
Differences between Human and Mouse IgM Fc Receptor (FcµR)



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Differences between Human and Mouse FcµRs
2.1. Identification of FcµR cDNAs, Functional Cloning vs Database Search
2.2. Cellular Distribution, Lymphocytes vs Only B Cells
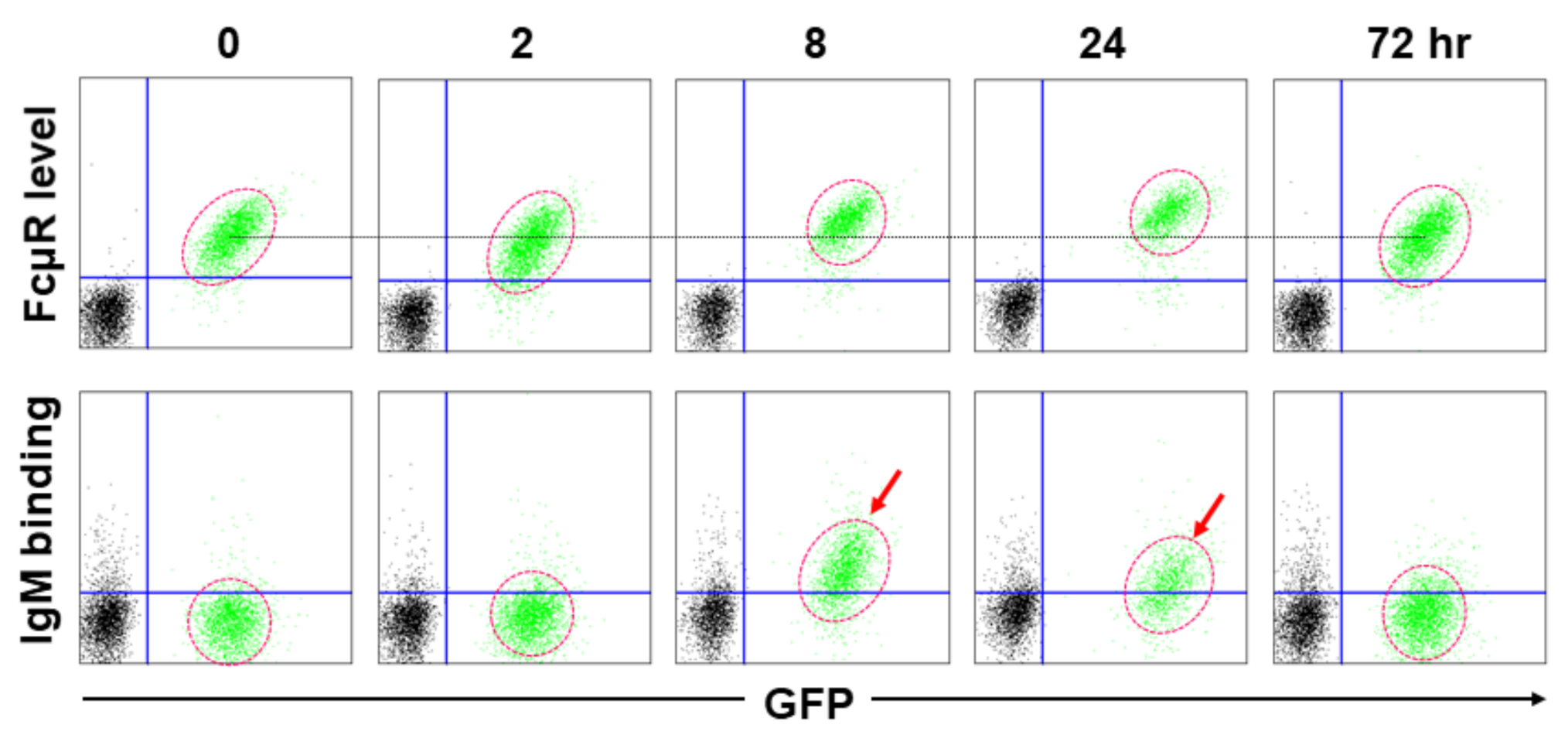
2.3. IgM Ligand Binding, Constitutive vs Transient Binding
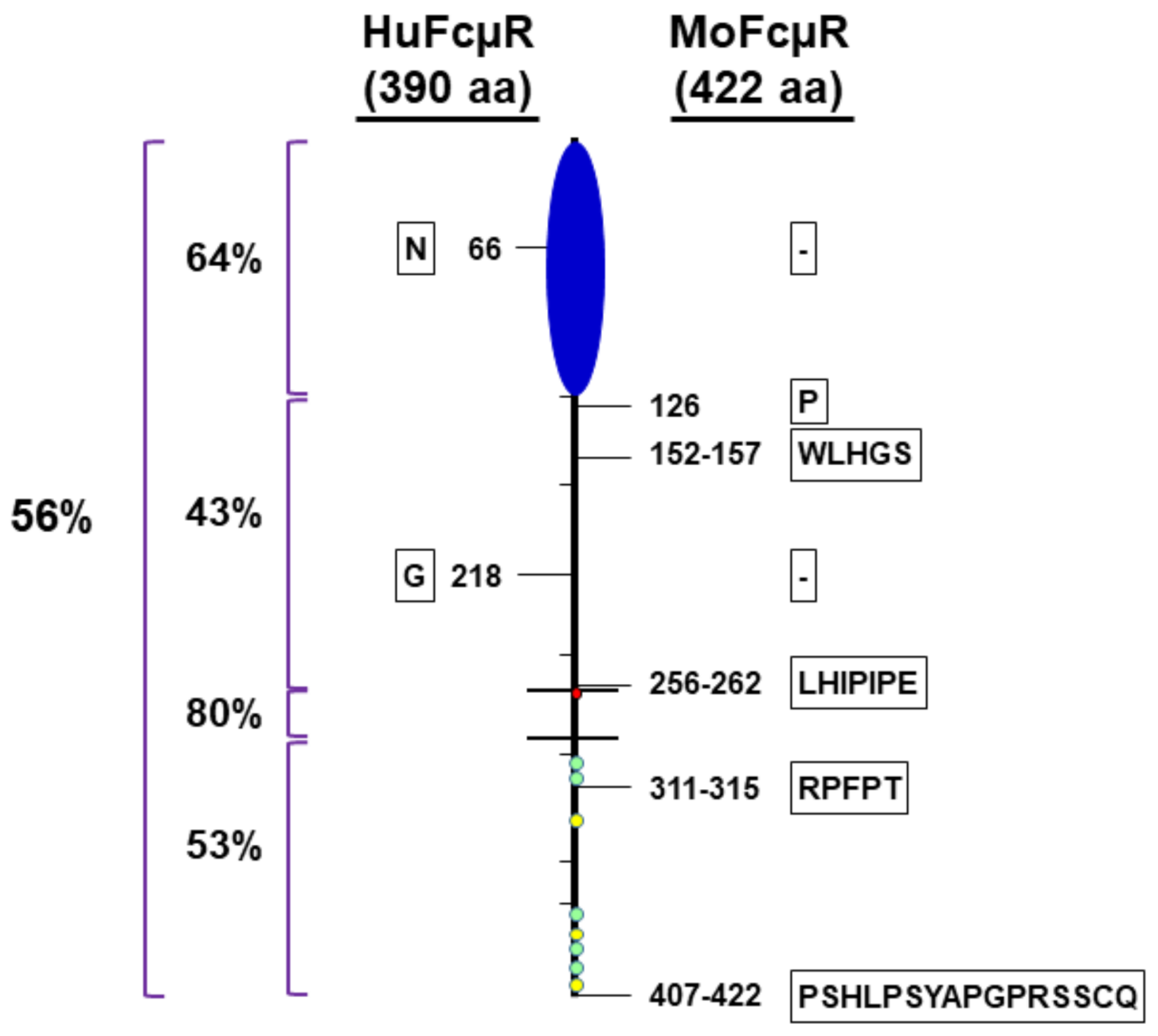
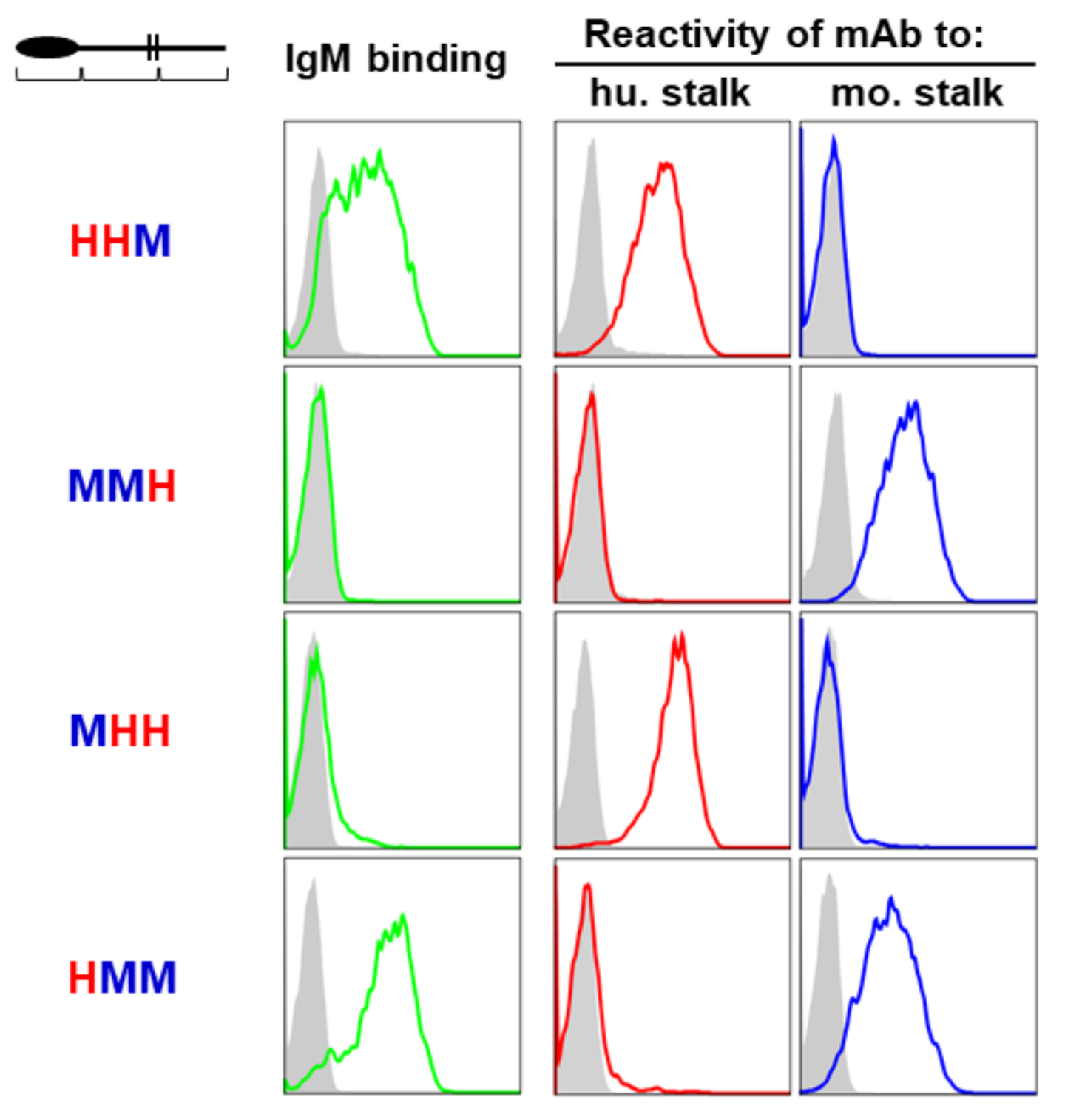
2.4. Dependence of the Ig-Like Domain of FcµR
3. Mutational Analysis of FcµR
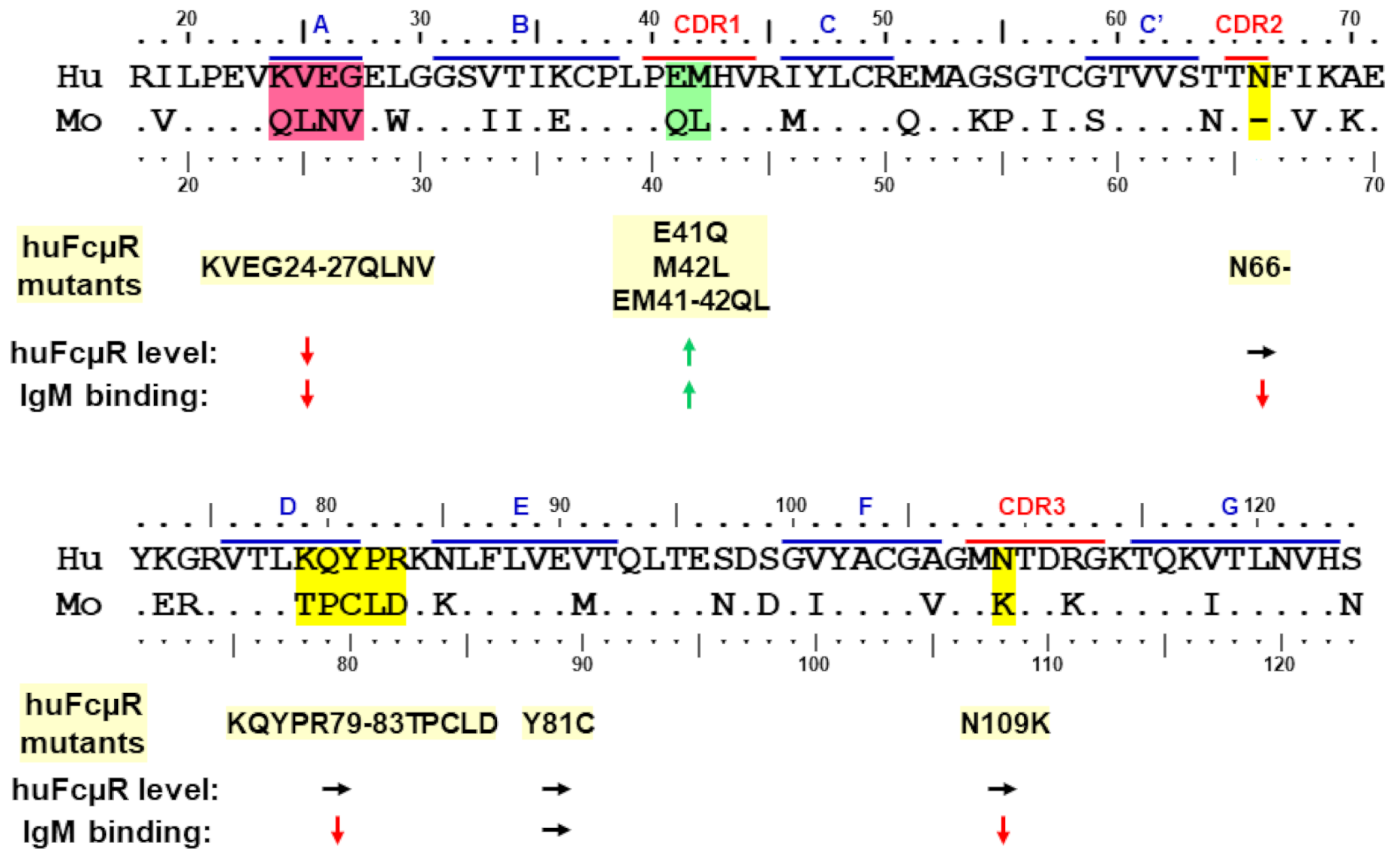
3.1. Amino Acid Sequence Alignment of the Ig-Like Domain of Human and Mouse FcµRs
3.2. Site-Directed Mutagenesis
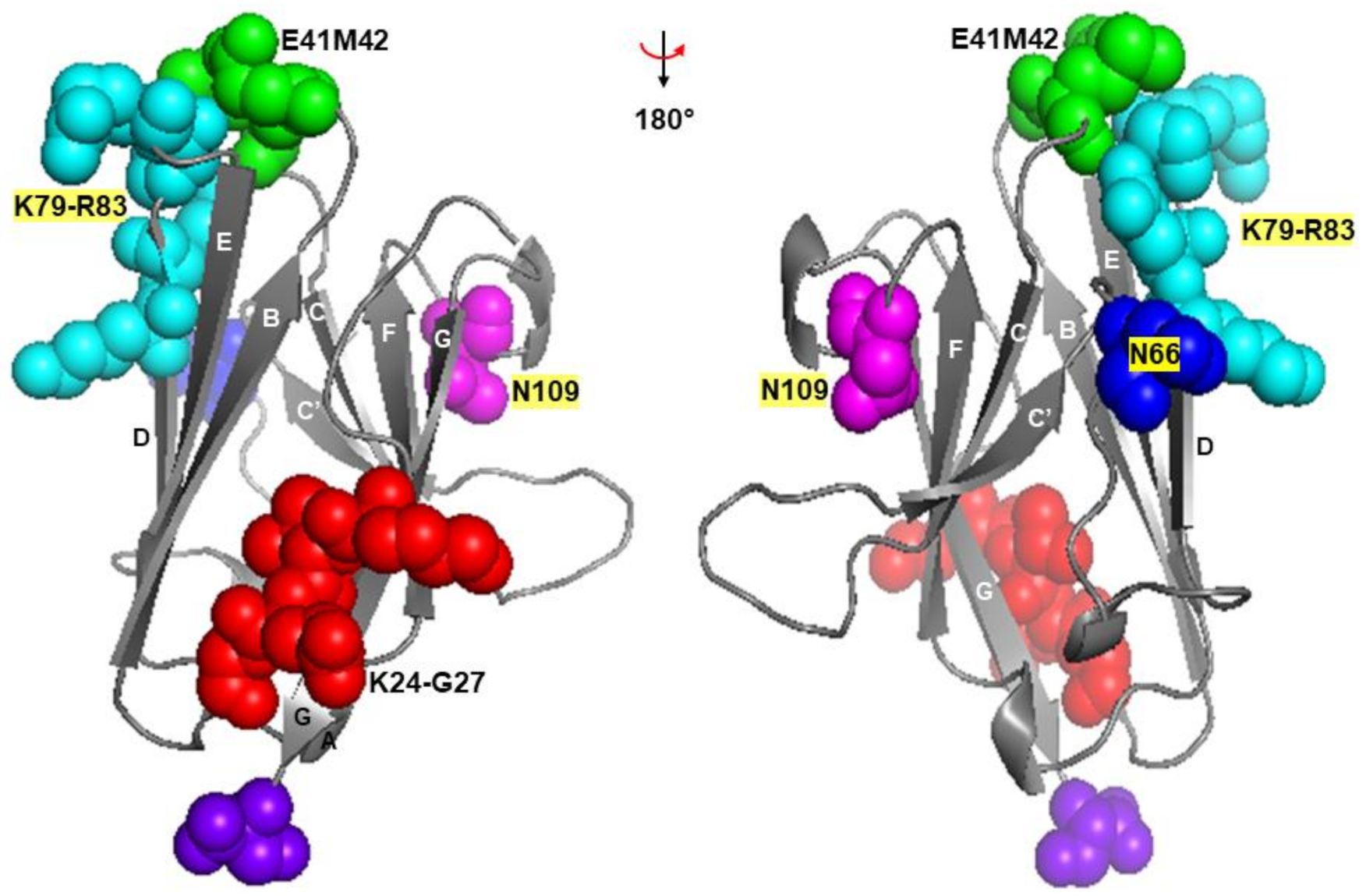
3.2.1. Asn66 in CDR2, Lys79-Arg83 in DE Loop and Asn109 in CDR3
3.2.2. Glu41 and Met42 in CDR1
3.2.3. Lys24-Gly27 in A Strand
4. Structural Aspects of FcµR and IgM
4.1. Computational Structural Modeling of Human FcµR
4.2. Recent Structural Aspects of IgM Ligand
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cooper, M.D.; Miller, J.F.A.P. Discovery of two distinctive lineages of lymphocytes, T cells and B cells, as the basis of the adaptive immune system and immunologic function: 2019 Albert Lasker Basic Medical Research Award. JAMA 2019, 322, 1247–1248. [Google Scholar] [CrossRef] [PubMed]
- Ehrenstein, M.R.; Notley, C.A. The importance of natural IgM: Scavenger, protector and regulator. Nat. Rev. Immunol. 2010, 10, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Bournazos, S.; Gupta, A.; Ravetch, J.V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 2020, 20, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Tsai, M. IgE and mast cells in allergic disease. Nat. Med. 2012, 18, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, R.C.; van de Winkel, J.G. IgA Fc receptors. Annu. Rev. Immunol. 2003, 21, 177–204. [Google Scholar] [CrossRef]
- Keyt, B.A.; Baliga, R.; Sinclair, A.M.; Carroll, S.F.; Peterson, M.S. Structure, function, and therapeutic use of IgM antibodies. Antibodies 2020, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Sutton, B.J.; Davies, A.M.; Bax, H.J.; Karagiannis, S.N. IgE antibodies: From structure to function and clinical translation. Antibodies 2019, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Pyzik, M.; Sand, K.M.K.; Hubbard, J.J.; Andersen, J.T.; Sandlie, I.; Blumberg, R.S. The neonatal Fc receptor (FcRn): A misnomer? Front. Immunol. 2019, 10, 1540. [Google Scholar] [CrossRef]
- Kubagawa, H.; Oka, S.; Kubagawa, Y.; Torii, I.; Takayama, E.; Kang, D.W.; Gartland, G.L.; Bertoli, L.F.; Mori, H.; Takatsu, H.; et al. Identity of the elusive IgM Fc receptor (FcμR) in humans. J. Exp. Med. 2009, 206, 2779–2793. [Google Scholar] [CrossRef] [Green Version]
- Klimovich, V.B. IgM and its receptors: Structural and functional aspects. Biochemistry 2011, 76, 534–549. [Google Scholar] [CrossRef] [PubMed]
- Kubagawa, H.; Oka, S.; Kubagawa, Y.; Torii, I.; Takayama, E.; Kang, D.W.; Jones, D.; Nishida, N.; Miyawaki, T.; Bertoli, L.F.; et al. The long elusive IgM Fc receptor, FcmR. J. Clin. Immunol. 2014, 34 (Suppl. 1), S35–S45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Coligan, J.E.; Morse, H.C. Emerging functions of natural IgM and its Fc receptor FCMR in immune homeostasis. Front. Immunol. 2016, 7, 99. [Google Scholar] [CrossRef] [PubMed]
- Blandino, R.; Baumgarth, N. Secreted IgM: New tricks for an old molecule. J. Leukoc. Biol. 2019, 106, 1021–1034. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, Y.; Xiong, E.; Hong, R.; Lu, Q.; Ohno, H.; Wang, J.Y. Role of the IgM Fc receptor in immunity and tolerance. Front. Immunol. 2019, 10, 529. [Google Scholar] [CrossRef]
- Kubagawa, H.; Honjo, K.; Ohkura, N.; Sakaguchi, S.; Radbruch, A.; Melchers, F.; Jani, P.K. Functional roles of the IgM Fc receptor in the immune system. Front. Immunol. 2019, 10, 945. [Google Scholar] [CrossRef]
- Hitoshi, Y.; Lorens, J.; Kitada, S.I.; Fisher, J.; LaBarge, M.; Ring, H.Z.; Francke, U.; Reed, J.C.; Kinoshita, S.; Nolan, G.P. Toso, a cell surface, specific regulator of Fas-induced apoptosis in T cells. Immunity 1998, 8, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Kubagawa, H.; Carroll, M.C.; Jacob, C.O.; Lang, K.S.; Lee, K.H.; Mak, T.; McAndrews, M.; Morse, H.C.; Nolan, G.P.; Ohno, H.; et al. Nomenclature of Toso, Fas apoptosis inhibitory molecule 3, and IgM FcR. J. Immunol. 2015, 194, 4055–4057. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Narayanan, S.; Su, S.; Childs, R.; Krzewski, K.; Borrego, F.; Weck, J.; Coligan, J.E. Toso, a functional IgM receptor, is regulated by IL-2 in T and NK cells. J. Immunol. 2012, 189, 587–597. [Google Scholar] [CrossRef]
- Shima, H.; Takatsu, H.; Fukuda, S.; Ohmae, M.; Hase, K.; Kubagawa, H.; Wang, J.Y.; Ohno, H. Identification of TOSO/FAIM3 as an Fc receptor for IgM. Int. Immunol. 2010, 22, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Honjo, K.; Kubagawa, Y.; Jones, D.M.; Dizon, B.; Zhu, Z.; Ohno, H.; Izui, S.; Kearney, J.F.; Kubagawa, H. Altered Ig levels and antibody responses in mice deficient for the Fc receptor for IgM (FcmR). Proc. Natl. Acad. Sci. USA 2012, 109, 15882–15887. [Google Scholar] [CrossRef] [Green Version]
- Ouchida, R.; Mori, H.; Hase, K.; Takatsu, H.; Kurosaki, T.; Tokuhisa, T.; Ohno, H.; Wang, J.Y. Critical role of the IgM Fc receptor in IgM homeostasis, B-cell survival, and humoral immune responses. Proc. Natl. Acad. Sci. USA 2012, 109, E2699–E2706. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, X.H.; Lang, P.A.; Lang, K.S.; Adam, D.; Fattakhova, G.; Foger, N.; Kamal, M.A.; Prilla, P.; Mathieu, S.; Wagner, C.; et al. Toso regulates the balance between apoptotic and nonapoptotic death receptor signaling by facilitating RIP1 ubiquitination. Blood 2011, 118, 598–608. [Google Scholar] [CrossRef]
- Lang, K.S.; Lang, P.A.; Meryk, A.; Pandyra, A.A.; Boucher, L.M.; Pozdeev, V.I.; Tusche, M.W.; Gothert, J.R.; Haight, J.; Wakeham, A.; et al. Involvement of Toso in activation of monocytes, macrophages, and granulocytes. Proc. Natl. Acad. Sci. USA 2013, 110, 2593–2598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, D.; Brustle, A.; Lin, G.H.; Lang, P.A.; Duncan, G.S.; Knobbe-Thomsen, C.B.; St, P.M.; Reardon, C.; Tusche, M.W.; Snow, B.; et al. Toso controls encephalitogenic immune responses by dendritic cells and regulatory T cells. Proc. Natl. Acad. Sci. USA 2014, 111, 1060–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, P.A.; Meryk, A.; Pandyra, A.A.; Brenner, D.; Brustle, A.; Xu, H.C.; Merches, K.; Lang, F.; Khairnar, V.; Sharma, P.; et al. Toso regulates differentiation and activation of inflammatory dendritic cells during persistence-prone virus infection. Cell Death Differ. 2015, 22, 164–173. [Google Scholar] [CrossRef] [Green Version]
- Gaublomme, J.T.; Yosef, N.; Lee, Y.; Gertner, R.S.; Yang, L.V.; Wu, C.; Pandolfi, P.P.; Mak, T.; Satija, R.; Shalek, A.K.; et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell 2015, 163, 1400–1412. [Google Scholar] [CrossRef] [Green Version]
- Kubli, S.P.; Vornholz, L.; Duncan, G.; Zhou, W.; Ramachandran, P.; Fortin, J.; Cox, M.; Han, S.; Nechanitzky, R.; Nechanitzky, D.; et al. Fcmr regulates mononuclear phagocyte control of anti-tumor immunity. Nat. Commun. 2019, 10, 2678. [Google Scholar] [CrossRef]
- Ravetch, J.V.; Nimmerjahn, F. Fc receptors and their role in immune regulation and inflammation. In Fundamental Immunology; Paul, W.E., Ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2008; pp. 684–705. [Google Scholar]
- Nguyen, T.T.; Klasener, K.; Zurn, C.; Castillo, P.A.; Brust-Mascher, I.; Imai, D.M.; Bevins, C.L.; Reardon, C.; Reth, M.; Baumgarth, N. The IgM receptor FcmR limits tonic BCR signaling by regulating expression of the IgM BCR. Nat. Immunol. 2017, 18, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Bergman, A.; Rutemark, C.; Ouchida, R.; Ohno, H.; Wang, J.Y.; Heyman, B. Complement-activating IgM enhances the humoral but not the T cell immune response in mice. PLoS ONE 2013, 8, e81299. [Google Scholar] [CrossRef] [PubMed]
- Horton, R.M.; Hunt, H.D.; Ho, S.N.; Pullen, J.K.; Pease., L.R. Engineering hybrid genes without the use of restriction enzymes: Gene splicing by overlap extension. Gene 1989, 77, 61–68. [Google Scholar] [CrossRef]
- Hamburger, A.E.; West, A.P., Jr.; Bjorkman, P.J. Crystal structure of a polymeric immunoglobulin binding fragment of the human polymeric immunoglobulin receptor. Structure 2004, 12, 1925–1935. [Google Scholar] [CrossRef] [Green Version]
- Stadtmueller, B.M.; Huey-Tubman, K.E.; Lopez, C.J.; Yang, Z.; Hubbell, W.L.; Bjorkman, P.J. The structure and dynamics of secretory component and its interactions with polymeric immunoglobulins. eLife 2016, 5, e10640. [Google Scholar] [CrossRef]
- Skopnik, C.M.; Al-Qaisi, K.; Calvert, R.A.; Enghard, P.; Radbruch, A.; Sutton, B.J.; Kubagawa, H. Identification of amino acid residues in human IgM Fc receptor (FcµR) critical for IgM binding. Front. Immunol. 2020, 11, 618327. [Google Scholar] [CrossRef]
- Griffin, M.; Casadio, R.; Bergamini, C.M. Transglutaminases: Nature’s biological glues. Biochem. J. 2002, 368, 377–396. [Google Scholar] [CrossRef] [Green Version]
- Al-Lazikani, B.; Lesk, A.M.; Chothia, C. Standard conformations for the canonical structures of immunoglobulins. J. Mol. Biol. 1997, 273, 927–948. [Google Scholar] [CrossRef]
- Lloyd, K.A.; Wang, J.; Urban, B.C.; Czajkowsky, D.M.; Pleass, R.J. Glycan-independent binding and internalization of human IgM to FCMR, its cognate cellular receptor. Sci. Rep. 2017, 7, 42989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, T.; Kubagawa, H.; Sanders, S.K.; Cooper, M.D. Biochemical nature of an Fcm receptor on human B-lineage cells. J. Exp. Med. 1990, 172, 1165–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Kubagawa, H.; Ohno, T.; Cooper, M.D. Characterization of an IgM Fc-binding receptor on human T cells. J. Immunol. 1993, 151, 6933–6941. [Google Scholar] [PubMed]
- Nyamboya, R.A.; Sutton, B.J.; Calvert, R.A. Mapping of the binding site for FcmR in human IgM-Fc. Biochim. Biophys. Acta Proteins Proteom. 2019, 1868, 140266. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Ruprecht, R.M. Immunoglobulin M: An ancient antiviral weapon—Rediscovered. Front. Immunol. 2020, 11, 1943. [Google Scholar] [CrossRef]
- Hiramoto, E.; Tsutsumi, A.; Suzuki, R.; Matsuoka, S.; Arai, S.; Kikkawa, M.; Miyazaki, T. The IgM pentamer is an asymmetric pentagon with an open groove that binds the AIM protein. Sci. Adv. 2018, 4, eaau1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wang, G.; Li, N.; Wang, Y.; Zhu, Q.; Chu, H.; Wu, W.; Tan, Y.; Yu, F.; Su, X.D.; et al. Structural insights into immunoglobulin M. Science 2020, 367, 1014–1017. [Google Scholar] [CrossRef] [PubMed]
- Kubagawa, H.; Skopnik, C.M.; Zimmermann, J.; Durek, P.; Chang, H.D.; Yoo, E.; Bertoli, L.F.; Honjo, K.; Radbruch, A. Authentic IgM Fc receptor (FcmR). Curr. Top. Microbiol. Immunol. 2017, 408, 25–45. [Google Scholar] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubagawa, H.; Skopnik, C.M.; Al-Qaisi, K.; Calvert, R.A.; Honjo, K.; Kubagawa, Y.; Teuber, R.; Aliabadi, P.M.; Enghard, P.; Radbruch, A.; et al. Differences between Human and Mouse IgM Fc Receptor (FcµR). Int. J. Mol. Sci. 2021, 22, 7024. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137024
Kubagawa H, Skopnik CM, Al-Qaisi K, Calvert RA, Honjo K, Kubagawa Y, Teuber R, Aliabadi PM, Enghard P, Radbruch A, et al. Differences between Human and Mouse IgM Fc Receptor (FcµR). International Journal of Molecular Sciences. 2021; 22(13):7024. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137024
Chicago/Turabian StyleKubagawa, Hiromi, Christopher M. Skopnik, Khlowd Al-Qaisi, Rosaleen A. Calvert, Kazuhito Honjo, Yoshiki Kubagawa, Ruth Teuber, Pedram Mahmoudi Aliabadi, Philipp Enghard, Andreas Radbruch, and et al. 2021. "Differences between Human and Mouse IgM Fc Receptor (FcµR)" International Journal of Molecular Sciences 22, no. 13: 7024. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137024