
The Role of Fc Receptors on the Effectiveness of Therapeutic Monoclonal Antibodies


Abstract
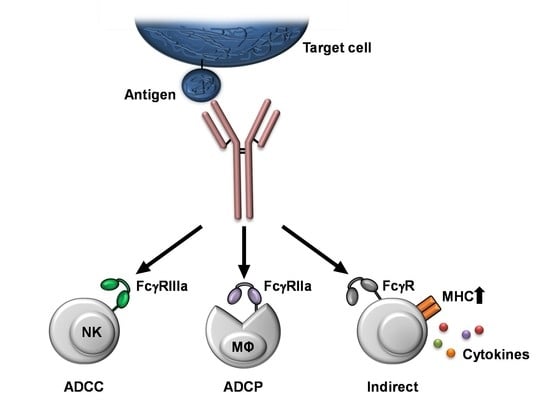
:
1. Introduction
2. Clinical Development and Approval of Therapeutic mAbs
2.1. Characterization of Therapeutic mAbs
2.2. Fc-Mediated Effector Functions as mAb Characteristic
2.3. Safety Assessment
2.4. Methods for Analyzing Fc:FcγR Interactions
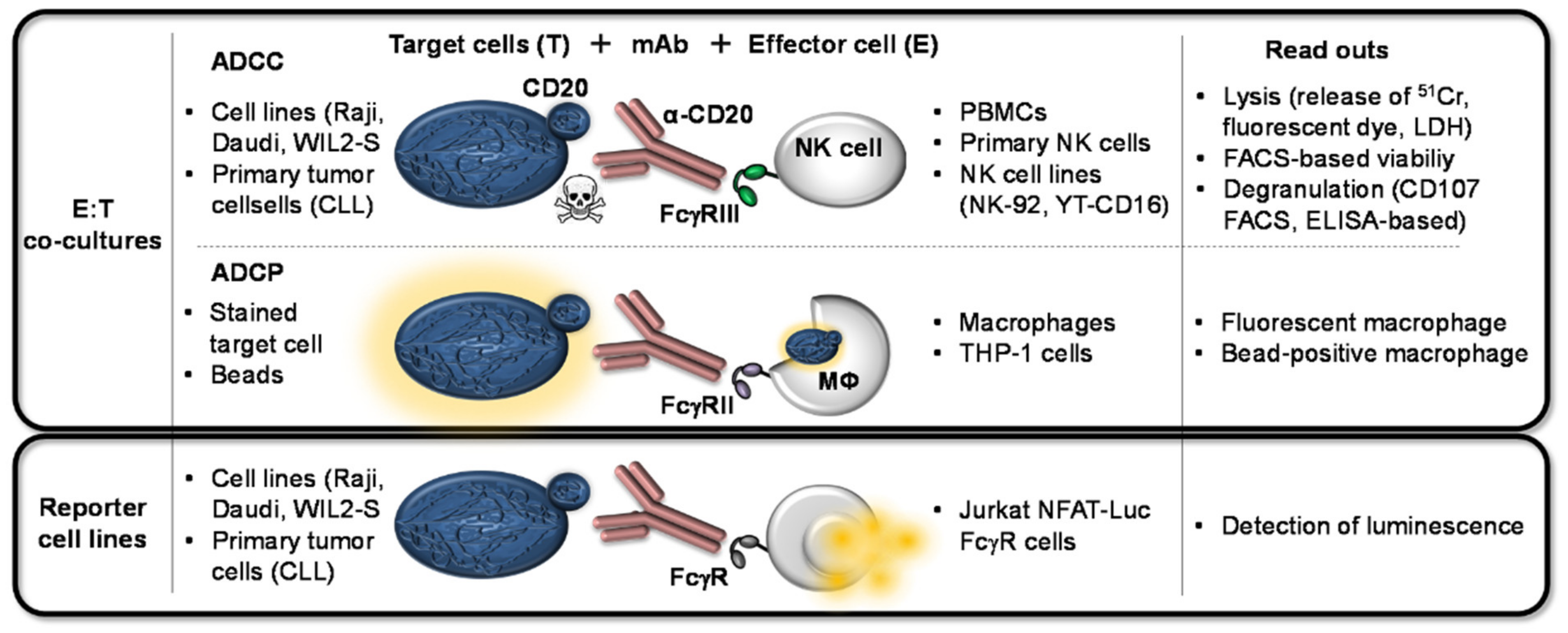
2.4.1. ADCC
2.4.2. ADCP
2.4.3. Other FcγR-Interaction/Binding Assays
2.4.4. FcRn Binding Assays
3. Fc:FcγR Interaction-Mediated Side Effects
4. Novel Strategies for the Development of Therapeutic mAbs
4.1. Engineering Methods to Address the FcγR Interaction
4.2. Alternative Antibody Formats
4.3. Further Strategies to Modify Fc:FcγR Interaction
5. Conclusions and Further Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- DiLillo, D.J.; Ravetch, J.V. Fc-Receptor Interactions Regulate Both Cytotoxic and Immunomodulatory Therapeutic Antibody Effector Functions. Cancer Immunol. Res. 2015, 3, 704–713. [Google Scholar] [CrossRef] [Green Version]
- Brandsma, A.M.; Jacobino, S.R.; Meyer, S.; Broeke, T.T.; Leusen, J.H.W. Fc receptor inside-out signaling and possible impact on antibody therapy. Immunol. Rev. 2015, 268, 74–87. [Google Scholar] [CrossRef]
- Hansel, T.T.; Kropshofer, H.; Singer, T.; Mitchell, J.A.; George, A.J.T. The safety and side effects of monoclonal antibodies. Nat. Rev. Drug Discov. 2010, 9, 325–338. [Google Scholar] [CrossRef]
- Teige, I.; Mårtensson, L.; Frendéus, B.L. Targeting the Antibody Checkpoints to Enhance Cancer Immunotherapy-Focus on FcγRIIB. Front. Immunol. 2019, 10, 481. [Google Scholar] [CrossRef] [Green Version]
- Kang, T.H.; Jung, S.T. Boosting therapeutic potency of antibodies by taming Fc domain functions. Exp. Mol. Med. 2019, 51, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Murphy, K.M.; Weaver, C. Janeway’s Immunobiology, 9th ed.; GS Garland Science Taylor & Francis Group: New York, NY, USA, 2017; ISBN 9780815345510. [Google Scholar]
- Saunders, K.O. Conceptual Approaches to Modulating Antibody Effector Functions and Circulation Half-Life. Front. Immunol. 2019, 10, 1296. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Iannello, A.; Ahmad, A. Role of antibody-dependent cell-mediated cytotoxicity in the efficacy of therapeutic anti-cancer monoclonal antibodies. Cancer Metastasis Rev. 2005, 24, 487–499. [Google Scholar] [CrossRef]
- Bruhns, P.; Jönsson, F. Mouse and human FcR effector functions. Immunol. Rev. 2015, 268, 25–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors: Old friends and new family members. Immunity 2006, 24, 19–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Ishii-Watabe, A.; Tada, M.; Kobayashi, T.; Kanayasu-Toyoda, T.; Kawanishi, T.; Yamaguchi, T. Importance of neonatal FcR in regulating the serum half-life of therapeutic proteins containing the Fc domain of human IgG1: A comparative study of the affinity of monoclonal antibodies and Fc-fusion proteins to human neonatal FcR. J. Immunol. 2010, 184, 1968–1976. [Google Scholar] [CrossRef]
- Brambell, F. The Transmission of Immunity From Mother To Young And The Catabolism Of Immunoglobulins. Lancet 1966, 288, 1087–1093. [Google Scholar] [CrossRef]
- Chauhan, A.K. Human CD4(+) T-Cells: A Role for Low-Affinity Fc Receptors. Front. Immunol. 2016, 7, 215. [Google Scholar] [CrossRef] [Green Version]
- Starbeck-Miller, G.R.; Badovinac, V.P.; Barber, D.L.; Harty, J.T. Cutting edge: Expression of FcγRIIB tempers memory CD8 T cell function in vivo. J. Immunol. 2014, 192, 35–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandor, M.; Lynch, R.G. Lymphocyte Fc receptors: The special case of T cells. Immunol. Today 1993, 14, 227–231. [Google Scholar] [CrossRef]
- Stewart, R.; Morrow, M.; Hammond, S.A.; Mulgrew, K.; Marcus, D.; Poon, E.; Watkins, A.; Mullins, S.; Chodorge, M.; Andrews, J.; et al. Identification and Characterization of MEDI4736, an Antagonistic Anti-PD-L1 Monoclonal Antibody. Cancer Immunol. Res. 2015, 3, 1052–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, G. Overview of the development of Orthoclone OKT3: Monoclonal antibody for therapeutic use in transplantation. Transplant. Proc. 1987, 19, 1–6. [Google Scholar] [PubMed]
- Cosimi, A.B. Clinical development of Orthoclone OKT3. Transplant. Proc. 1987, 19, 7–16. [Google Scholar] [PubMed]
- Norman, D.J. Mechanisms of action and overview of OKT3. Ther. Drug Monit. 1995, 17, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Paul-Ehrlich-Institut. Monoklonale Antikörper: Federal Institute for Vaccines and Biomedicines. Available online: www.pei.de/DE/arzneimittel/antikoerper/monoklonale-antikoerper/monoklonale-antikoerper-node.html (accessed on 19 July 2021).
- Strohl, W.R. Optimization of Fc-mediated effector functions of monoclonal antibodies. Curr. Opin. Biotechnol. 2009, 20, 685–691. [Google Scholar] [CrossRef]
- Wang, X.; An, Z.; Luo, W.; Xia, N.; Zhao, Q. Molecular and functional analysis of monoclonal antibodies in support of biologics development. Protein Cell 2018, 9, 74–85. [Google Scholar] [CrossRef]
- Lu, C.; Liu, D.; Liu, H.; Motchnik, P. Characterization of monoclonal antibody size variants containing extra light chains. MAbs 2013, 5, 102–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, F.R.; Morton, L.D.; Spindeldreher, S.; Kiessling, A.; Allenspach, R.; Hey, A.; Muller, P.Y.; Frings, W.; Sims, J. Safety and immunotoxicity assessment of immunomodulatory monoclonal antibodies. MAbs 2010, 2, 233–255. [Google Scholar] [CrossRef] [Green Version]
- Lynch, C.M.; Hart, B.W.; Grewal, I.S. Practical considerations for nonclinical safety evaluation of therapeutic monoclonal antibodies. MAbs 2009, 1, 2–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reusch, D.; Tejada, M.L. Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology 2015, 25, 1325–1334. [Google Scholar] [CrossRef] [Green Version]
- Beck, A.; Wagner-Rousset, E.; Bussat, M.-C.; Lokteff, M.; Klinguer-Hamour, C.; Haeuw, J.-F.; Goetsch, L.; Wurch, T.; van Dorsselaer, A.; Corvaïa, N. Trends in glycosylation, glycoanalysis and glycoengineering of therapeutic antibodies and Fc-fusion proteins. Curr. Pharm. Biotechnol. 2008, 9, 482–501. [Google Scholar] [CrossRef]
- Jefferis, R. Glycosylation of recombinant antibody therapeutics. Biotechnol. Prog. 2005, 21, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Thomann, M.; Reckermann, K.; Reusch, D.; Prasser, J.; Tejada, M.L. Fc-galactosylation modulates antibody-dependent cellular cytotoxicity of therapeutic antibodies. Mol. Immunol. 2016, 73, 69–75. [Google Scholar] [CrossRef] [PubMed]
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. Available online: www.ich.org/ (accessed on 19 July 2021).
- Cymer, F.; Thomann, M.; Wegele, H.; Avenal, C.; Schlothauer, T.; Gygax, D.; Beck, H. Oxidation of M252 but not M428 in hu-IgG1 is responsible for decreased binding to and activation of hu-FcγRIIa (His131). Biologicals 2017, 50, 125–128. [Google Scholar] [CrossRef]
- Mathur, A.; Arora, T.; Liu, L.; Crouse-Zeineddini, J.; Mukku, V. Qualification of a homogeneous cell-based neonatal Fc receptor (FcRn) binding assay and its application to studies on Fc functionality of IgG-based therapeutics. J. Immunol. Methods 2013, 390, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Ober, R.J.; Martinez, C.; Lai, X.; Zhou, J.; Ward, E.S. Exocytosis of IgG as mediated by the receptor, FcRn: An analysis at the single-molecule level. Proc. Natl. Acad. Sci. USA 2004, 101, 11076–11081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrichts, P.; Guglietta, A.; Dreier, T.; van Bragt, T.; Hanssens, V.; Hofman, E.; Vankerckhoven, B.; Verheesen, P.; Ongenae, N.; Lykhopiy, V.; et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J. Clin. Investig. 2018, 128, 4372–4386. [Google Scholar] [CrossRef] [Green Version]
- Jefferis, R. Isotype and glycoform selection for antibody therapeutics. Arch. Biochem. Biophys. 2012, 526, 159–166. [Google Scholar] [CrossRef]
- Bussiere, J.L.; Martin, P.; Horner, M.; Couch, J.; Flaherty, M.; Andrews, L.; Beyer, J.; Horvath, C. Alternative strategies for toxicity testing of species-specific biopharmaceuticals. Int. J. Toxicol. 2009, 28, 230–253. [Google Scholar] [CrossRef] [PubMed]
- Bugelski, P.J.; Martin, P.L. Concordance of preclinical and clinical pharmacology and toxicology of therapeutic monoclonal antibodies and fusion proteins: Cell surface targets. Br. J. Pharmacol. 2012, 166, 823–846. [Google Scholar] [CrossRef]
- Lo, M.; Kim, H.S.; Tong, R.K.; Bainbridge, T.W.; Vernes, J.-M.; Zhang, Y.; Lin, Y.L.; Chung, S.; Dennis, M.S.; Zuchero, Y.J.Y.; et al. Effector-attenuating Substitutions That Maintain Antibody Stability and Reduce Toxicity in Mice. J. Biol. Chem. 2017, 292, 3900–3908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein, S.L.; Gottlieb, P.D. Quantitative measurement of mouse IgG subclasses with the use of heteroantisera: The importance of allotype considerations. J. Immunol. 1977, 118, 935–942. [Google Scholar] [PubMed]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef]
- McCamish, M.; Woollett, G. The state of the art in the development of biosimilars. Clin. Pharmacol. Ther. 2012, 91, 405–417. [Google Scholar] [CrossRef]
- Prior, S.; Hufton, S.E.; Fox, B.; Dougall, T.; Rigsby, P.; Bristow, A. International standards for monoclonal antibodies to support pre- and post-marketing product consistency: Evaluation of a candidate international standard for the bioactivities of rituximab. MAbs 2018, 10, 129–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency. 20 Years of Sampling and Testing of Centrally Authorised Products. Available online: https://www.ema.europa.eu/en/news/20-years-sampling-testing-programme-medicines-authorised-eu (accessed on 20 July 2021).
- OMCL Network of the Council of Europe. The Advantages and Benefits of the CAP Surveillance Project: General Document. PA/PH/CAP (13) 32 2R. Available online: https://www.edqm.eu/medias/fichiers/the_advantages_and_benefits_of_the_cap_surveillance_project_paphcap_13_32_2r.pdf: (accessed on 20 July 2021).
- Chung, S.; Lin, Y.L.; Reed, C.; Ng, C.; Cheng, Z.J.; Malavasi, F.; Yang, J.; Quarmby, V.; Song, A.N. Characterization of in vitro antibody-dependent cell-mediated cytotoxicity activity of therapeutic antibodies–impact of effector cells. J. Immunol. Methods 2014, 407, 63–75. [Google Scholar] [CrossRef]
- Parekh, B.S.; Berger, E.; Sibley, S.; Cahya, S.; Xiao, L.; LaCerte, M.A.; Vaillancourt, P.; Wooden, S.; Gately, D. Development and validation of an antibody-dependent cell-mediated cytotoxicity-reporter gene assay. MAbs 2012, 4, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Schnueriger, A.; Grau, R.; Sondermann, P.; Schreitmueller, T.; Marti, S.; Zocher, M. Development of a quantitative, cell-line based assay to measure ADCC activity mediated by therapeutic antibodies. Mol. Immunol. 2011, 48, 1512–1517. [Google Scholar] [CrossRef] [PubMed]
- De Taeye, S.W.; Rispens, T.; Vidarsson, G. The Ligands for Human IgG and Their Effector Functions. Antibodies 2019, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- VanDerMeid, K.R.; Elliott, M.R.; Baran, A.M.; Barr, P.M.; Chu, C.C.; Zent, C.S. Cellular Cytotoxicity of Next-Generation CD20 Monoclonal Antibodies. Cancer Immunol. Res. 2018, 6, 1150–1160. [Google Scholar] [CrossRef] [Green Version]
- Kavian, N.; Hachim, A.; Poon, L.L.M.; Valkenburg, S.A. Vaccination with ADCC activating HA peptide epitopes provides partial protection from influenza infection. Vaccine 2020, 38, 5885–5890. [Google Scholar] [CrossRef]
- Asokan, M.; Dias, J.; Liu, C.; Maximova, A.; Ernste, K.; Pegu, A.; McKee, K.; Shi, W.; Chen, X.; Almasri, C.; et al. Fc-mediated effector function contributes to the in vivo antiviral effect of an HIV neutralizing antibody. Proc. Natl. Acad. Sci. USA 2020, 117, 18754–18763. [Google Scholar] [CrossRef] [PubMed]
- Mata, M.M.; Mahmood, F.; Sowell, R.T.; Baum, L.L. Effects of cryopreservation on effector cells for antibody dependent cell-mediated cytotoxicity (ADCC) and natural killer (NK) cell activity in (51)Cr-release and CD107a assays. J. Immunol. Methods 2014, 406, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Tada, M.; Ishii-Watabe, A.; Suzuki, T.; Kawasaki, N. Development of a cell-based assay measuring the activation of FcγRIIa for the characterization of therapeutic monoclonal antibodies. PLoS ONE 2014, 9, e95787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pross, H.F.; Maroun, J.A. The standardization of NK cell assays for use in studies of biological response modifiers. J. Immunol. Methods 1984, 68, 235–249. [Google Scholar] [CrossRef]
- Davis, Z.B.; Ward, J.P.; Barker, E. Preparation and use of HIV-1 infected primary CD4+ T-cells as target cells in natural killer cell cytotoxic assays. J. Vis. Exp. 2011, 49, e2668. [Google Scholar] [CrossRef]
- Krzewski, K.; Gil-Krzewska, A.; Nguyen, V.; Peruzzi, G.; Coligan, J.E. LAMP1/CD107a is required for efficient perforin delivery to lytic granules and NK-cell cytotoxicity. Blood 2013, 121, 4672–4683. [Google Scholar] [CrossRef]
- Miller, A.S.; Tejada, M.L.; Gazzano-Santoro, H. Development of an ELISA based bridging assay as a surrogate measure of ADCC. J. Immunol. Methods 2012, 385, 45–50. [Google Scholar] [CrossRef]
- Ackerman, M.E.; Moldt, B.; Wyatt, R.T.; Dugast, A.-S.; McAndrew, E.; Tsoukas, S.; Jost, S.; Berger, C.T.; Sciaranghella, G.; Liu, Q.; et al. A robust, high-throughput assay to determine the phagocytic activity of clinical antibody samples. J. Immunol. Methods 2011, 366, 8–19. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.M.; Roy, V.; Gunn, B.M.; Huang, L.; Edeling, M.A.; Mack, M.; Fremont, D.H.; Doranz, B.J.; Johnson, S.; Alter, G.; et al. Optimal therapeutic activity of monoclonal antibodies against chikungunya virus requires Fc-FcγR interaction on monocytes. Sci. Immunol. 2019, 4, eaav5062. [Google Scholar] [CrossRef] [PubMed]
- Rijkers, M.; Saris, A.; Heidt, S.; Mulder, A.; Porcelijn, L.; Claas, F.H.J.; Bierings, R.; Leebeek, F.W.G.; Jansen, A.J.G.; Vidarsson, G.; et al. A subset of anti-HLA antibodies induces FcγRIIa-dependent platelet activation. Haematologica 2018, 103, 1741–1752. [Google Scholar] [CrossRef] [Green Version]
- Corrales-Aguilar, E.; Trilling, M.; Hunold, K.; Fiedler, M.; Le, V.T.K.; Reinhard, H.; Ehrhardt, K.; Mercé-Maldonado, E.; Aliyev, E.; Zimmermann, A.; et al. Human cytomegalovirus Fcγ binding proteins gp34 and gp68 antagonize Fcγ receptors I, II and III. PLoS Pathog. 2014, 10, e1004131. [Google Scholar] [CrossRef] [Green Version]
- Corrales-Aguilar, E.; Trilling, M.; Reinhard, H.; Mercé-Maldonado, E.; Widera, M.; Schaal, H.; Zimmermann, A.; Mandelboim, O.; Hengel, H. A novel assay for detecting virus-specific antibodies triggering activation of Fcγ receptors. J. Immunol. Methods 2013, 387, 21–35. [Google Scholar] [CrossRef]
- Chen, H.; Maul-Pavicic, A.; Holzer, M.; Salzer, U.; Chevalier, N.; Voll, R.E.; Hengel, H.; Kolb, P. Immune complex solubility and size govern Fc-gamma receptor responses: A scalable cell-based reporter system. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zuercher, A.W.; Spirig, R.; Baz Morelli, A.; Rowe, T.; Käsermann, F. Next-generation Fc receptor-targeting biologics for autoimmune diseases. Autoimmun. Rev. 2019, 18, 102366. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Lee, H.Y.; Wong, P.Y.; Jiang, G.; Gazzano-Santoro, H. Development and applications of AlphaScreen-based FcRn binding assay to characterize monoclonal antibodies. J. Immunol. Methods 2015, 420, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Nanovskaya, T.; Patrikeeva, S.; Cochran, E.; Parge, V.; Guess, J.; Schaeck, J.; Choudhury, A.; Ahmed, M.; Ling, L.E. M281, an anti-FcRn antibody, inhibits IgG transfer in a human ex vivo placental perfusion model. Am. J. Obstet. Gynecol. 2019, 220, 498.e1–498.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.; Barnes, L.; Sheahan, M.; Tsai, H.; Goldstein, G. Orthoclone OKT3 treatment of acute renal allograft rejection. Transplant. Proc. 1987, 19, 46. [Google Scholar] [PubMed]
- Goldstein, G.; Norman, D.J.; Henell, K.R.; Smith, I.L. Pharmacokinetic study of orthoclone OKT3 serum levels during treatment of acute renal allograft rejection. Transplantation 1988, 46, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Alegre, M.L.; Collins, A.M.; Pulito, V.L.; Brosius, R.A.; Olson, W.C.; Zivin, R.A.; Knowles, R.; Thistlethwaite, J.R.; Jolliffe, L.K.; Bluestone, J.A. Effect of a single amino acid mutation on the activating and immunosuppressive properties of a "humanized" OKT3 monoclonal antibody. J. Immunol. 1992, 148, 3461–3468. [Google Scholar]
- Masharani, U.B.; Becker, J. Teplizumab therapy for type 1 diabetes. Expert Opin. Biol. Ther. 2010, 10, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Herold, K.C.; Gitelman, S.E.; Masharani, U.; Hagopian, W.; Bisikirska, B.; Donaldson, D.; Rother, K.; Diamond, B.; Harlan, D.M.; Bluestone, J.A. A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes 2005, 54, 1763–1769. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Alegre, M.L.; Varga, S.S.; Rothermel, A.L.; Collins, A.M.; Pulito, V.L.; Hanna, L.S.; Dolan, K.P.; Parren, P.W.; Bluestone, J.A.; et al. In vitro characterization of five humanized OKT3 effector function variant antibodies. Cell. Immunol. 2000, 200, 16–26. [Google Scholar] [CrossRef]
- TeGenero Immuno Therapeutics. Investigator’s Brochure: TGN1412 Humanized Agonistic Anti CD28 Monoclonal Antibody. Available online: www.circare.org/foia5/tgn1412.htm#docs (accessed on 18 July 2021).
- Waibler, Z.; Sender, L.Y.; Merten, C.; Hartig, R.; Kliche, S.; Gunzer, M.; Reichardt, P.; Kalinke, U.; Schraven, B. Signaling signatures and functional properties of anti-human CD28 superagonistic antibodies. PLoS ONE 2008, 3, e1708. [Google Scholar] [CrossRef]
- Dudek, S.; Weißmüller, S.; Anzaghe, M.; Miller, L.; Sterr, S.; Hoffmann, K.; Hengel, H.; Waibler, Z. Human Fcγ receptors compete for TGN1412 binding that determines the antibody’s effector function. Eur. J. Immunol. 2019, 49, 1117–1126. [Google Scholar] [CrossRef]
- Jefferis, R.; Lund, J.; Pound, J. Molecular definition of interaction sites on human IgG for Fc receptors (huFcγR). Mol. Immunol. 1990, 27, 1237–1240. [Google Scholar] [CrossRef]
- Yamane-Ohnuki, N.; Kinoshita, S.; Inoue-Urakubo, M.; Kusunoki, M.; Iida, S.; Nakano, R.; Wakitani, M.; Niwa, R.; Sakurada, M.; Uchida, K.; et al. Establishment of FUT8 knockout Chinese hamster ovary cells: An ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol. Bioeng. 2004, 87, 614–622. [Google Scholar] [CrossRef]
- Niwa, R.; Natsume, A.; Uehara, A.; Wakitani, M.; Iida, S.; Uchida, K.; Satoh, M.; Shitara, K. IgG subclass-independent improvement of antibody-dependent cellular cytotoxicity by fucose removal from Asn297-linked oligosaccharides. J. Immunol. Methods 2005, 306, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Imai-Nishiya, H. Double knockdown of α1,6-fucosyltransferase (FUT8) and GDP-mannose 4,6-dehydratase (GMD) in antibody-producing cells: A new strategy for generating fully non-fucosylated therapeutic antibodies with enhanced ADCC. BMC Biotechnol. 2007, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheney, C.M.; Stephens, D.M.; Mo, X.; Rafiq, S.; Butchar, J.; Flynn, J.M.; Jones, J.A.; Maddocks, K.; O’Reilly, A.; Ramachandran, A.; et al. Ocaratuzumab, an Fc-engineered antibody demonstrates enhanced antibody-dependent cell-mediated cytotoxicity in chronic lymphocytic leukemia. MAbs 2014, 6, 749–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jefferis, R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat. Rev. Drug Discov. 2009, 8, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S.; Daëron, M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009, 113, 3716–3725. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [Green Version]
- Ball, C.; Fox, B.; Hufton, S.; Sharp, G.; Poole, S.; Stebbings, R.; Eastwood, D.; Findlay, L.; Parren, P.W.H.I.; Thorpe, R.; et al. Antibody C region influences TGN1412-like functional activity in vitro. J. Immunol. 2012, 189, 5831–5840. [Google Scholar] [CrossRef] [Green Version]
- Maggi, E.; Vultaggio, A.; Matucci, A. Acute infusion reactions induced by monoclonal antibody therapy. Expert Rev. Clin. Immunol. 2011, 7, 55–63. [Google Scholar] [CrossRef]
- Seifert, M.; Lubitz, A.; Trommer, J.; Könnig, D.; Korus, G.; Marx, U.; Volk, H.-D.; Duda, G.; Kasper, G.; Lehmann, K.; et al. Crosstalk between immune cells and mesenchymal stromal cells in a 3D bioreactor system. Int. J. Artif. Organs 2012, 35, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, H.S.; Kasi, P.M. Rituximab and cytokine release syndrome. Case Rep. Oncol. 2012, 5, 134–141. [Google Scholar] [CrossRef]
- Makino, K. Treatment strategy for reducing the risk of rituximab-induced cytokine release syndrome in patients with intravascular large B-cell lymphoma: A case report and review of the literature. J. Med. Case Rep. 2013, 7, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Panelli, M.C.; White, R.; Foster, M.; Martin, B.; Wang, E.; Smith, K.; Marincola, F.M. Forecasting the cytokine storm following systemic interleukin (IL)-2 administration. J. Transl. Med. 2004, 2, 17. [Google Scholar] [CrossRef] [Green Version]
- Wing, M. Monoclonal antibody first dose cytokine release syndromes-mechanisms and prediction. J. Immunotoxicol. 2008, 5, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Stebbings, R.; Findlay, L.; Edwards, C.; Eastwood, D.; Bird, C.; North, D.; Mistry, Y.; Dilger, P.; Liefooghe, E.; Cludts, I.; et al. "Cytokine storm" in the phase I trial of monoclonal antibody TGN1412: Better understanding the causes to improve preclinical testing of immunotherapeutics. J. Immunol. 2007, 179, 3325–3331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, M.H.; Smith, R.I.; Morrison, S.L. Structural features of human immunoglobulin G that determine isotype-specific differences in complement activation. J. Exp. Med. 1993, 178, 661–667. [Google Scholar] [CrossRef]
- Jefferis, R. Recombinant antibody therapeutics: The impact of glycosylation on mechanisms of action. Trends Pharmacol. Sci. 2009, 30, 356–362. [Google Scholar] [CrossRef]
- Salfeld, J.G. Isotype selection in antibody engineering. Nat. Biotechnol. 2007, 25, 1369–1372. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Gao, X.; Gong, R. Engineering of Fc Fragments with Optimized Physicochemical Properties Implying Improvement of Clinical Potentials for Fc-Based Therapeutics. Front. Immunol. 2017, 8, 1860. [Google Scholar] [CrossRef] [PubMed]
- Jefferis, R. Antibody therapeutics: Isotype and glycoform selection. Expert Opin. Biol. Ther. 2007, 7, 1401–1413. [Google Scholar] [CrossRef]
- Liu, R.; Oldham, R.J.; Teal, E.; Beers, S.A.; Cragg, M.S. Fc-Engineering for Modulated Effector Functions-Improving Antibodies for Cancer Treatment. Antibodies 2020, 9, 64. [Google Scholar] [CrossRef]
- Sterlin, D.; Gorochov, G. When Therapeutic IgA Antibodies Might Come of Age. Pharmacology 2021, 106, 9–19. [Google Scholar] [CrossRef]
- Wallace, A.L.; Schneider, M.I.; Toomey, J.R.; Schneider, R.M.; Klempner, M.S.; Wang, Y.; Cavacini, L.A. IgA as a potential candidate for enteric monoclonal antibody therapeutics with improved gastrointestinal stability. Vaccine 2020, 38, 7490–7497. [Google Scholar] [CrossRef]
- Matson, D.O.; O’Ryan, M.L.; Herrera, I.; Pickering, L.K.; Estes, M.K. Fecal antibody responses to symptomatic and asymptomatic rotavirus infections. J. Infect. Dis. 1993, 167, 577–583. [Google Scholar] [CrossRef] [PubMed]
- O’Ryan, M.L.; Matson, D.O.; Estes, M.K.; Pickering, L.K. Acquisition of serum isotype-specific and G type-specific antirotavirus antibodies among children in day care centers. Pediatric Infect. Dis. J. 1994, 13, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Saif, L.; Yuan, L.; Ward, L.; To, T. Comparative studies of the pathogenesis, antibody immune responses, and homologous protection to porcine and human rotaviruses in gnotobiotic piglets. Adv. Exp. Med. Biol. 1997, 412, 397–403. [Google Scholar] [CrossRef]
- Tô, T.L.; Ward, L.A.; Yuan, L.; Saif, L.J. Serum and intestinal isotype antibody responses and correlates of protective immunity to human rotavirus in a gnotobiotic pig model of disease. J. Gen. Virol. 1998, 79, 2661–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, N.; Burns, J.W.; Bracy, L.; Greenberg, H.B. Comparison of mucosal and systemic humoral immune responses and subsequent protection in mice orally inoculated with a homologous or a heterologous rotavirus. J. Virol. 1994, 68, 7766–7773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onorato, I.M.; Modlin, J.F.; McBean, A.M.; Thoms, M.L.; Losonsky, G.A.; Bernier, R.H. Mucosal immunity induced by enhance-potency inactivated and oral polio vaccines. J. Infect. Dis. 1991, 163, 1–6. [Google Scholar] [CrossRef]
- Ogra, P.L.; Karzon, D.T.; Righthand, F.; MacGillivray, M. Immunoglobulin response in serum and secretions after immunization with live and inactivated poliovaccine and natural infection. N. Engl. J. Med. 1968, 279, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Resik, S.; Tejeda, A.; Sutter, R.W.; Diaz, M.; Sarmiento, L.; Alemañi, N.; Garcia, G.; Fonseca, M.; Hung, L.H.; Kahn, A.-L.; et al. Priming after a fractional dose of inactivated poliovirus vaccine. N. Engl. J. Med. 2013, 368, 416–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazanec, M.B.; Coudret, C.L.; Fletcher, D.R. Intracellular neutralization of influenza virus by immunoglobulin A anti-hemagglutinin monoclonal antibodies. J. Virol. 1995, 69, 1339–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazanec, M.B.; Kaetzel, C.S.; Lamm, M.E.; Fletcher, D.; Peterra, J.; Nedrud, J.G. Intracellular neutralization of Sendai and influenza viruses by IgA monoclonal antibodies. Adv. Exp. Med. Biol. 1995, 371, 651–654. [Google Scholar] [CrossRef]
- Sterlin, D.; Mathian, A.; Miyara, M.; Mohr, A.; Anna, F.; Claër, L.; Quentric, P.; Fadlallah, J.; Devilliers, H.; Ghillani, P.; et al. IgA dominates the early neutralizing antibody response to SARS-CoV-2. Sci. Transl. Med. 2020, 13, eabd2223. [Google Scholar] [CrossRef]
- Burton, D.R.; Jefferis, R.; Partridge, L.J.; Woof, J.M. Molecular recognition of antibody (IgG) by cellular Fc receptor (FcRI). Mol. Immunol. 1988, 25, 1175–1181. [Google Scholar] [CrossRef]
- Canfield, S.M.; Morrison, S.L. The binding affinity of human IgG for its high affinity Fc receptor is determined by multiple amino acids in the CH2 domain and is modulated by the hinge region. J. Exp. Med. 1991, 173, 1483–1491. [Google Scholar] [CrossRef] [Green Version]
- Chappel, M.S.; Isenman, D.E.; Everett, M.; Xu, Y.Y.; Dorrington, K.J.; Klein, M.H. Identification of the Fc gamma receptor class I binding site in human IgG through the use of recombinant IgG1/IgG2 hybrid and point-mutated antibodies. Proc. Natl. Acad. Sci. USA 1991, 88, 9036–9040. [Google Scholar] [CrossRef] [Green Version]
- Duncan, A.R.; Woof, J.M.; Partridge, L.J.; Burton, D.R.; Winter, G. Localization of the binding site for the human high-affinity Fc receptor on IgG. Nature 1988, 332, 563–564. [Google Scholar] [CrossRef]
- Hulett, M.D.; Witort, E.; Brinkworth, R.I.; McKenzie, I.F.; Hogarth, P.M. Identification of the IgG binding site of the human low affinity receptor for IgG Fc gamma RII. Enhancement and ablation of binding by site-directed mutagenesis. J. Biol. Chem. 1994, 269, 15287–15293. [Google Scholar] [CrossRef]
- Partridge, L.; Woof, J.; Jefferis, R.; Burton, D. The use of anti-IgG monoclonal antibodies in mapping the monocyte receptor site on IgG. Mol. Immunol. 1986, 23, 1365–1372. [Google Scholar] [CrossRef]
- Brüggemann, M.; Williams, G.T.; Bindon, C.I.; Clark, M.R.; Walker, M.R.; Jefferis, R.; Waldmann, H.; Neuberger, M.S. Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J. Exp. Med. 1987, 166, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Natsume, A.; In, M.; Takamura, H.; Nakagawa, T.; Shimizu, Y.; Kitajima, K.; Wakitani, M.; Ohta, S.; Satoh, M.; Shitara, K.; et al. Engineered antibodies of IgG1/IgG3 mixed isotype with enhanced cytotoxic activities. Cancer Res. 2008, 68, 3863–3872. [Google Scholar] [CrossRef] [Green Version]
- Van der Horst, H.J.; Nijhof, I.S.; Mutis, T.; Chamuleau, M.E.D. Fc-Engineered Antibodies with Enhanced Fc-Effector Function for the Treatment of B-Cell Malignancies. Cancers 2020, 12, 3041. [Google Scholar] [CrossRef] [PubMed]
- An, Z.; Forrest, G.; Moore, R.; Cukan, M.; Haytko, P.; Huang, L.; Vitelli, S.; Zhao, J.Z.; Lu, P.; Hua, J.; et al. IgG2m4, an engineered antibody isotype with reduced Fc function. MAbs 2009, 1, 572–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- US20150337053A1–Antibody Fc Mutants with Ablated Effector Functions–Google Patents. Available online: https://patents.google.com/patent/US20150337053A1/en (accessed on 27 July 2021).
- Vafa, O.; Gilliland, G.L.; Brezski, R.J.; Strake, B.; Wilkinson, T.; Lacy, E.R.; Scallon, B.; Teplyakov, A.; Malia, T.J.; Strohl, W.R. An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods 2014, 65, 114–126. [Google Scholar] [CrossRef]
- Rother, R.P.; Rollins, S.A.; Mojcik, C.F.; Brodsky, R.A.; Bell, L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat. Biotechnol. 2007, 25, 1256–1264. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Keremane, S.R.; Vielmetter, J.; Bjorkman, P.J. Structural characterization of GASDALIE Fc bound to the activating Fc receptor FcγRIIIa. J. Struct. Biol. 2016, 194, 78–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazar, G.A.; Dang, W.; Karki, S.; Vafa, O.; Peng, J.S.; Hyun, L.; Chan, C.; Chung, H.S.; Eivazi, A.; Yoder, S.C.; et al. Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. USA 2006, 103, 4005–4010. [Google Scholar] [CrossRef] [Green Version]
- Oganesyan, V.; Damschroder, M.M.; Leach, W.; Wu, H.; Dall’Acqua, W.F. Structural characterization of a mutated, ADCC-enhanced human Fc fragment. Mol. Immunol. 2008, 45, 1872–1882. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.; DiLillo, D.J.; Bournazos, S.; Li, F.; Ravetch, J.V. Mouse model recapitulating human Fcγ receptor structural and functional diversity. Proc. Natl. Acad. Sci. USA 2012, 109, 6181–6186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavenhagen, J.B.; Gorlatov, S.; Tuaillon, N.; Rankin, C.T.; Li, H.; Burke, S.; Huang, L.; Vijh, S.; Johnson, S.; Bonvini, E.; et al. Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcgamma receptors. Cancer Res. 2007, 67, 8882–8890. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.L.B.; Czerniecki, B.J. Clinical development of immunotherapies for HER2+ breast cancer: A review of HER2-directed monoclonal antibodies and beyond. NPJ Breast Cancer 2020, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oganesyan, V.; Damschroder, M.M.; Woods, R.M.; Cook, K.E.; Wu, H.; Dall’Acqua, W.F. Structural characterization of a human Fc fragment engineered for extended serum half-life. Mol. Immunol. 2009, 46, 1750–1755. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.; Hammond, S.A.; Oberst, M.; Wilkinson, R.W. The role of Fc gamma receptors in the activity of immunomodulatory antibodies for cancer. J. Immunother. Cancer 2014, 2, 29. [Google Scholar] [CrossRef] [Green Version]
- Shields, R.L.; Namenuk, A.K.; Hong, K.; Meng, Y.G.; Rae, J.; Briggs, J.; Xie, D.; Lai, J.; Stadlen, A.; Li, B.; et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J. Biol. Chem. 2001, 276, 6591–6604. [Google Scholar] [CrossRef] [Green Version]
- Inman, B.A.; Longo, T.A.; Ramalingam, S.; Harrison, M.R. Atezolizumab: A PD-L1-Blocking Antibody for Bladder Cancer. Clin. Cancer Res. 2017, 23, 1886–1890. [Google Scholar] [CrossRef] [Green Version]
- Arnold, J.N.; Wormald, M.R.; Sim, R.B.; Rudd, P.M.; Dwek, R.A. The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu. Rev. Immunol. 2007, 25, 21–50. [Google Scholar] [CrossRef] [PubMed]
- Forthal, D.N.; Gach, J.S.; Landucci, G.; Jez, J.; Strasser, R.; Kunert, R.; Steinkellner, H. Fc-glycosylation influences Fcγ receptor binding and cell-mediated anti-HIV activity of monoclonal antibody 2G12. J. Immunol. 2010, 185, 6876–6882. [Google Scholar] [CrossRef] [Green Version]
- Kao, D.; Danzer, H.; Collin, M.; Groß, A.; Eichler, J.; Stambuk, J.; Lauc, G.; Lux, A.; Nimmerjahn, F. A Monosaccharide Residue Is Sufficient to Maintain Mouse and Human IgG Subclass Activity and Directs IgG Effector Functions to Cellular Fc Receptors. Cell Rep. 2015, 13, 2376–2385. [Google Scholar] [CrossRef] [Green Version]
- Bolt, S.; Routledge, E.; Lloyd, I.; Chatenoud, L.; Pope, H.; Gorman, S.D.; Clark, M.; Waldmann, H. The generation of a humanized, non-mitogenic CD3 monoclonal antibody which retains in vitro immunosuppressive properties. Eur. J. Immunol. 1993, 23, 403–411. [Google Scholar] [CrossRef]
- Leabman, M.K.; Meng, Y.G.; Kelley, R.F.; DeForge, L.E.; Cowan, K.J.; Iyer, S. Effects of altered FcγR binding on antibody pharmacokinetics in cynomolgus monkeys. MAbs 2013, 5, 896–903. [Google Scholar] [CrossRef] [Green Version]
- Tao, M.H.; Morrison, S.L. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J. Immunol. 1989, 143, 2595–2601. [Google Scholar]
- Walker, M.R.; Lund, J.; Thompson, K.M.; Jefferis, R. Aglycosylation of human IgG1 and IgG3 monoclonal antibodies can eliminate recognition by human cells expressing Fc gamma RI and/or Fc gamma RII receptors. Biochem. J. 1989, 259, 347–353. [Google Scholar] [CrossRef]
- Sazinsky, S.L.; Ott, R.G.; Silver, N.W.; Tidor, B.; Ravetch, J.V.; Wittrup, K.D. Aglycosylated immunoglobulin G1 variants productively engage activating Fc receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 20167–20172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, M.-S.; Na, J.-H.; Yu, Y.G.; Kim, J.-Y.; Jeong, C.; Jung, S.T. Structural consequences of aglycosylated IgG Fc variants evolved for FcγRI binding. Mol. Immunol. 2015, 67, 350–356. [Google Scholar] [CrossRef]
- Jung, S.T.; Reddy, S.T.; Kang, T.H.; Borrok, M.J.; Sandlie, I.; Tucker, P.W.; Georgiou, G. Aglycosylated IgG variants expressed in bacteria that selectively bind FcgammaRI potentiate tumor cell killing by monocyte-dendritic cells. Proc. Natl. Acad. Sci. USA 2010, 107, 604–609. [Google Scholar] [CrossRef] [Green Version]
- Jo, M.; Kwon, H.S.; Lee, K.-H.; Lee, J.C.; Jung, S.T. Engineered aglycosylated full-length IgG Fc variants exhibiting improved FcγRIIIa binding and tumor cell clearance. MAbs 2018, 10, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.W.; Jo, M.; Ko, S.; Kwon, H.S.; Lim, C.S.; Ko, B.J.; Lee, J.C.; Jung, S.T. Optimal combination of beneficial mutations for improved ADCC effector function of aglycosylated antibodies. Mol. Immunol. 2019, 114, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Shields, R.L.; Lai, J.; Keck, R.; O’Connell, L.Y.; Hong, K.; Meng, Y.G.; Weikert, S.H.A.; Presta, L.G. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J. Biol. Chem. 2002, 277, 26733–26740. [Google Scholar] [CrossRef] [Green Version]
- Shinkawa, T.; Nakamura, K.; Yamane, N.; Shoji-Hosaka, E.; Kanda, Y.; Sakurada, M.; Uchida, K.; Anazawa, H.; Satoh, M.; Yamasaki, M.; et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J. Biol. Chem. 2003, 278, 3466–3473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, A.; Shoji-Hosaka, E.; Nakamura, K.; Wakitani, M.; Uchida, K.; Kakita, S.; Tsumoto, K.; Kumagai, I.; Shitara, K. Fucose depletion from human IgG1 oligosaccharide enhances binding enthalpy and association rate between IgG1 and FcgammaRIIIa. J. Mol. Biol. 2004, 336, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Matsumiya, S.; Yamaguchi, Y.; Saito, J.; Nagano, M.; Sasakawa, H.; Otaki, S.; Satoh, M.; Shitara, K.; Kato, K. Structural comparison of fucosylated and nonfucosylated Fc fragments of human immunoglobulin G1. J. Mol. Biol. 2007, 368, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, C.; Grau, S.; Jäger, C.; Sondermann, P.; Brünker, P.; Waldhauer, I.; Hennig, M.; Ruf, A.; Rufer, A.C.; Stihle, M.; et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc. Natl. Acad. Sci. USA 2011, 108, 12669–12674. [Google Scholar] [CrossRef] [Green Version]
- Subedi, G.P.; Barb, A.W. The Structural Role of Antibody N-Glycosylation in Receptor Interactions. Structure 2015, 23, 1573–1583. [Google Scholar] [CrossRef] [Green Version]
- Beck, A.; Reichert, J.M. Marketing approval of mogamulizumab: A triumph for glyco-engineering. MAbs 2012, 4, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Utsunomiya, A.; Tobinai, K.; Tsukasaki, K.; Uike, N.; Uozumi, K.; Yamaguchi, K.; Yamada, Y.; Hanada, S.; Tamura, K.; et al. Phase I study of KW-0761, a defucosylated humanized anti-CCR4 antibody, in relapsed patients with adult T-cell leukemia-lymphoma and peripheral T-cell lymphoma. J. Clin. Oncol. 2010, 28, 1591–1598. [Google Scholar] [CrossRef]
- Wang, B.; Yan, L.; Yao, Z.; Roskos, L.K. Population Pharmacokinetics and Pharmacodynamics of Benralizumab in Healthy Volunteers and Patients With Asthma. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 249–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elias, S.; Kahlon, S.; Kotzur, R.; Kaynan, N.; Mandelboim, O. Obinutuzumab activates FcγRI more potently than other anti-CD20 antibodies in chronic lymphocytic leukemia (CLL). Onco. Immunol. 2018, 7, e1428158. [Google Scholar] [CrossRef]
- Marcus, R.; Davies, A.; Ando, K.; Klapper, W.; Opat, S.; Owen, C.; Phillips, E.; Sangha, R.; Schlag, R.; Seymour, J.F.; et al. Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. N. Engl. J. Med. 2017, 377, 1331–1344. [Google Scholar] [CrossRef]
- Marcus, R.E.; Davies, A.J.; Ando, K.; Klapper, W.; Opat, S.; Owen, C.J.; Phillips, E.H.; Sangha, R.; Schlag, R.; Seymour, J.F.; et al. Obinutuzumab-Based Induction and Maintenance Prolongs Progression-Free Survival (PFS) in Patients with Previously Untreated Follicular Lymphoma: Primary Results of the Randomized Phase 3 GALLIUM Study. Blood 2016, 128, 6. [Google Scholar] [CrossRef]
- Alduaij, W.; Ivanov, A.; Honeychurch, J.; Cheadle, E.J.; Potluri, S.; Lim, S.H.; Shimada, K.; Chan, C.H.T.; Tutt, A.; Beers, S.A.; et al. Novel type II anti-CD20 monoclonal antibody (GA101) evokes homotypic adhesion and actin-dependent, lysosome-mediated cell death in B-cell malignancies. Blood 2011, 117, 4519–4529. [Google Scholar] [CrossRef]
- Mössner, E.; Brünker, P.; Moser, S.; Püntener, U.; Schmidt, C.; Herter, S.; Grau, R.; Gerdes, C.; Nopora, A.; van Puijenbroek, E.; et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood 2010, 115, 4393–4402. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Lammens, A.; Schäfer, W.; Georges, G.; Schwaiger, M.; Mössner, E.; Hopfner, K.-P.; Umaña, P.; Niederfellner, G. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. MAbs 2013, 5, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Niederfellner, G.; Lammens, A.; Mundigl, O.; Georges, G.J.; Schaefer, W.; Schwaiger, M.; Franke, A.; Wiechmann, K.; Jenewein, S.; Slootstra, J.W.; et al. Epitope characterization and crystal structure of GA101 provide insights into the molecular basis for type I/II distinction of CD20 antibodies. Blood 2011, 118, 358–367. [Google Scholar] [CrossRef] [Green Version]
- Iida, S.; Misaka, H.; Inoue, M.; Shibata, M.; Nakano, R.; Yamane-Ohnuki, N.; Wakitani, M.; Yano, K.; Shitara, K.; Satoh, M. Nonfucosylated therapeutic IgG1 antibody can evade the inhibitory effect of serum immunoglobulin G on antibody-dependent cellular cytotoxicity through its high binding to FcgammaRIIIa. Clin. Cancer Res. 2006, 12, 2879–2887. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, Y.; Nimmerjahn, F.; Ravetch, J.V. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 2006, 313, 670–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scallon, B.J.; Tam, S.H.; McCarthy, S.G.; Cai, A.N.; Raju, T.S. Higher levels of sialylated Fc glycans in immunoglobulin G molecules can adversely impact functionality. Mol. Immunol. 2007, 44, 1524–1534. [Google Scholar] [CrossRef]
- Anthony, R.M.; Nimmerjahn, F.; Ashline, D.J.; Reinhold, V.N.; Paulson, J.C.; Ravetch, J.V. Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science 2008, 320, 373–376. [Google Scholar] [CrossRef] [Green Version]
- Quast, I.; Keller, C.W.; Maurer, M.A.; Giddens, J.P.; Tackenberg, B.; Wang, L.-X.; Münz, C.; Nimmerjahn, F.; Dalakas, M.C.; Lünemann, J.D. Sialylation of IgG Fc domain impairs complement-dependent cytotoxicity. J. Clin. Investig. 2015, 125, 4160–4170. [Google Scholar] [CrossRef] [Green Version]
- Sondermann, P.; Pincetic, A.; Maamary, J.; Lammens, K.; Ravetch, J.V. General mechanism for modulating immunoglobulin effector function. Proc. Natl. Acad. Sci. USA 2013, 110, 9868–9872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Shah, B.; Richardson, J. Impact of Fc N-glycan sialylation on IgG structure. MAbs 2019, 11, 1381–1390. [Google Scholar] [CrossRef] [Green Version]
- Bas, M.; Terrier, A.; Jacque, E.; Dehenne, A.; Pochet-Béghin, V.; Beghin, C.; Dezetter, A.-S.; Dupont, G.; Engrand, A.; Beaufils, B.; et al. Fc Sialylation Prolongs Serum Half-Life of Therapeutic Antibodies. J. Immunol. 2019, 202, 1582–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.; Golding, B.; Strome, S.E.; Sauna, Z.E. Fc fusion as a platform technology: Potential for modulating immunogenicity. Trends Biotechnol. 2015, 33, 27–34. [Google Scholar] [CrossRef]
- Farajpour, Z.; Rahbarizadeh, F.; Kazemi, B.; Ahmadvand, D. A nanobody directed to a functional epitope on VEGF, as a novel strategy for cancer treatment. Biochem. Biophys. Res. Commun. 2014, 446, 132–136. [Google Scholar] [CrossRef]
- Gleason, M.K.; Verneris, M.R.; Todhunter, D.A.; Zhang, B.; McCullar, V.; Zhou, S.X.; Panoskaltsis-Mortari, A.; Weiner, L.M.; Vallera, D.A.; Miller, J.S. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol. Cancer Ther. 2012, 11, 2674–2684. [Google Scholar] [CrossRef] [Green Version]
- Felices, M.; Lenvik, T.R.; Davis, Z.B.; Miller, J.S.; Vallera, D.A. Generation of BiKEs and TriKEs to Improve NK Cell-Mediated Targeting of Tumor Cells. Methods Mol. Biol. 2016, 1441, 333–346. [Google Scholar] [CrossRef] [Green Version]
- Congy-Jolivet, N.; Bolzec, A.; Ternant, D.; Ohresser, M.; Watier, H.; Thibault, G. Fc gamma RIIIa expression is not increased on natural killer cells expressing the Fc gamma RIIIa-158V allotype. Cancer Res. 2008, 68, 976–980. [Google Scholar] [CrossRef] [Green Version]
- Moore, G.L.; Bautista, C.; Pong, E.; Nguyen, D.-H.T.; Jacinto, J.; Eivazi, A.; Muchhal, U.S.; Karki, S.; Chu, S.Y.; Lazar, G.A. A novel bispecific antibody format enables simultaneous bivalent and monovalent co-engagement of distinct target antigens. MAbs 2011, 3, 546–557. [Google Scholar] [CrossRef]
- Preithner, S.; Elm, S.; Lippold, S.; Locher, M.; Wolf, A.; da Silva, A.J.; Baeuerle, P.A.; Prang, N.S. High concentrations of therapeutic IgG1 antibodies are needed to compensate for inhibition of antibody-dependent cellular cytotoxicity by excess endogenous immunoglobulin G. Mol. Immunol. 2006, 43, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Berinstein, N.L.; Grillo-López, A.J.; White, C.A.; Bence-Bruckler, I.; Maloney, D.; Czuczman, M.; Green, D.; Rosenberg, J.; McLaughlin, P.; Shen, D. Association of serum Rituximab (IDEC-C2B8) concentration and anti-tumor response in the treatment of recurrent low-grade or follicular non-Hodgkin’s lymphoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1998, 9, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Albanell, J. Mechanism of action of anti-HER2 monoclonal antibodies. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2001, 12, S35–S41. [Google Scholar] [CrossRef] [PubMed]
- Huston, J.S.; Levinson, D.; Mudgett-Hunter, M.; Tai, M.S.; Novotný, J.; Margolies, M.N.; Ridge, R.J.; Bruccoleri, R.E.; Haber, E.; Crea, R. Protein engineering of antibody binding sites: Recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc. Natl. Acad. Sci. USA 1988, 85, 5879–5883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef]
- Liu, H.; Saxena, A.; Sidhu, S.S.; Wu, D. Fc Engineering for Developing Therapeutic Bispecific Antibodies and Novel Scaffolds. Front. Immunol. 2017, 8, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiernik, A.; Foley, B.; Zhang, B.; Verneris, M.R.; Warlick, E.; Gleason, M.K.; Ross, J.A.; Luo, X.; Weisdorf, D.J.; Walcheck, B.; et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clin. Cancer Res. 2013, 19, 3844–3855. [Google Scholar] [CrossRef] [Green Version]
- Thurber, G.M.; Schmidt, M.M.; Wittrup, K.D. Factors determining antibody distribution in tumors. Trends Pharmacol. Sci. 2008, 29, 57–61. [Google Scholar] [CrossRef] [Green Version]
- Chapman, A.P.; Antoniw, P.; Spitali, M.; West, S.; Stephens, S.; King, D.J. Therapeutic antibody fragments with prolonged in vivo half-lives. Nat. Biotechnol. 1999, 17, 780–783. [Google Scholar] [CrossRef]
- Czajkowsky, D.M.; Hu, J.; Shao, Z.; Pleass, R.J. Fc-fusion proteins: New developments and future perspectives. EMBO Mol. Med. 2012, 4, 1015–1028. [Google Scholar] [CrossRef]
- Keizer, R.J.; Huitema, A.D.R.; Schellens, J.H.M.; Beijnen, J.H. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin. Pharmacokinet. 2010, 49, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Zalevsky, J.; Chamberlain, A.K.; Horton, H.M.; Karki, S.; Leung, I.W.L.; Sproule, T.J.; Lazar, G.A.; Roopenian, D.C.; Desjarlais, J.R. Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 2010, 28, 157–159. [Google Scholar] [CrossRef] [Green Version]
- Dall’Acqua, W.F.; Woods, R.M.; Ward, E.S.; Palaszynski, S.R.; Patel, N.K.; Brewah, Y.A.; Wu, H.; Kiener, P.A.; Langermann, S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: Biological consequences. J. Immunol. 2002, 169, 5171–5180. [Google Scholar] [CrossRef] [Green Version]
- Yamane-Ohnuki, N.; Satoh, M. Production of therapeutic antibodies with controlled fucosylation. MAbs 2009, 1, 230–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Chen, Z.Y.; Li, M.; Shang, H.; Li, N.; Li, F.; Wang, W.; Wang, Y.; Jin, R.; Liu, S.; Zhang, H.; et al. A general Fc engineering platform for the next generation of antibody therapeutics. Theranostics 2021, 11, 1901–1917. [Google Scholar] [CrossRef] [PubMed]
- De Romeuf, C.; Dutertre, C.-A.; le Garff-Tavernier, M.; Fournier, N.; Gaucher, C.; Glacet, A.; Jorieux, S.; Bihoreau, N.; Behrens, C.K.; Béliard, R.; et al. Chronic lymphocytic leukaemia cells are efficiently killed by an anti-CD20 monoclonal antibody selected for improved engagement of FcgammaRIIIA/CD16. Br. J. Haematol. 2008, 140, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Le Garff-Tavernier, M.; Herbi, L.; de Romeuf, C.; Nguyen-Khac, F.; Davi, F.; Grelier, A.; Boudjoghra, M.; Maloum, K.; Choquet, S.; Urbain, R.; et al. Antibody-dependent cellular cytotoxicity of the optimized anti-CD20 monoclonal antibody ublituximab on chronic lymphocytic leukemia cells with the 17p deletion. Leukemia 2014, 28, 230–233. [Google Scholar] [CrossRef]
- Lux, A.; Yu, X.; Scanlan, C.N.; Nimmerjahn, F. Impact of immune complex size and glycosylation on IgG binding to human FcγRs. J. Immunol. 2013, 190, 4315–4323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edberg, J.C.; Kimberly, R.P. Cell type-specific glycoforms of Fc gamma RIIIa (CD16): Differential ligand binding. J. Immunol. 1997, 159, 3849–3857. [Google Scholar]
- Koene, H.R.; Kleijer, M.; Algra, J.; Roos, D.; von dem Borne, A.E.; de Haas, M. Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa-48L/R/H phenotype. Blood 1997, 90, 1109–1114. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Edberg, J.C.; Redecha, P.B.; Bansal, V.; Guyre, P.M.; Coleman, K.; Salmon, J.E.; Kimberly, R.P. A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J. Clin. Investig. 1997, 100, 1059–1070. [Google Scholar] [CrossRef]
- Gavin, P.G.; Song, N.; Kim, S.R.; Lipchik, C.; Johnson, N.L.; Bandos, H.; Finnigan, M.; Rastogi, P.; Fehrenbacher, L.; Mamounas, E.P.; et al. Association of Polymorphisms in FCGR2A and FCGR3A With Degree of Trastuzumab Benefit in the Adjuvant Treatment of ERBB2/HER2-Positive Breast Cancer: Analysis of the NSABP B-31 Trial. JAMA Oncol. 2017, 3, 335–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibeau, F.; Lopez-Crapez, E.; di Fiore, F.; Thezenas, S.; Ychou, M.; Blanchard, F.; Lamy, A.; Penault-Llorca, F.; Frébourg, T.; Michel, P.; et al. Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J. Clin. Oncol. 2009, 27, 1122–1129. [Google Scholar] [CrossRef]
- Cartron, G.; Dacheux, L.; Salles, G.; Solal-Celigny, P.; Bardos, P.; Colombat, P.; Watier, H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 2002, 99, 754–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barb, A.W. Fc γ receptor compositional heterogeneity: Considerations for immunotherapy development. J. Biol. Chem. 2020, 296, 100057. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Rezvani, K.; Rouce, R.H. The Application of Natural Killer Cell Immunotherapy for the Treatment of Cancer. Front. Immunol. 2015, 6, 578. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity. Front. Immunol. 2017, 8, 533. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Ni, Z.; Wu, J.; Higgins, L.; Markowski, T.W.; Kaufman, D.S.; Walcheck, B. Identification of an ADAM17 cleavage region in human CD16 (FcγRIII) and the engineering of a non-cleavable version of the receptor in NK cells. PLoS ONE 2015, 10, e0121788. [Google Scholar] [CrossRef] [Green Version]
- Srpan, K.; Ambrose, A.; Karampatzakis, A.; Saeed, M.; Cartwright, A.N.R.; Guldevall, K.; de Matos, G.D.S.C.; Önfelt, B.; Davis, D.M. Shedding of CD16 disassembles the NK cell immune synapse and boosts serial engagement of target cells. J. Cell Biol. 2018, 217, 3267–3283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomeroy, E.J.; Hunzeker, J.T.; Kluesner, M.G.; Lahr, W.S.; Smeester, B.A.; Crosby, M.R.; Lonetree, C.-L.; Yamamoto, K.; Bendzick, L.; Miller, J.S.; et al. A Genetically Engineered Primary Human Natural Killer Cell Platform for Cancer Immunotherapy. Mol. Ther. 2020, 28, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Blum, R.H.; Bjordahl, R.; Gaidarova, S.; Rogers, P.; Lee, T.T.; Abujarour, R.; Bonello, G.B.; Wu, J.; Tsai, P.-F.; et al. Pluripotent stem cell-derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood 2020, 135, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Dixon, K.J.; Wu, J.; Walcheck, B. Engineering Anti-Tumor Monoclonal Antibodies and Fc Receptors to Enhance ADCC by Human NK Cells. Cancers 2021, 13, 312. [Google Scholar] [CrossRef] [PubMed]
- Jochems, C.; Hodge, J.W.; Fantini, M.; Fujii, R.; Morillon, Y.M.; Greiner, J.W.; Padget, M.R.; Tritsch, S.R.; Tsang, K.Y.; Campbell, K.S.; et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget 2016, 7, 86359–86373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walcheck, B.; Wu, J. iNK-CD64/16A cells: A promising approach for ADCC? Expert Opin. Biol. Ther. 2019, 19, 1229–1232. [Google Scholar] [CrossRef] [PubMed]





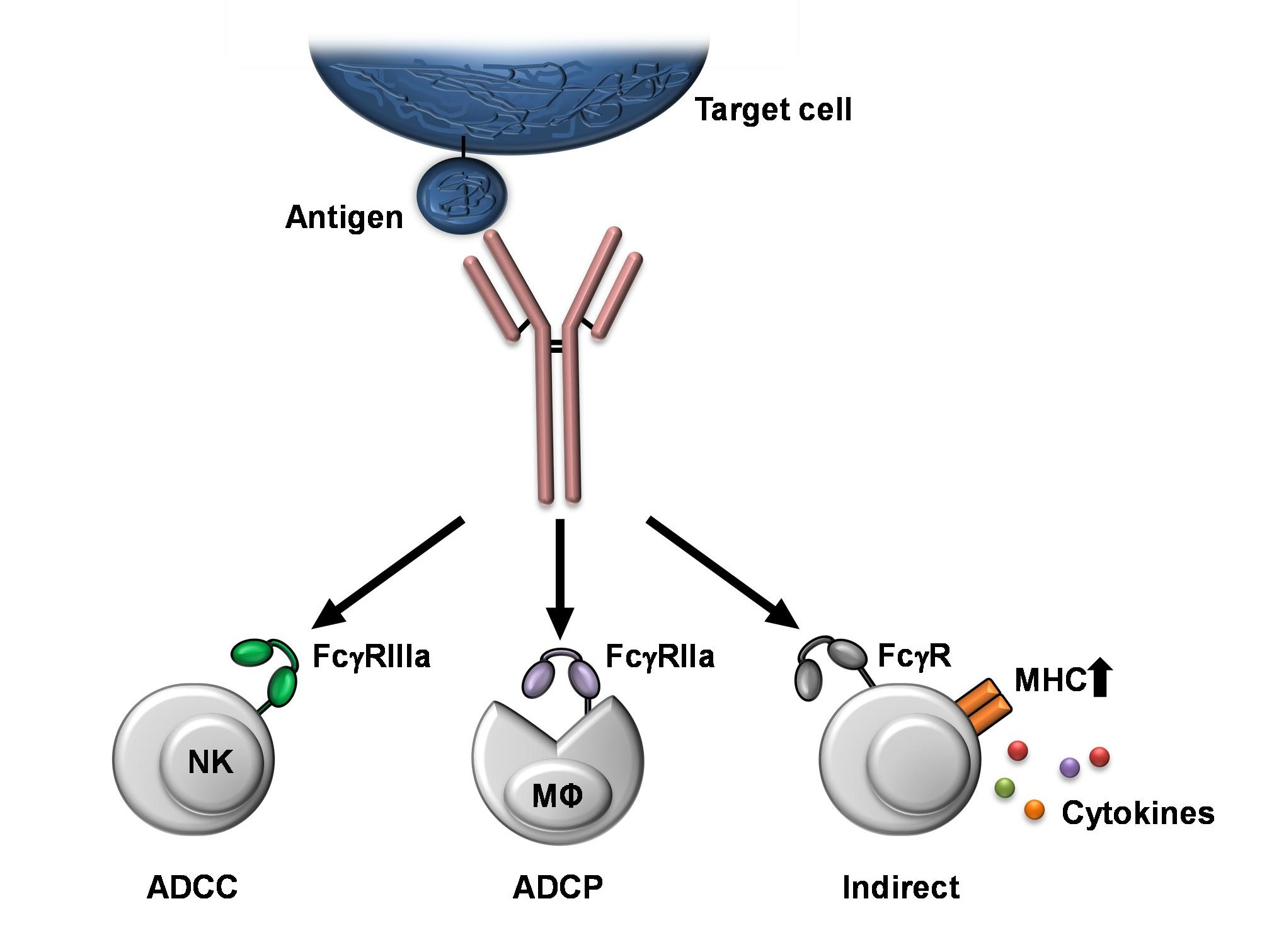{kind=link}
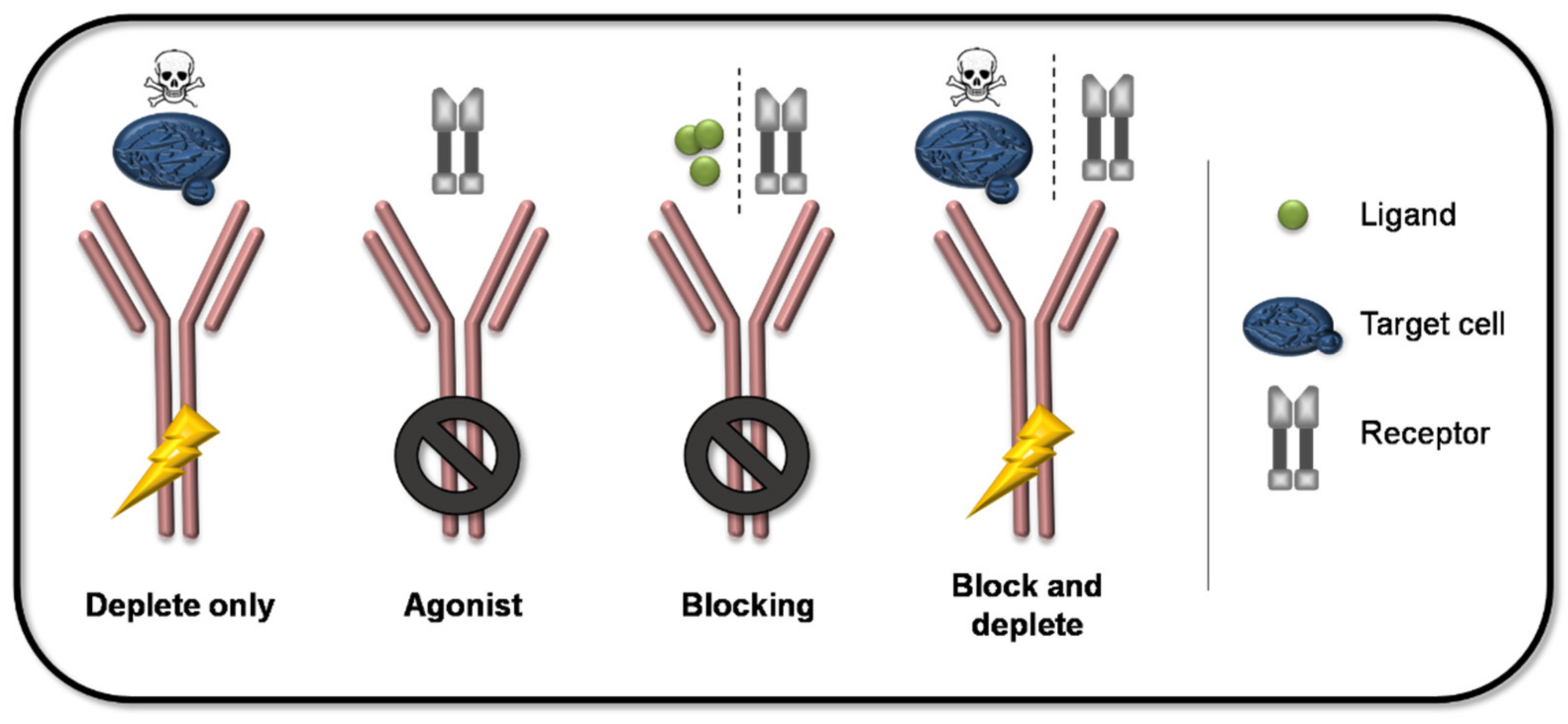{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |  |  |  |  |  | |
|---|---|---|---|---|---|---|
| Name | FcγRI | FcγRIIa | FcγRIIb | FcγRIIc | FcγRIIIa | FcγRIIIb |
| CD | CD64 | CD32a | CD32b | CD32c | CD16a | CD16b |
| Gene | FCGR1A | FCGR2A | FCGR2B | FCGR2B | FCGR3A | FCGR3B |
| Affinity | High | Low to medium | Low to medium | Low to medium | Low to medium | Low to medium |
| Major role | Activation | Activation | Inhibition | Activation | Activation | Decoy Activation |
| Human IgG interaction | IgG1, IgG3, IgG4 | IgG3, IgG1, IgG2, IgG4 | IgG3, IgG1, IgG4 | IgG3, IgG1 | IgG1, IgG3, IgG4 | IgG1, IgG3 |
| Cell type | Monocytes DCs (Neutrophils) (Mast cells) (Macrophages) | Monocytes Neutrophils DCs Macrophages Basophils Eosinophils Mast cells Platelets | B cells Monocytes DCs Macrophages Eosinophils Basophiles (Neutrophils) (Activated T cells) | NK cells Monocytes Neutrophils | NK cells Monocytes (Macrophages) (Activated T cells) | Neutrophils (Basophils) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gogesch, P.; Dudek, S.; van Zandbergen, G.; Waibler, Z.; Anzaghe, M. The Role of Fc Receptors on the Effectiveness of Therapeutic Monoclonal Antibodies. Int. J. Mol. Sci. 2021, 22, 8947. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168947
Gogesch P, Dudek S, van Zandbergen G, Waibler Z, Anzaghe M. The Role of Fc Receptors on the Effectiveness of Therapeutic Monoclonal Antibodies. International Journal of Molecular Sciences. 2021; 22(16):8947. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168947
Chicago/Turabian StyleGogesch, Patricia, Simone Dudek, Ger van Zandbergen, Zoe Waibler, and Martina Anzaghe. 2021. "The Role of Fc Receptors on the Effectiveness of Therapeutic Monoclonal Antibodies" International Journal of Molecular Sciences 22, no. 16: 8947. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168947