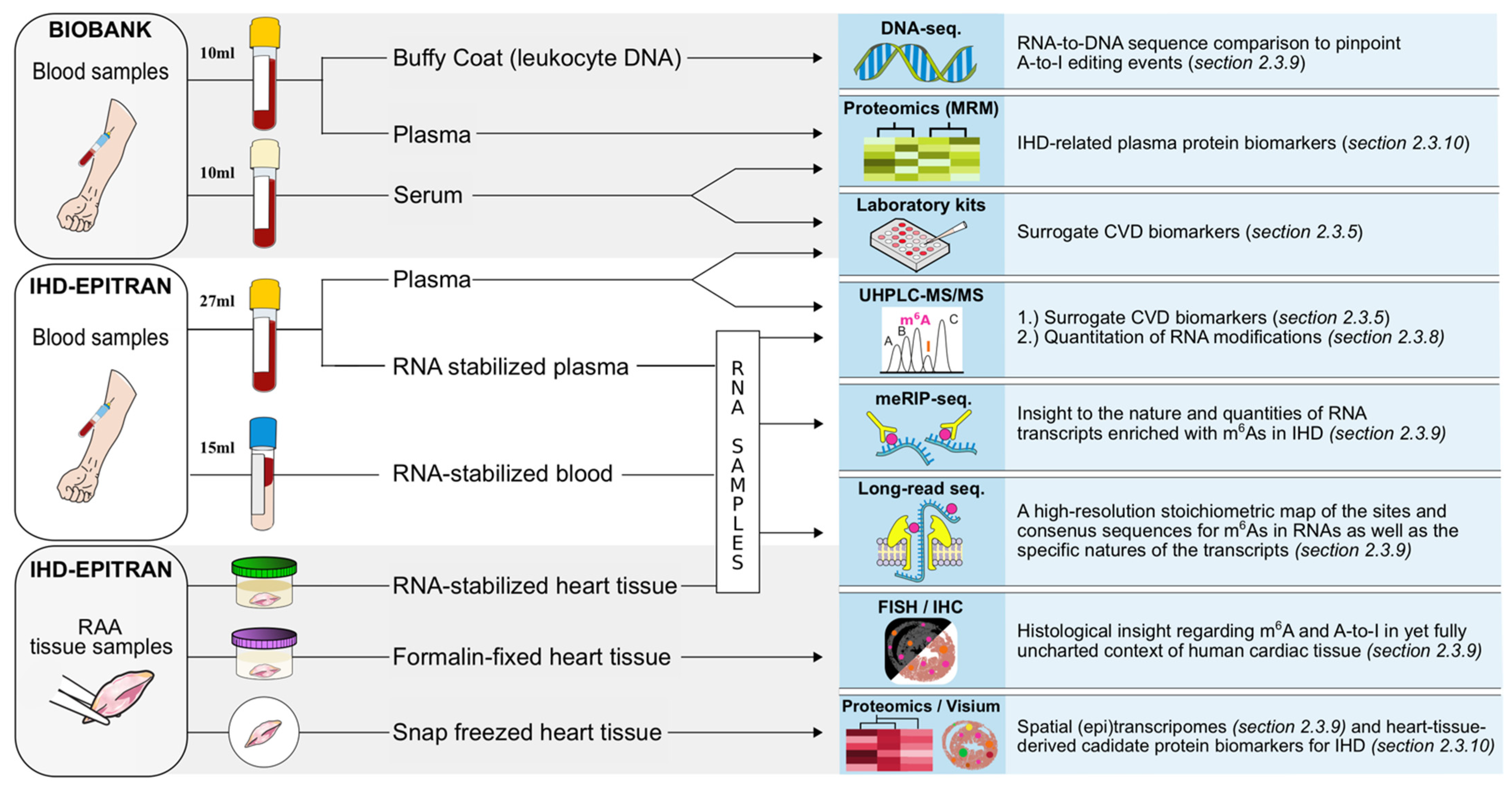
2.3.5. Study Blood Samples
2.3.6. Right Atrial Appendage Tissue Samples
2.3.7. RNA-Stabilized Blood and RAA Tissue RNA Fractionation
2.3.8. Quantitative RNA Modification Analysis with UHPLC-LC-MS/MS
2.3.9. Qualitative Analyses of RNA Modifications
2.3.10. Proteomics
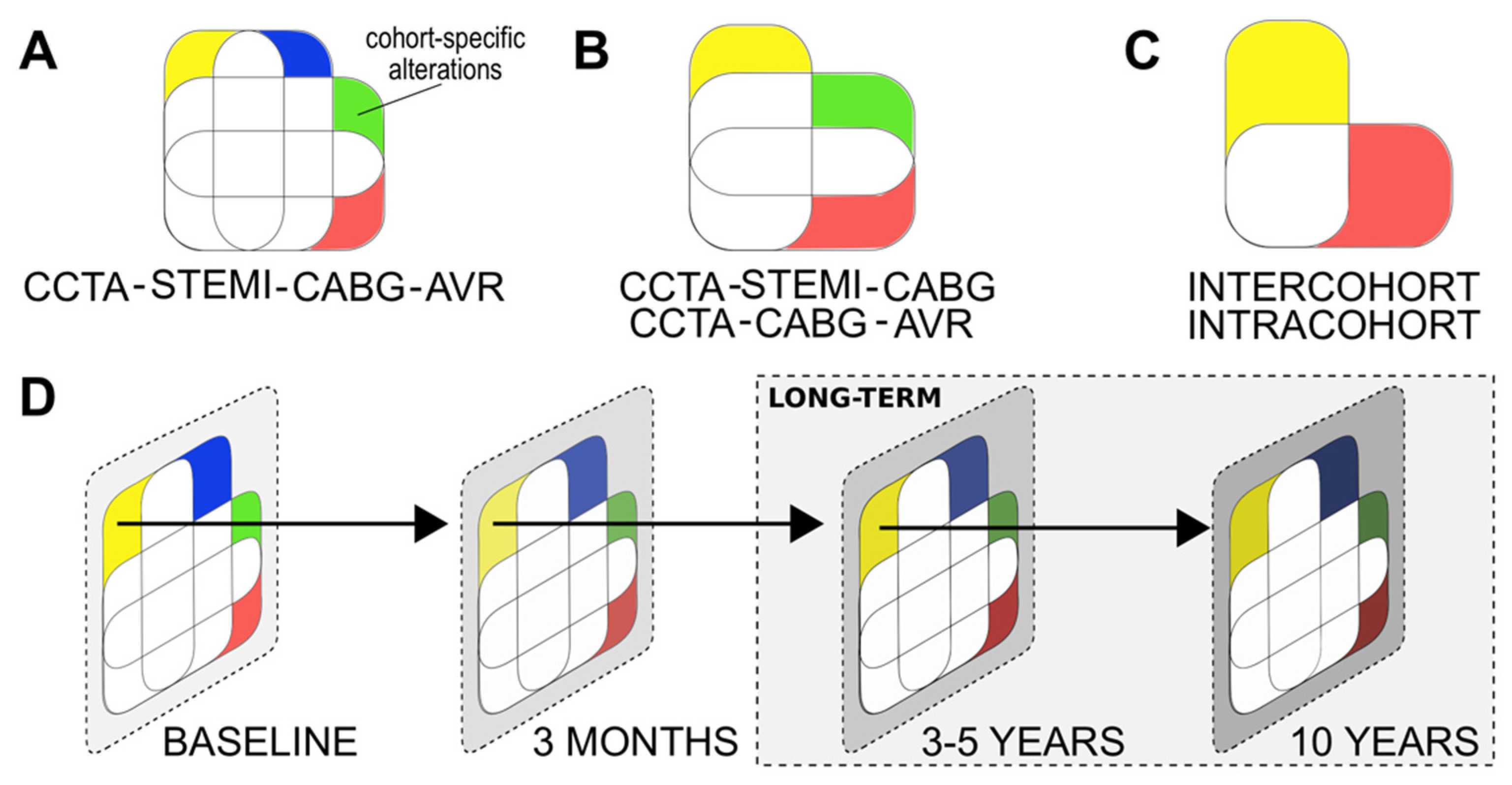
2.4. Power Calculations, Cohort Comparisons, Outcomes, and Data Management
2.5. Collaborators, the IHD-EPITRAN Consortium
2.6. Long-Term Follow-Up, IHD-EPITRAN Extensions
3. Expected Results
4. Discussion
5. Conclusions
6. Contact Us
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Prim. 2019, 5, 56. [Google Scholar] [CrossRef]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for Cardiomyocyte Renewal in Humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrigo, M.; Jessup, M.; Mullens, W.; Reza, N.; Shah, A.M.; Sliwa, K.; Mebazaa, A. Acute heart failure. Nat. Rev. Dis. Prim. 2020, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A. Coronary heart disease: Overview. Lancet 1996, 348, S1–S4. [Google Scholar] [CrossRef]
- Isbister, J.; Semsarian, C. Sudden cardiac death: An update. Intern. Med. J. 2019, 49, 826–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell. Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef] [PubMed]
- Solly, E.L.; Di Masi, C.G.; Bursill, C.A.; Psaltis, P.J.; Tan, J.T.M. MicroRNAs as Therapeutic Targets and Clinical Biomarkers in Atherosclerosis. J. Clin. Med. 2019, 8, 2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aimo, A.; Januzzi, J.L., Jr.; Bayes-Genis, A.; Vergaro, G.; Sciarrone, P.; Passino, C.; Emdin, M. Clinical and Prognostic Significance of sST2 in Heart Failure: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 2193–2203. [Google Scholar] [CrossRef]
- Tasevska, I.; Enhörning, S.; Persson, M.; Nilsson, P.M.; Melander, O. Copeptin predicts coronary artery disease cardiovascular and total mortality. Heart 2015, 102, 127–132. [Google Scholar] [CrossRef]
- Tang, W.W.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemet, I.; Saha, P.P.; Gupta, N.; Zhu, W.; Romano, K.A.; Skye, S.M.; Cajka, T.; Mohan, M.L.; Li, L.; Wu, Y.; et al. A Cardiovascular Disease-Linked Gut Microbial Metabolite Acts via Adrenergic Receptors. Cell 2020, 180, 862–877.e22. [Google Scholar] [CrossRef] [PubMed]
- Quispe, R.; Michos, E.D.; Martin, S.S.; Puri, R.; Toth, P.P.; Al Suwaidi, J.; Banach, M.; Virani, S.S.; Blumenthal, R.S.; Jones, S.R.; et al. High-Sensitivity C-Reactive Protein Discordance With Atherogenic Lipid Measures and Incidence of Atherosclerotic Cardiovascular Disease in Primary Prevention: The ARIC Study. J. Am. Heart Assoc. 2020, 9, e013600. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.-P.; Thiele, H.; Barbato, E.; Barthélémy, O.; Bauersachs, J.; Bhatt, D.L.; Dendale, P.; Dorobantu, M.; Edvardsen, T.; Folliguet, T.; et al. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur. Heart J. 2021, 42, ehaa575. [Google Scholar] [CrossRef] [PubMed]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. Eur. Heart J. 2020, 41, 407–477. [Google Scholar] [CrossRef] [PubMed]
- Diamond, G.A.; Forrester, J.S. Analysis of Probability as an Aid in the Clinical Diagnosis of Coronary-Artery Disease. N. Engl. J. Med. 1979, 300, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.T.; Corra, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts)Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Saletore, Y.; Meyer, K.; Korlach, J.; Vilfan, I.D.; Jaffrey, S.; Mason, C.E. The birth of the Epitranscriptome: Deciphering the function of RNA modifications. Genome Biol. 2012, 13, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, K.; Cai, J.; Zhang, M.; Zhang, X.; Xiong, X.; Meng, H.; Xu, X.; Huang, Z.; Peng, J.; et al. Landscape and Regulation of m6A and m6Am Methylome across Human and Mouse Tissues. Mol. Cell 2020, 77, 426–440.e6. [Google Scholar] [CrossRef] [PubMed]
- Christofi, T.; Zaravinos, A. RNA editing in the forefront of epitranscriptomics and human health. J. Transl. Med. 2019, 17, 319. [Google Scholar] [CrossRef] [PubMed]
- Vik, E.S.; Nawaz, M.S.; Andersen, P.S.; Fladeby, C.; Bjørås, M.; Dalhus, B.; Alseth, I. Endonuclease V cleaves at inosines in RNA. Nat. Commun. 2013, 4, 2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, Y.; Ooshio, I.; Fusamae, Y.; Kitae, K.; Kawaguchi, M.; Jingushi, K.; Hase, H.; Harada, K.; Hirata, K.; Tsujikawa, K. AlkB homolog 3-mediated tRNA demethylation promotes protein synthesis in cancer cells. Sci. Rep. 2017, 7, 42271. [Google Scholar] [CrossRef]
- Stellos, K.; Gatsiou, A.; Stamatelopoulos, K.; Matic, L.P.; John, D.; Lunella, F.F.; Jaé, N.; Rossbach, O.; Amrhein, C.; Sigala, F.; et al. Adenosine-to-inosine RNA editing controls cathepsin S expression in atherosclerosis by enabling HuR-mediated post-transcriptional regulation. Nat. Med. 2016, 22, 1140–1150. [Google Scholar] [CrossRef]
- Frye, M.; Jaffrey, S.R.; Pan, T.; Rechavi, G.; Suzuki, T. RNA modifications: What have we learned and where are we headed? Nat. Rev. Genet. 2016, 17, 365–372. [Google Scholar] [CrossRef]
- Alarcón, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485. [Google Scholar] [CrossRef]
- Li, J.; Yang, X.; Qi, Z.; Sang, Y.; Liu, Y.; Xu, B.; Liu, W.; Xu, Z.; Deng, Y. The role of mRNA m6A methylation in the nervous system. Cell Biosci. 2019, 9, 66. [Google Scholar] [CrossRef]
- Wu, J.; Frazier, K.; Zhang, J.; Gan, Z.; Wang, T.; Zhong, X. Emerging role of m 6 A RNA methylation in nutritional physiology and metabolism. Obes. Rev. 2020, 21, e12942. [Google Scholar] [CrossRef]
- Wang, T.; Kong, S.; Tao, M.; Ju, S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol. Cancer 2020, 19, 88. [Google Scholar] [CrossRef]
- Vu, L.P.; Cheng, Y.; Kharas, M.G. The Biology of m6A RNA Methylation in Normal and Malignant Hematopoiesis. Cancer Discov. 2019, 9, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Li, L.; Luo, E.; Hou, J.; Yan, G.; Wang, D.; Qiao, Y.; Tang, C. Role of m6A RNA methylation in cardiovascular disease (Review). Int. J. Mol. Med. 2020, 46, 1958–1972. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, S.; Wu, X.; Zhou, X. m6A RNA Methylation in Cardiovascular Diseases. Mol. Ther. 2020, 28, 2111–2119. [Google Scholar] [CrossRef]
- Longenecker, J.Z.; Gilbert, C.J.; Golubeva, V.A.; Martens, C.R.; Accornero, F. Epitranscriptomics in the Heart: A Focus on m6A. Curr. Heart Fail. Rep. 2020, 17, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.-Q.; Zhang, Y.-H.; Liu, F.; Ponnusamy, M.; Zhao, X.-M.; Zhou, L.-Y.; Zhai, M.; Liu, C.-Y.; Li, X.-M.; Wang, M.; et al. The piRNA CHAPIR regulates cardiac hypertrophy by controlling METTL3-dependent N6-methyladenosine methylation of Parp10 mRNA. Nat. Cell Biol. 2020, 22, 1319–1331. [Google Scholar] [CrossRef]
- Hinger, S.A.; Wei, J.; Dorn, L.E.; Whitson, B.A.; Janssen, P.M.; He, C.; Accornero, F. Remodeling of the m6A landscape in the heart reveals few conserved post-transcriptional events underlying cardiomyocyte hypertrophy. J. Mol. Cell. Cardiol. 2021, 151, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Fan, Z.; Yang, J. m6A modification of LncRNA MALAT1: A novel therapeutic target for myocardial ischemia-reperfusion injury. Int. J. Cardiol. 2020, 306, 162. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Li, H.; Su, H.; Chen, K.; Yan, J. FTO overexpression inhibits apoptosis of hypoxia/reoxygenation-treated myocardial cells by regulating m6A modification of Mhrt. Mol. Cell. Biochem. 2021, 476, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, J.; Ma, Y.; Zeng, Y.; Lu, C.; Yang, F.; Jiang, N.; Zhang, X.; Wang, Y.; Xu, Y.; et al. WTAP promotes myocardial ischemia/reperfusion injury by increasing endoplasmic reticulum stress via regulating m6A modification of ATF4 mRNA. Aging 2021, 13, 11135–11149. [Google Scholar] [CrossRef]
- Han, Z.; Wang, X.; Xu, Z.; Cao, Y.; Gong, R.; Yu, Y.; Yu, Y.; Guo, X.; Liu, S.; Yu, M.; et al. ALKBH5 regulates cardiomyocyte proliferation and heart regeneration by demethylating the mRNA of YTHDF1. Theranostics 2021, 11, 3000–3016. [Google Scholar] [CrossRef]
- Li, T.; Zhuang, Y.; Yang, W.; Xie, Y.; Shang, W.; Su, S.; Dong, X.; Wu, J.; Jiang, W.; Zhou, Y.; et al. Silencing of METTL3 attenuates cardiac fibrosis induced by myocardial infarction via inhibiting the activation of cardiac fibroblasts. FASEB J. 2021, 35, e21162. [Google Scholar] [CrossRef]
- Zhang, B.; Xu, Y.; Cui, X.; Jiang, H.; Luo, W.; Weng, X.; Wang, Y.; Zhao, Y.; Sun, A.; Ge, J. Alteration of m6A RNA Methylation in Heart Failure With Preserved Ejection Fraction. Front. Cardiovasc. Med. 2021, 8, 647806. [Google Scholar] [CrossRef] [PubMed]
- Jian, D.; Wang, Y.; Jian, L.; Tang, H.; Rao, L.; Chen, K.; Jia, Z.; Zhang, W.; Liu, Y.; Chen, X.; et al. METTL14 aggravates endothelial inflammation and atherosclerosis by increasing FOXO1 N6-methyladeosine modifications. Theranostics 2020, 10, 8939–8956. [Google Scholar] [CrossRef]
- Quiles-Jiménez, A.; Gregersen, I.; de Sousa, M.M.L.; Abbas, A.; Kong, X.Y.; Alseth, I.; Holm, S.; Dahl, T.B.; Skagen, K.; Skjelland, M.; et al. N6-methyladenosine in RNA of atherosclerotic plaques: An epitranscriptomic signature of human carotid atherosclerosis. Biochem. Biophys. Res. Commun. 2020, 533, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Xue, Y.; Li, H.; Huo, R.; Yan, Z.; Wang, J.; Xu, H.; Wang, J.; Cao, Y.; Zhao, J. Wilms’ tumour 1-associating protein inhibits endothelial cell angiogenesis by m6A-dependent epigenetic silencing of desmoplakin in brain arteriovenous malformation. J. Cell. Mol. Med. 2020, 24, 4981–4991. [Google Scholar] [CrossRef] [Green Version]
- Yao, M.-D.; Jiang, Q.; Ma, Y.; Liu, C.; Zhu, C.-Y.; Sun, Y.-N.; Shan, K.; Ge, H.-M.; Zhang, Q.-Y.; Zhang, H.-Y.; et al. Role of METTL3-Dependent N6-Methyladenosine mRNA Modification in the Promotion of Angiogenesis. Mol. Ther. 2020, 28, 2191–2202. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-J.; Xue, Y.; Huo, R.; Yan, Z.; Xu, H.; Li, H.; Wang, J.; Zhang, Q.; Cao, Y.; Zhao, J.-Z. N6-methyladenosine methyltransferase METTL3 affects the phenotype of cerebral arteriovenous malformation via modulating Notch signaling pathway. J. Biomed. Sci. 2020, 27, 62. [Google Scholar] [CrossRef]
- Shan, K.; Zhou, R.-M.; Xiang, J.; Sun, Y.-N.; Liu, C.; Lv, M.-W.; Xu, J.-J. FTO regulates ocular angiogenesis via m6A-YTHDF2-dependent mechanism. Exp. Eye Res. 2020, 197, 108107. [Google Scholar] [CrossRef]
- Picardi, E.; Manzari, C.; Mastropasqua, F.; Aiello, I.; D’Erchia, A.M.; Pesole, G. Profiling RNA editing in human tissues: Towards the inosinome Atlas. Sci. Rep. 2015, 5, 14941. [Google Scholar] [CrossRef] [Green Version]
- Samuel, C.E. Adenosine deaminase acting on RNA (ADAR1), a suppressor of double-stranded RNA–triggered innate immune responses. J. Biol. Chem. 2019, 294, 1710–1720. [Google Scholar] [CrossRef] [Green Version]
- Van Der Kwast, R.V.; Quax, P.H.; Nossent, A.Y. An Emerging Role for isomiRs and the microRNA Epitranscriptome in Neovascularization. Cells 2019, 9, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Kwast, R.V.; Parma, L.; van der Bent, M.L.; van Ingen, E.; Baganha, F.; Peters, H.A.; Goossens, E.A.; Simons, K.H.; Palmen, M.; de Vries, M.R.; et al. Adenosine-to-Inosine Editing of Vasoactive MicroRNAs Alters Their Targetome and Function in Ischemia. Mol. Ther. Nucleic Acids 2020, 21, 932–953. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Mann, T.D.; Stulić, M.; Rao, S.P.; Kirsch, A.; Pullirsch, D.; Strobl, X.; Rath, C.; Reissig, L.; Moreth, K.; et al. RNA editing of Filamin A pre- mRNA regulates vascular contraction and diastolic blood pressure. EMBO J. 2018, 37, e94813. [Google Scholar] [CrossRef] [PubMed]
- Borik, S.; Simon, A.J.; Nevo-Caspi, Y.; Mishali, D.; Amariglio, N.; Rechavi, G.; Paret, G. Increased RNA editing in children with cyanotic congenital heart disease. Intensiv Care Med. 2011, 37, 1664–1671. [Google Scholar] [CrossRef]
- Altaf, F.; Vesely, C.; Sheikh, A.M.; Munir, R.; Shah, S.T.A.; Tariq, A. Modulation of ADAR mRNA expression in patients with congenital heart defects. PLoS ONE 2019, 14, e0200968. [Google Scholar] [CrossRef]
- El Azzouzi, H.; Vilaça, A.P.; Feyen, D.A.M.; Gommans, W.M.; De Weger, R.A.; Doevendans, P.A.F.; Sluijter, J.P.G. Cardiomyocyte Specific Deletion of ADAR1 Causes Severe Cardiac Dysfunction and Increased Lethality. Front. Cardiovasc. Med. 2020, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Swirski, F.K.; Nahrendorf, M. Leukocyte Behavior in Atherosclerosis, Myocardial Infarction, and Heart Failure. Science 2013, 339, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Robbins, C.S.; Hilgendorf, I.; Weber, G.F.; Theurl, I.; Iwamoto, Y.; Figueiredo, J.-L.; Gorbatov, R.; Sukhova, G.K.; Gerhardt, L.M.S.; Smyth, D.; et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 2013, 19, 1166–1172. [Google Scholar] [CrossRef]
- Mallat, Z.; Hugel, B.; Ohan, J.; Lesèche, G.; Freyssinet, J.-M.; Tedgui, A. Shed Membrane Microparticles with Procoagulant Potential in Human Atherosclerotic Plaques. Circulation 1999, 99, 348–353. [Google Scholar] [CrossRef] [Green Version]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, E.; Flammer, A.J.; Lerman, L.O.; Elízaga, J.; Lerman, A.; Fernández-Avilés, F. Endothelial dysfunction over the course of coronary artery disease. Eur. Heart J. 2013, 34, 3175–3181. [Google Scholar] [CrossRef] [Green Version]
- Heyde, A.; Rohde, D.; McAlpine, C.S.; Zhang, S.; Hoyer, F.F.; Gerold, J.M.; Cheek, D.; Iwamoto, Y.; Schloss, M.J.; Vandoorne, K.; et al. Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell 2021, 184, 1348–1361.e22. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Nahrendorf, M.; Swirski, F. Leukocytes Link Local and Systemic Inflammation in Ischemic Cardiovascular Disease: An Expanded “Cardiovascular Continuum”. J. Am. Coll. Cardiol. 2016, 67, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Nahrendorf, M. Bone Marrow Takes Center Stage in Cardiovascular Disease. Circ. Res. 2016, 119, 701–703. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Pagler, T.; Gautier, E.L.; Avagyan, S.; Siry, R.L.; Han, S.; Welch, C.L.; Wang, N.; Randolph, G.J.; Snoeck, H.W.; et al. ATP-Binding Cassette Transporters and HDL Suppress Hematopoietic Stem Cell Proliferation. Science 2010, 328, 1689–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, A.J.; Akhtari, M.; Tolani, S.; Pagler, T.; Bijl, N.; Kuo, C.-L.; Wang, M.; Sanson, M.; Abramowicz, S.; Welch, C.; et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J. Clin. Investig. 2011, 121, 4138–4149. [Google Scholar] [CrossRef] [Green Version]
- Robbins, C.S.; Chudnovskiy, A.; Rauch, P.J.; Figueiredo, J.; Iwamoto, Y.; Gorbatov, R.; Etzrodt, M.; Weber, G.F.; Ueno, T.; van Rooijen, N.; et al. Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation 2012, 125, 364–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swirski, F.; Libby, P.; Aikawa, E.; Alcaide, P.; Luscinskas, F.W.; Weissleder, R.; Pittet, M.J. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Investig. 2007, 117, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.J.; Sheedy, F.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Schloss, M.J.; Hilby, M.; Nitz, K.; Prats, R.G.; Ferraro, B.; Leoni, G.; Soehnlein, O.; Kessler, T.; He, W.; Luckow, B.; et al. Ly6C high Monocytes Oscillate in the Heart During Homeostasis and After Myocardial Infarction—Brief Report. Arter. Thromb. Vasc. Biol. 2017, 37, 1640–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swirski, F.K.; Nahrendorf, M. Cardioimmunology: The immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 733–744. [Google Scholar] [CrossRef]
- Neto, A.A.; Mansur, A.D.P.; Avakian, S.D.; Gomes, E.P.S.G.; Ramires, J.A.F. Monocitose é um marcador de risco independente para a doença arterial coronariana. Arq. Bras. Cardiol. 2006, 86, 240–244. [Google Scholar] [CrossRef]
- Welsh, C.; Welsh, P.; Mark, P.B.; Celis-Morales, C.A.; Lewsey, J.; Gray, S.R.; Lyall, D.M.; Iliodromiti, S.; Gill, J.M.; Pell, J.; et al. Association of Total and Differential Leukocyte Counts With Cardiovascular Disease and Mortality in the UK Biobank. Arter. Thromb. Vasc. Biol. 2018, 38, 1415–1423. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Hu, X.; Huang, M.; Liu, J.; Gu, Y.; Ma, L.; Zhou, Q.; Cao, X. Mettl3-mediated mRNA m6A methylation promotes dendritic cell activation. Nat. Commun. 2019, 10, 1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, C.-S.; Li, J.Y.-S.; Chien, Y.; Wang, M.-L.; Yarmishyn, A.A.; Tsai, P.-H.; Juan, C.-C.; Nguyen, P.; Cheng, H.-M.; Huo, T.-I.; et al. METTL3-dependent N6-methyladenosine RNA modification mediates the atherogenic inflammatory cascades in vascular endothelium. Proc. Natl. Acad. Sci. USA 2021, 118, e2025070118. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Cardiac Extracellular Vesicles in Normal and Infarcted Heart. Int. J. Mol. Sci. 2016, 17, 63. [Google Scholar] [CrossRef] [PubMed]
- Rezaie, J.; Rahbarghazi, R.; Pezeshki, M.; Mazhar, M.; Yekani, F.; Khaksar, M.; Shokrollahi, E.; Amini, H.; Hashemzadeh, S.; Sokullu, S.E.; et al. Cardioprotective role of extracellular vesicles: A highlight on exosome beneficial effects in cardiovascular diseases. J. Cell. Physiol. 2019, 234, 21732–21745. [Google Scholar] [CrossRef] [PubMed]
- Preston, R.A.; Jy, W.; Jimenez, J.J.; Mauro, L.M.; Horstman, L.L.; Valle, M.; Aime, G.; Ahn, Y.S. Effects of Severe Hypertension on Endothelial and Platelet Microparticles. Hypertension 2003, 41, 211–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnouf, T.; Goubran, H.A.; Chou, M.-L.; Devos, D.; Radosevic, M. Platelet microparticles: Detection and assessment of their paradoxical functional roles in disease and regenerative medicine. Blood Rev. 2014, 28, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.T.; Lip, G.Y.H. The potential role of platelet microparticles in atherosclerosis. Thromb. Haemost. 2005, 94, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Hafiane, A.; Daskalopoulou, S.S. Extracellular vesicles characteristics and emerging roles in atherosclerotic cardiovascular disease. Metabolism 2018, 85, 213–222. [Google Scholar] [CrossRef]
- Xia, H.; Wu, Y.; Zhao, J.; Li, W.; Lu, L.; Ma, H.; Cheng, C.; Sun, J.; Xiang, Q.; Bian, T.; et al. The aberrant cross-talk of epithelium–macrophages via METTL3-regulated extracellular vesicle miR-93 in smoking-induced emphysema. Cell Biol. Toxicol. 2021, 4, 1–17. [Google Scholar] [CrossRef]
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130, 742–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartner, J.C.; Walkley, C.; Lu, J.; Orkin, S.H. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat. Immunol. 2008, 10, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Vu, L.P.; Pickering, B.F.; Cheng, Y.; Zaccara, S.; Nguyen, D.; Minuesa, G.; Chou, T.; Chow, A.; Saletore, Y.; Mackay, M.; et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017, 23, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Weng, H.; Huang, H.; Wu, H.; Qin, X.; Zhao, B.S.; Dong, L.; Shi, H.; Skibbe, J.; Shen, C.; Hu, C.; et al. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m6A Modification. Cell Stem Cell 2018, 22, 191–205.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Qian, P.; Shao, W.; Shi, H.; He, X.C.; Gogol, M.; Yu, Z.; Wang, Y.; Qi, M.; Zhu, Y.; et al. Suppression of m6A reader Ythdf2 promotes hematopoietic stem cell expansion. Cell Res. 2018, 28, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Mapperley, C.; Van De Lagemaat, L.N.; Lawson, H.; Tavosanis, A.; Paris, J.; Campos, J.; Wotherspoon, D.; Durko, J.; Sarapuu, A.; Choe, J.; et al. The mRNA m6A reader YTHDF2 suppresses proinflammatory pathways and sustains hematopoietic stem cell function. J. Exp. Med. 2021, 218, 20200829. [Google Scholar] [CrossRef] [PubMed]
- WMA Declaration of Helsinki—Ethical Principles for Medical Research Involving Human Subjects. 2018. Available online: https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ (accessed on 28 January 2021).
- D’Agostino, S.R.B.; Vasan, R.S.; Pencina, M.J.; Wolf, P.A.; Cobain, M.; Massaro, J.M.; Kannel, W.B. General Cardiovascular Risk Profile for Use in Primary Care: The Framingham Heart Study. Circulation 2008, 117, 743–753. [Google Scholar] [CrossRef] [Green Version]
- Russell, S.D.; Saval, M.A.; Robbins, J.L.; Ellestad, M.H.; Gottlieb, S.S.; Handberg, E.M.; Zhou, Y.; Chandler, B.; HF-ACTION Investigators. New York Heart Association functional class predicts exercise parameters in the current era. Am Heart J. 2009, 158, S24–S30. [Google Scholar] [CrossRef] [Green Version]
- Huber, A.; Oldridge, N.; Höfer, S. International SF-36 reference values in patients with ischemic heart disease. Qual. Life Res. 2016, 25, 2787–2798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bundhun, P.K.; Yanamala, C.M.; Huang, F. Percutaneous Coronary Intervention, Coronary Artery Bypass Surgery and the SYNTAX score: A systematic review and meta-analysis. Sci. Rep. 2017, 7, 43801. [Google Scholar] [CrossRef] [Green Version]
- Nagueh, S.F.; Smiseth, O.A.; Appleton, C.P.; Byrd, B.F.; Dokainish, H.; Edvardsen, T.; Flachskampf, F.A.; Gillebert, T.; Klein, A.L.; Lancellotti, P.; et al. Recommendations for the Evaluation of Left Ventricular Diastolic Function by Echocardiography: An Update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 2016, 29, 277–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didier, R.; Morice, M.C.; Barragan, P.; Noryani, A.A.; Noor, H.A.; Majwal, T.; Hovasse, T.; Castellant, P.; Schneeberger, M.; Maillard, L.; et al. 6- Versus 24-Month Dual Antiplatelet Therapy After Implantation of Drug-Eluting Stents in Patients Nonresistant to Aspirin. JACC Cardiovasc. Interv. 2017, 10, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Vranckx, P.; White, H.D.; Huang, Z.; Mahaffey, K.W.; Armstrong, P.; Van de Werf, F.; Moliterno, D.J.; Wallentin, L.; Held, C.; Aylward, P.E.; et al. Validation of BARC Bleeding Criteria in Patients With Acute Coronary Syndromes: The TRACER Trial. J. Am. Coll. Cardiol. 2016, 67, 2135–2144. [Google Scholar] [CrossRef]
- Shiotsu, H.; Okada, K.; Shibuta, T.; Kobayashi, Y.; Shirahama, S.; Kuroki, C.; Ueda, S.; Ohkuma, M.; Ikeda, K.; Ando, Y.; et al. The Influence of Pre-analytical Factors on the Analysis of Circulating MicroRNA. MicroRNA 2018, 7, 195–203. [Google Scholar] [CrossRef]
- McKie, P.M.; Burnett, J.C. NT-proBNP: The Gold Standard Biomarker in Heart Failure. J. Am. Coll. Cardiol. 2016, 68, 2437–2439. [Google Scholar] [CrossRef]
- Aimo, A.; Vergaro, G.; Ripoli, A.; Bayes-Genis, A.; Figal, D.A.P.; de Boer, R.A.; Lassus, J.; Mebazaa, A.; Gayat, E.; Breidthardt, T.; et al. Meta-Analysis of Soluble Suppression of Tumorigenicity-2 and Prognosis in Acute Heart Failure. JACC Heart Fail. 2017, 5, 287–296. [Google Scholar] [CrossRef]
- Huang, D.-H.; Sun, H.; Shi, J.-P. Diagnostic Value of Soluble Suppression of Tumorigenicity-2 for Heart Failure. Chin. Med. J. 2016, 129, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Aimo, A.; Vergaro, G.; Passino, C.; Ripoli, A.; Ky, B.; Miller, W.L.; Bayes-Genis, A.; Anand, I.; Januzzi, J.L.; Emdin, M. Prognostic Value of Soluble Suppression of Tumorigenicity-2 in Chronic Heart Failure. JACC Heart Fail. 2017, 5, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Morrow, D.A.; Higgins, L.J.; MacGillivray, C.; Guo, W.; Bode, C.; Rifai, N.; Cannon, C.P.; Gerszten, R.E.; Lee, R.T. Complementary Roles for Biomarkers of Biomechanical Strain ST2 and N-Terminal Prohormone B-Type Natriuretic Peptide in Patients With ST-Elevation Myocardial Infarction. Circulation 2008, 117, 1936–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Vark, L.C.; Lesman-Leegte, I.; Baart, S.J.; Postmus, D.; Pinto, Y.M.; Orsel, J.G.; Westenbrink, B.D.; Rocca, H.P.B.-L.; van Miltenburg, A.J.; Boersma, E.; et al. Prognostic Value of Serial ST2 Measurements in Patients with Acute Heart Failure. J. Am. Coll. Cardiol. 2017, 70, 2378–2388. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.A.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senthong, V.; Wang, Z.; Li, X.S.; Fan, Y.; Wu, Y.; Tang, W.H.W.; Hazen, S.L. Intestinal Microbiota-Generated Metabolite Trimethylamine- N- Oxide and 5-Year Mortality Risk in Stable Coronary Artery Disease: The Contributory Role of Intestinal Microbiota in a COURAGE-Like Patient Cohort. J. Am. Heart Assoc. 2016, 5, e002816. [Google Scholar] [CrossRef] [Green Version]
- Zile, M.R.; Claggett, B.L.; Prescott, M.F.; McMurray, J.J.; Packer, M.; Rouleau, J.L.; Swedberg, K.; Desai, A.S.; Gong, J.; Shi, V.C.; et al. Prognostic Implications of Changes in N-Terminal Pro-B-Type Natriuretic Peptide in Patients With Heart Failure. J. Am. Coll. Cardiol. 2016, 68, 2425–2436. [Google Scholar] [CrossRef] [Green Version]
- McKie, P.M.; Cataliotti, A.; Lahr, B.D.; Martin, F.L.; Redfield, M.M.; Bailey, K.R.; Rodeheffer, R.J.; Burnett, J.C. The Prognostic Value of N-Terminal Pro–B-Type Natriuretic Peptide for Death and Cardiovascular Events in Healthy Normal and Stage A/B Heart Failure Subjects. J. Am. Coll. Cardiol. 2010, 55, 2140–2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lainchbury, J.G.; Campbell, E.; Frampton, C.M.; Yandle, T.G.; Nicholls, M.; Richards, A. Brain natriuretic peptide and n-terminal brain natriuretic peptide in the diagnosis of heart failure in patients with acute shortness of breath. J. Am. Coll. Cardiol. 2003, 42, 728–735. [Google Scholar] [CrossRef] [Green Version]
- Clerico, A.; Fontana, M.; Zyw, L.; Passino, C.; Emdin, M. Comparison of the Diagnostic Accuracy of Brain Natriuretic Peptide (BNP) and the N-Terminal Part of the Propeptide of BNP Immunoassays in Chronic and Acute Heart Failure: A Systematic Review. Clin. Chem. 2007, 53, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Buckley, D.I.; Fu, R.; Freeman, M.; Rogers, K.; Helfand, M. C-Reactive Protein as a Risk Factor for Coronary Heart Disease: A Systematic Review and Meta-analyses for the U.S. Preventive Services Task Force. Ann. Intern. Med. 2009, 151, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Januzzi, J.L.; Camargo, C.A.; Anwaruddin, S.; Baggish, A.L.; Chen, A.A.; Krauser, D.G.; Tung, R.; Cameron, R.; Nagurney, J.T.; Chae, C.U.; et al. The N-terminal Pro-BNP Investigation of Dyspnea in the Emergency department (PRIDE) study. Am. J. Cardiol. 2005, 95, 948–954. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin–Neprilysin Inhibition versus Enalapril in Heart Failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [Green Version]
- The Atherosclerosis Risk in Communities (ARIC) Study: Design and objectives. The ARIC investigators. Am. J. Epidemiol. 1989, 129, 687–702. [CrossRef]
- Nummi, A.; Nieminen, T.; Pätilä, T.; Lampinen, M.; Lehtinen, M.L.; Kivistö, S.; Holmström, M.; Wilkman, E.; Teittinen, K.; Laine, M.; et al. Epicardial delivery of autologous atrial appendage micrografts during coronary artery bypass surgery—Safety and feasibility study. Pilot Feasibility Stud. 2017, 3, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Lampinen, M.; Takala, J.; Sikorski, V.; Soliymani, R.; Tarkia, M.; Lalowski, M.; Mervaala, E.; Kupari, M.; Zheng, Z.; et al. Epicardial transplantation of atrial appendage micrograft patch salvages myocardium after infarction. J. Heart Lung Transplant. 2020, 39, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Selberg, S.; Blokhina, D.; Aatonen, M.; Koivisto, P.; Siltanen, A.; Mervaala, E.; Kankuri, E.; Karelson, M. Discovery of Small Molecules that Activate RNA Methylation through Cooperative Binding to the METTL3-14-WTAP Complex Active Site. Cell Rep. 2019, 26, 3762–3771.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Meyer, K.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Begik, O.; Lucas, M.C.; Ramirez, J.M.; Mason, C.E.; Wiener, D.; Schwartz, S.; Mattick, J.S.; Smith, M.A.; Novoa, E.M. Accurate detection of m6A RNA modifications in native RNA sequences. Nat. Commun. 2019, 10, 4079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Campos, M.A.; Edelheit, S.; Toth, U.; Safra, M.; Shachar, R.; Viukov, S.; Winkler, R.; Nir, R.; Lasman, L.; Brandis, A.; et al. Deciphering the “m6A Code” via Antibody-Independent Quantitative Profiling. Cell 2019, 178, 731–747.e16. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Wang, Y.; Tang, Q.; Wei, L.; Zhang, X.; Jia, G. An Elongation- and Ligation-Based qPCR Amplification Method for the Radiolabeling-Free Detection of Locus-Specific N 6 -Methyladenosine Modification. Angew. Chem. Int. Ed. Engl. 2018, 57, 15995–16000. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, A.B.R.; Gokhale, N.; Cerchietti, L.; Jaffrey, S.R.; Horner, S.M.; Mason, C.E. Limits in the detection of m6A changes using MeRIP/m6A-seq. Sci. Rep. 2020, 10, 6590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinie, B.; Wang, J.; Lim, K.S.; Hillebrand, R.; Lu, Z.-X.; Van Wittenberghe, N.; Howard, B.D.; Daneshvar, K.; Mullen, A.C.; Dedon, P.; et al. m6A-LAIC-seq reveals the census and complexity of the m6A epitranscriptome. Nat. Methods 2016, 13, 692–698. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Toth, J.I.; Petroski, M.D.; Zhang, Z.; Zhao, J.C. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014, 16, 191–198. [Google Scholar] [CrossRef]
- Ke, S.; Alemu, E.A.; Mertens, C.; Gantman, E.C.; Fak, J.J.; Mele, A.; Haripal, B.; Zucker-Scharff, I.; Moore, M.J.; Park, C.Y.; et al. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev. 2015, 29, 2037–2053. [Google Scholar] [CrossRef] [Green Version]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef]
- Bringmann, P.; Lührmann, R. Antibodies specific for N6-methyladenosine react with intact snRNPs U2 and U4/U6. FEBS Lett. 1987, 213, 309–315. [Google Scholar] [CrossRef] [Green Version]
- Thüring, K.; Schmid, K.; Keller, P.; Helm, M. LC-MS Analysis of Methylated RNA. Methods Mol. Biol. 2017, 1562, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Pan, T. Probing RNA Modification Status at Single-Nucleotide Resolution in Total RNA. Methods Enzymol. 2015, 560, 149–159. [Google Scholar] [CrossRef]
- Imanishi, M.; Tsuji, S.; Suda, A.; Futaki, S. Detection of N6-methyladenosine based on the methyl-sensitivity of MazF RNA endonuclease. Chem. Commun. 2017, 53, 12930–12933. [Google Scholar] [CrossRef]
- Bodi, Z.; Fray, R.G. Detection and Quantification of N 6-Methyladenosine in Messenger RNA by TLC. Methods Mol. Biol. 2017, 1562, 79–87. [Google Scholar] [CrossRef]
- Sednev, M.V.; Mykhailiuk, V.; Choudhury, P.; Halang, J.; Sloan, K.E.; Bohnsack, M.T.; Höbartner, C. N 6 -Methyladenosine-Sensitive RNA-Cleaving Deoxyribozymes. Angew. Chem. Int. Ed. 2018, 57, 15117–15121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, T.; Yuan, Y.; Chen, Z.; Xi, K.; Wang, T.; Xie, Y.; He, Z.; Su, H.; Zhou, Y.; Tan, Z.-J.; et al. Precise Antibody-Independent m6A Identification via 4SedTTP-Involved and FTO-Assisted Strategy at Single-Nucleotide Resolution. J. Am. Chem. Soc. 2018, 140, 5886–5889. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, E.; Ehrenschwender, T.; Batista, P.J.; Chang, H.Y.; Kool, E.T. Identification of a Selective Polymerase Enables Detection of N6-Methyladenosine in RNA. J. Am. Chem. Soc. 2013, 135, 19079–19082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aschenbrenner, J.; Werner, S.; Marchand, V.; Adam, M.; Motorin, Y.; Helm, M.; Marx, A. Engineering of a DNA Polymerase for Direct m6A Sequencing. Angew. Chem. Int. Ed. 2018, 57, 417–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Yan, J.; Zhang, Z.; Pian, H.; Liu, C.; Li, Z. Identification of a selective DNA ligase for accurate recognition and ultrasensitive quantification of N6-methyladenosine in RNA at one-nucleotide resolution. Chem. Sci. 2018, 9, 3354–3359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellanos-Rubio, A.; Santin, I.; Olazagoitia-Garmendia, A.; Romero-Garmendia, I.; Jauregi-Miguel, A.; Legarda, M.; Bilbao, J.R. A novel RT-QPCR-based assay for the relative quantification of residue specific m6A RNA methylation. Sci. Rep. 2019, 9, 4220. [Google Scholar] [CrossRef] [PubMed]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, Y.; Liu, L.; Feng, J.; Zhang, T.; Qin, S.; Zhao, X.; Wang, C.; Li, D.; Han, W.; et al. Study of the whole genome, methylome and transcriptome of Cordyceps militaris. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, Y.; Dong, S.; Yu, Q.; Jia, G. Antibody-free enzyme-assisted chemical approach for detection of N6-methyladenosine. Nat. Chem. Biol. 2020, 16, 896–903. [Google Scholar] [CrossRef]
- Meyer, K.D. DART-seq: An antibody-free method for global m6A detection. Nat. Methods 2019, 16, 1275–1280. [Google Scholar] [CrossRef]
- Chen, K.; Lu, Z.; Wang, X.; Fu, Y.; Luo, G.-Z.; Liu, N.; Han, D.; Dominissini, D.; Dai, Q.; Pan, T.; et al. High-ResolutionN6-Methyladenosine (m6A) Map Using Photo-Crosslinking-Assisted m6A Sequencing. Angew. Chem. Int. Ed. Engl. 2015, 54, 1587–1590. [Google Scholar] [CrossRef] [Green Version]
- Bahn, J.H.; Lee, J.-H.; Li, G.; Greer, C.; Peng, G.; Xiao, X. Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res. 2011, 22, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Ranasinghe, R.T.; Challand, M.R.; Ganzinger, K.A.; Lewis, B.W.; Softley, C.; Schmied, W.H.; Horrocks, M.H.; Shivji, N.; Chin, J.W.; Spencer, J.; et al. Detecting RNA base methylations in single cells by in situ hybridization. Nat. Commun. 2018, 9, 655. [Google Scholar] [CrossRef]
- Kontostathi, G.; Makridakis, M.; Zoidakis, J.; Vlahou, A. Applications of multiple reaction monitoring targeted proteomics assays in human plasma. Expert Rev. Mol. Diagn. 2019, 19, 499–515. [Google Scholar] [CrossRef]
- Gholizadeh, E.; Karbalaei, R.; Khaleghian, A.; Salimi, M.; Gilany, K.; Soliymani, R.; Tanoli, Z.; Rezadoost, H.; Baumann, M.; Jafari, M.; et al. Identification of Celecoxib targeted proteins using label-free thermal proteome profiling on rat hippocampus. Mol. Pharmacol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhao, S.; Li, C.-I.; Sheng, Q.; Shyr, Y. RNAseqPS: A Web Tool for Estimating Sample Size and Power for RNAseq Experiment. Cancer Inform. 2014, 13, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Berulava, T.; Buchholz, E.; Elerdashvili, V.; Pena, T.; Islam, R.; Lbik, D.; Mohamed, B.A.; Renner, A.; Von Lewinski, D.; Sacherer, M.; et al. Changes in m6A RNA methylation contribute to heart failure progression by modulating translation. Eur. J. Heart Fail. 2020, 22, 54–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.; Nam, D. Gene dispersion is the key determinant of the read count bias in differential expression analysis of RNA-seq data. BMC Genom. 2017, 18, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.M.; Jungner, Y.G. Principios y metodos del examen colectivo para identificar enfermedades [Principles and practice of mass screening for disease]. Bol. Oficina Sanit. Panam. 1968, 65, 281–393. [Google Scholar] [PubMed]
- Deng, K.; Ning, X.; Ren, X.; Yang, B.; Li, J.; Cao, J.; Chen, J.; Lu, X.; Chen, S.; Wang, L. Transcriptome-wide N6-methyladenosine methylation landscape of coronary artery disease. Epigenomics 2021, 13, 793–808. [Google Scholar] [CrossRef]
- Jantsch, M.F.; Quattrone, A.; O’Connell, M.; Helm, M.; Frye, M.; Gonzales, M.M.; Öhman, M.; Ameres, S.; Willems, L.; Fuks, F.; et al. Positioning Europe for the Epitranscriptomics challenge. RNA Biol. 2018, 15, 829–831. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C. Lipid and macrophage accumulations in arteries of children and the development of atherosclerosis. Am. J. Clin. Nutr. 2000, 72, 1297s–1306s. [Google Scholar] [CrossRef] [PubMed]
- Paulson, K.E.; Zhu, S.-N.; Chen, M.; Nurmohamed, S.; Jongstra-Bilen, J.; Cybulsky, M.I. Resident Intimal Dendritic Cells Accumulate Lipid and Contribute to the Initiation of Atherosclerosis. Circ. Res. 2010, 106, 383–390. [Google Scholar] [CrossRef] [Green Version]
- Zuchtriegel, G.; Uhl, B.; Puhr-Westerheide, D.; Pörnbacher, M.; Lauber, K.; Krombach, F.; Reichel, C.A. Platelets Guide Leukocytes to Their Sites of Extravasation. PLoS Biol. 2016, 14, e1002459. [Google Scholar] [CrossRef] [Green Version]
- Mause, S.F.; von Hundelshausen, P.; Zernecke, A.; Koenen, R.R.; Weber, C. Platelet Microparticles. Arter. Thromb. Vasc. Biol. 2005, 25, 1512–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Gils, J.; Derby, M.C.; Fernandes, L.R.; Ramkhelawon, B.; Ray, T.D.; Rayner, K.; Parathath, S.; Distel, E.; Feig, J.L.; Alvarez-Leite, J.I.; et al. The neuroimmune guidance cue netrin-1 promotes atherosclerosis by inhibiting the emigration of macrophages from plaques. Nat. Immunol. 2012, 13, 136–143. [Google Scholar] [CrossRef] [Green Version]
- Geissler, A.; Ryzhov, S.; Sawyer, D.B. Neuregulins: Protective and reparative growth factors in multiple forms of cardiovascular disease. Clin. Sci. 2020, 134, 2623–2643. [Google Scholar] [CrossRef]
- Zhao, B.S.; Nachtergaele, S.; Roundtree, I.A.; He, C. Our views of dynamic N6-methyladenosine RNA methylation. RNA 2017, 24, 268–272. [Google Scholar] [CrossRef]
- Schaefer, M. The Regulation of RNA Modification Systems: The Next Frontier in Epitranscriptomics? Genes 2021, 12, 345. [Google Scholar] [CrossRef]
- Ke, S.; Pandya-Jones, A.; Saito, Y.; Fak, J.J.; Vågbø, C.B.; Geula, S.; Hanna, J.H.; Black, D.L.; Darnell, J.E., Jr.; Darnell, R. m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017, 31, 990–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Laurent, B.; Hsu, C.-H.; Nachtergaele, S.; Lu, Z.; Sheng, W.; Xu, C.; Chen, H.; Ouyang, J.; Wang, S.; et al. RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature 2017, 543, 573–576. [Google Scholar] [CrossRef]
- Darnell, R.; Ke, S.; Darnell, J.E., Jr. Pre-mRNA processing includesN6methylation of adenosine residues that are retained in mRNA exons and the fallacy of “RNA epigenetics”. RNA 2018, 24, 262–267. [Google Scholar] [CrossRef] [Green Version]
- Rosa-Mercado, N.A.; Withers, J.B.; Steitz, J.A. Settling the m6A debate: Methylation of mature mRNA is not dynamic but accelerates turnover. Genes Dev. 2017, 31, 957–958. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Fan, X.; Zhang, R.; Wu, G. Upregulated N6-Methyladenosine RNA in Peripheral Blood: Potential Diagnostic Biomarker for Breast Cancer. Cancer Res. Treat. 2021, 53, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Zhang, N.; Chen, Z.; Song, J.; Wu, Y.; Li, Z.; Chen, F.; Wu, J.; Li, D.; Li, J.; et al. Level of N6-Methyladenosine in Peripheral Blood RNA: A Novel Predictive Biomarker for Gastric Cancer. Clin. Chem. 2020, 66, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Lou, X.; Li, K.; Xu, X.; Guo, Y.; Xu, D.; Yang, Z.; Xu, D.; Cui, W.; Zhang, D. Peripheral Blood Leukocyte N6-methyladenosine is a Noninvasive Biomarker for Non-small-cell Lung Carcinoma. OncoTargets Ther. 2020, 13, 11913–11921. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Rao, J.; Zhang, L.; Fu, B.; Guo, Y.; Huang, Z.; Li, J. The study of METTL14, ALKBH5, and YTHDF2 in peripheral blood mononuclear cells from systemic lupus erythematosus. Mol. Genet. Genom. Med. 2020, 8, e1298. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Gao, Y.; Zhang, L.; Rao, J.; Guo, Y.; Huang, Z.; Li, J. Decreased ALKBH5, FTO, and YTHDF2 in Peripheral Blood Are as Risk Factors for Rheumatoid Arthritis. BioMed Res. Int. 2020, 20, 5735279. [Google Scholar] [CrossRef] [PubMed]
- Selberg, S.; Yu, L.-Y.; Bondarenko, O.; Kankuri, E.; Seli, N.; Kovaleva, V.; Herodes, K.; Saarma, M.; Karelson, M. Small-Molecule Inhibitors of the RNA M6A Demethylases FTO Potently Support the Survival of Dopamine Neurons. Int. J. Mol. Sci. 2021, 22, 4537. [Google Scholar] [CrossRef] [PubMed]
- Selberg, S.; Seli, N.; Kankuri, E.; Karelson, M. Rational Design of Novel Anticancer Small-Molecule RNA m6A Demethylase ALKBH5 Inhibitors. ACS Omega 2021, 6, 13310–13320. [Google Scholar] [CrossRef] [PubMed]
- Yankova, E.; Blackaby, W.; Albertella, M.; Rak, J.; De Braekeleer, E.; Tsagkogeorga, G.; Pilka, E.S.; Aspris, D.; Leggate, D.; Hendrick, A.G.; et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 2021, 593, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Nummi, A.; Mulari, S.; Stewart, J.A.; Kivistö, S.; Teittinen, K.; Nieminen, T.; Lampinen, M.; Pätilä, T.; Sintonen, H.; Juvonen, T.; et al. Autologous cardiac micrografts as support therapy to coronary artery bypass surgery. medRxiv 2021. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Goal | Description |
|---|---|
| To identify IHD-specific candidate biomarkers and lay the foundation for both clinical and diagnostic efficacy studies in the future | Corss-sectional cohort study design Comparsion of blood RNA modifications of IHD cohorts to non-IHD cohorts Porspective cohort study design Comparsion of circulating RNA modifications of IHD-positive cohorts to thoes in IHD-negative cohort |
| To establish protocals and modify detection methods and workflows for RNA modications | Optimization of RNA isolation, type-fragmentation for modification-targeted RNA sequencing using both second and third generation methodologies |
| To increase the pathophysiologic knowledge of IHD | The cross-sectional and prospective comparsions of blood and right atrial appendage epitramscriptomes is expectable to provide novel insight to the IHD pathophysiology |
| To open venues for therapeutic development previously been disregarded due to the lack of research and methodological limitations | Identification of novel epotanscriptomic candidate blood biomarkers for IHD can provide potenial targets also for future drug development |
| General | Justification |
|---|---|
| Condition that limits life expectancy | May modify blood epitranscriptomes hampering reliable biomarker identification |
| Active inflammatory state (i.e., gout, systemic lupus erythematosus) | Cimplement, cytokines and leukocyte activation may infuence blood epitranscriptomes |
| Primary desease of blood or bone marrow | Expectable to alter epotranscriptomic regulation precluding reliable biomarker discovery |
| Major congenital heart disease of atrial fibrillation | To exclude the effects of remodelling in atrial appendges |
| Renal insufficiency (GTReEPI < 45 mL/min) | To exclude the effects of altered blood solute dynamics for reliable biomarker discovery |
| Uncontrolled hypertension (RR > 180/100 mmHg) or diabetes (HbA1c > 60 mmol/L), insulin us diabetes | Hight blood pressure and significant hyperglycemia damage endothelium and blood cells |
| Prior open-heart surgery (i.e., coronary artery bypass surgery) | To exclude very high-morbidity IHD and support the goal to identify biomarkers for early-to-moderate IHD |
| Other manifestations of atherosclerosis: a. Arteriosclerosis oblitierans/claudication b. Earlier stroke, cerebral hemorrhage or transient ischemic attach (TIA) c. Vascular or mixed type dementia d. Clinically releveant carotid artery stenosis e. Mesenteric ischemia | To exclude effects of other manifestations of atherosclerosis in blood epitranscriptomes as extensively as possible; Except for surveying vascular claudication, prospective investigations to exclude these manifestations will not be performed; Ankle-brachial index is recorded for any asymptomatic peripheral artery disease in cohort II |
| Transthoracic echocardiography: a. Cardiomyopathy (Hypertrophic/Dilated) b. Left ventricular hypertrophy (LVH) c. Clear heart failure (i.e., LVEF < 25%) d. Indication of atrial remodeling e. Functionally significant valve defects | To exclude remodeling effects due to intrinsic myocardial pathology, significant heart failure or valve defects; LVH is considered as an exception for the cohort III (part of pathophysiology) |
| Study cohort I, patients with myocardial infarction revascularized with urgent PCI | |
| Stent thrombosis, vasospastic coronary occlusion | This is IHD-focused study, myocardial infartions of other than atherothrombotic etiology are excluded |
| MI complications (e.g., chordal rupture, aorthic disserction, acute heart failure, cardiogenic shock) | To focus biomarker discovery to the IHD-induced infarction-specific epitranscriptomic alterations |
| Global ischemia on electrocardiogram | High rish of insufficient PCI and ischemia relievement |
| Study cohort II, patients with stable IHD undergoing coronary artery bypass surgery | |
| Duration of stable angina pectoris or exertional dyspneas < 1-month, crescendo angina | To exclude acute and subacute IHD related alterations in blood epitranscriptomes |
| Complex surgeries (e.g., valve/aneurysm repair) | To exclude other major cardiac remodeling effects |
| Study cohort III, patients with stable aortic valve stenosis undergoing valve replacement surgery | |
| Clinically mild-to-moderate stenosis with mild symptoms (NYHA/CCA 0-I) | Indicate less pronounced pathophysiology that might be reflected with blunted changes in the alterations of the blood epitranscriptomes regarding AVS |
| Documented IHD or complex operations | As a control cohort with non-IHD cardiac pathology, any indication of IHD will lead to exclusion |
| Transcatheter asortic valve implantations | To enable right atrial appendage sample collection |
| Study cohort IV, patients screened negative for IHD with coronary computed tomography | |
| Any prior cardiovascular disease or medication currently or in history | As critical non-IHD controls, the aim is to also recruit patients that represent overall “cardiovascular health” |
| Primary | Secondary |
|---|---|
| 1. Study sample m6A and A-to-I profiles from the recruitment stage associating with IHD and AVS pathophysiology 1.1. Acute IR controlled for stable ishcemia, pressure overload, and homeostasis (I vs. II-III-IV) 1.2. Stable ishcemia controlled for acute IR, pressure overload, and homeostasis (II vs. I-III-IV) 1.3. Acute IR controlled for homeostasis (I vs. IV) 1.4. Pressure overload controlled for acute IR, stable ishcemia, and hemeostasis (III vs. I-II-IV) | 4. Changes in the study sample m6A and A-to-I profiles (Recruitment vs. 3-months) associating to 4.1 Therapy effects on Pathophysiology at 3-month follow-up 4.1.1. STEMI-PCI effects (Resolution and relievement acute IR, remodelingl I vs. I) 4.1.2. IHD-CABG effects (Relievement of stable ischemia, remodeling; II vs III) 4.1.3. AVS-AVR effects (Relievement of pressure overload, remodeling; III vs. III) 4.2 All-cause mortality during 3-month follow-up 4.3 Beneficial/adverse/no-response for therapy as measured via echocardiography at 3 months 4.4 Surrogate (non-)metabolite CVD biomarker levels at recruitment, 3 months, changes between |
| 2. Study sample m6A and A-to-I profiles from the recruitment stage associating with 3-moth follow-up clinicial parmeters 2.1. Cardiovascular mortality 2.2 Cardiovascular morbidity 2.3. MACCE 2.4 Cardiovascular medication increases or reductions | 5. study sample m6A and A-to-I profiles from the recruitment stage as in outcomes 4.2.–4.5. |
| 3. Changes in the sample m6A and A-to-I profiles (recruitment vs. 3-months) as in outcome 2 | 6. Study sample quantitative alterations in other RNA modifications as in outcomes 1–5 (2.3.8.) |
| 7. Plasma metabolute and non-metabolite CVD biomarkers as in outcomes 1–5 (excl.4.4) (2.3.5.) | |
| 8. Biobank plasma and RAA sample proteomic profiles as in outcomes 1–5 (2.3.10.) | |
| 9. Whole blood sample transcriptome-based leukpcyte profiles as in outcomes 1–5 |
| Controllers | Main responsibility area/task |
| Heart and Lung Center & Cardiac Unit, Helsinki University Hospital (HUS), Finland | Patient recruitment; clinicial evaluations; operations (PCA, CABG, AVR); imagings (ICA, CCTA, TTE); control visits; study sample collection; registry data storage and governace; sample storage |
| Department of Pharmacology, University of Helsinki (UH), Finland | Coordination of (1) collaboration, (2)funding acquisition and (3) competitve tendering; RNA exctraction and initial sample quality measurements; academic analyses; scientific publishing |
| Collaborator centers | |
| Heart Hospital, Tampere Unviersity Hospital (TAYS), Finland | Patients recruitment (cohort III); clinical evaluations; operations (AVR); imagings (ICA, TTE); control visits; sample collection; clinical data and sample storage |
| Helsinki and Tampere Biobanks, Finland | Additional biobank sample collection and storage; DNA extraction; protocal designing |
| Meilahti Clinical Proteomics Core Faculity, UH, Finland | Targeted proteomics from plasma and snap-frozen RAA tissue samples; protocal designing |
| Chemistry Unit, Ginnish Food Authority, Finland | Specific measurements and analyses of modified RNA necleotides from via UHPLC-MS.MS methodolgy |
| Folkhälsan Research Center, Finland | Leading bioinformatics of second and third generation RNA sequencing targeting modifications (m6A) as well as DNA-to-RNA sequence matching (A-to-I) |
| Middle East Technical Univeristy, Ankara, Turkey | Ollaboration in the bioinformatics with Folkhälsan, specific share of responsiblities is decided later |
| Koç University, Istanbul, Turkey | See above |
| University of Tartu, Estonia | Leading the collaboration for the development of potential binding molecules as novel drugs for IHD |
| Strengths |
| 1. Interdisciplinary collaboration enables simultaneous recruitment, sample handling and measurements. |
| 2. A relevant IHD vs. non-IHD comparison is incorporated with a follow-up dimension as well. |
| 3. Coronary artery status is visualized from all participants recruited. |
| 4. Cardiac function is assessed by TTE from all participants recruited. |
| 5. Exclusion of patients with prior clinically relevant manifestations of atherosclerosis than IHD. |
| 6. Participants are surveyed for vascular claudication. ABI for the cohort II to record any asymptomatic PAD. |
| 7. Cohort morbidity an dother background characteristics are meticulously registered, assessed and reported. |
| 8. Study sample collection is designed to minimize the timespan for RNA vulnerable for degradation. |
| 9. Sample use is optimized for as comprehensive measurements as possible. RNA is fractionated for analyses. |
| Limitations and management consideration |
| 1. Coronary evaluation is non-uniform across cohorts. SYNTAX with ICA for complexity assesment of IHD. The CCTA cohort IV is a primary non-IHD control. |
| 2. Complete rule out of the effects of systemic atherosclerosis cannot be achieved Most earlier manifestations of systemic atherosclerosis lead to exclusion, vascular claudication is surveyed (exclusion criterion) and ABI is measure from CABG cohort to record any asymptomatic PAD. |
| 3. Limited possiblity to adjust medication effects (cohort IV has neither any CVD pathology nor medication). ATC and DDT alterations are recorded and reported. Immunomodulatry, edication leads to exclusion. |
| 4. Limited ability to pinpoint precise origins of the forthecoming epitranscriptomic alterations. Buffy coat leukpcytes can be used for validation, plasma cfRNA is principally derived from EVs. |
| 5. Incapability to identify biomarkers directly for subclinical IHD due to required cohort morbidity. (1) Current study assesses the harnessing potential of epitranscriptomics for a source of IHD biomarkers (2) Long-term follow-up of cohort IV can address the task. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sikorski, V.; Karjalainen, P.; Blokhina, D.; Oksaharju, K.; Khan, J.; Katayama, S.; Rajala, H.; Suihko, S.; Tuohinen, S.; Teittinen, K.; et al. Epitranscriptomics of Ischemic Heart Disease—The IHD-EPITRAN Study Design and Objectives. Int. J. Mol. Sci. 2021, 22, 6630. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126630
Sikorski V, Karjalainen P, Blokhina D, Oksaharju K, Khan J, Katayama S, Rajala H, Suihko S, Tuohinen S, Teittinen K, et al. Epitranscriptomics of Ischemic Heart Disease—The IHD-EPITRAN Study Design and Objectives. International Journal of Molecular Sciences. 2021; 22(12):6630. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126630
Chicago/Turabian StyleSikorski, Vilbert, Pasi Karjalainen, Daria Blokhina, Kati Oksaharju, Jahangir Khan, Shintaro Katayama, Helena Rajala, Satu Suihko, Suvi Tuohinen, Kari Teittinen, and et al. 2021. "Epitranscriptomics of Ischemic Heart Disease—The IHD-EPITRAN Study Design and Objectives" International Journal of Molecular Sciences 22, no. 12: 6630. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126630