
In Vitro and In Silico Characterization of an Antimalarial Compound with Antitumor Activity Targeting Human DNA Topoisomerase IB

 ,
,  ,
,  ,
,  , and
, and
Abstract
:1. Introduction
2. Results
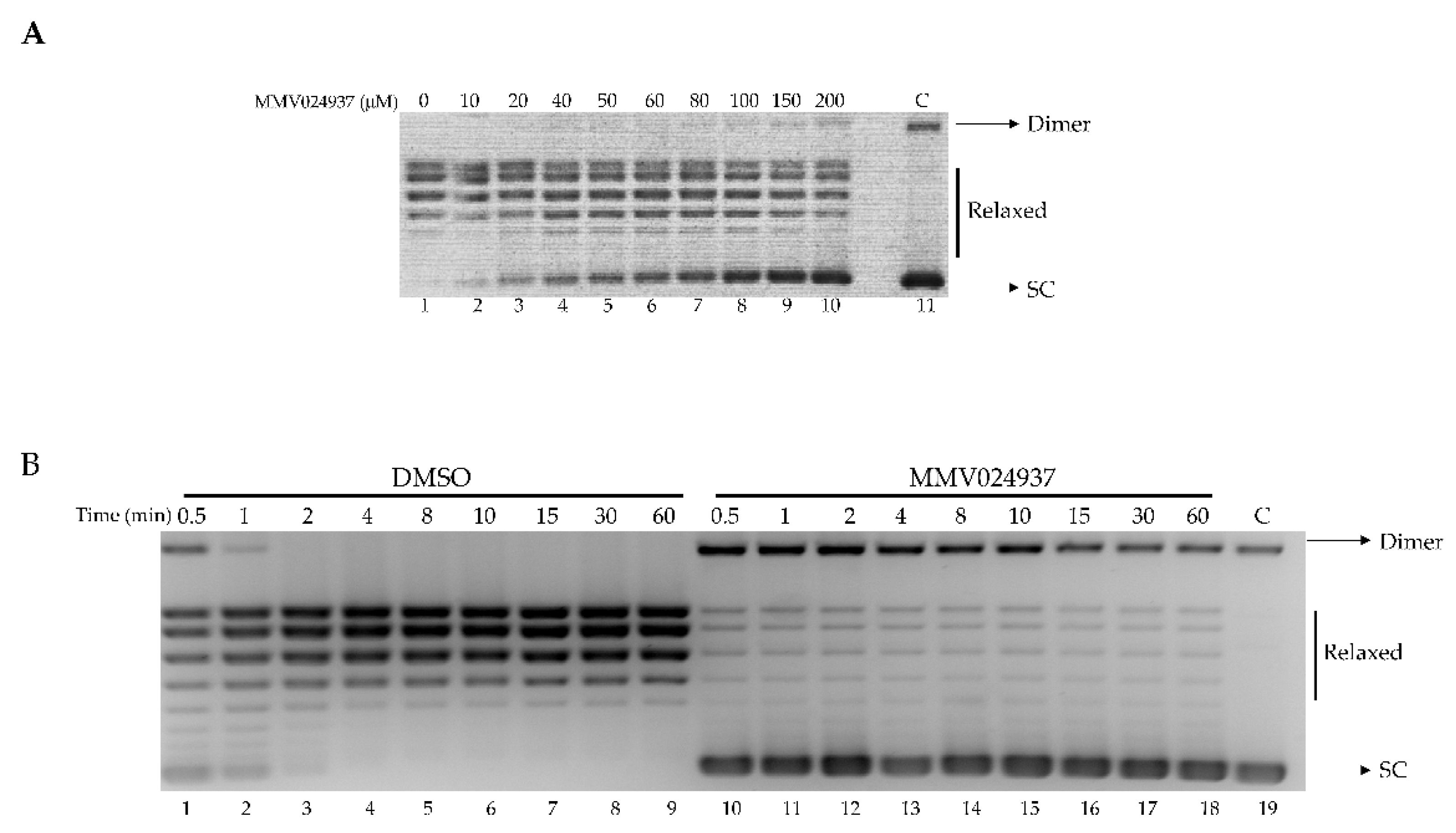
2.1. MMV024937 Inhibits the Catalytic Activity of htopIB
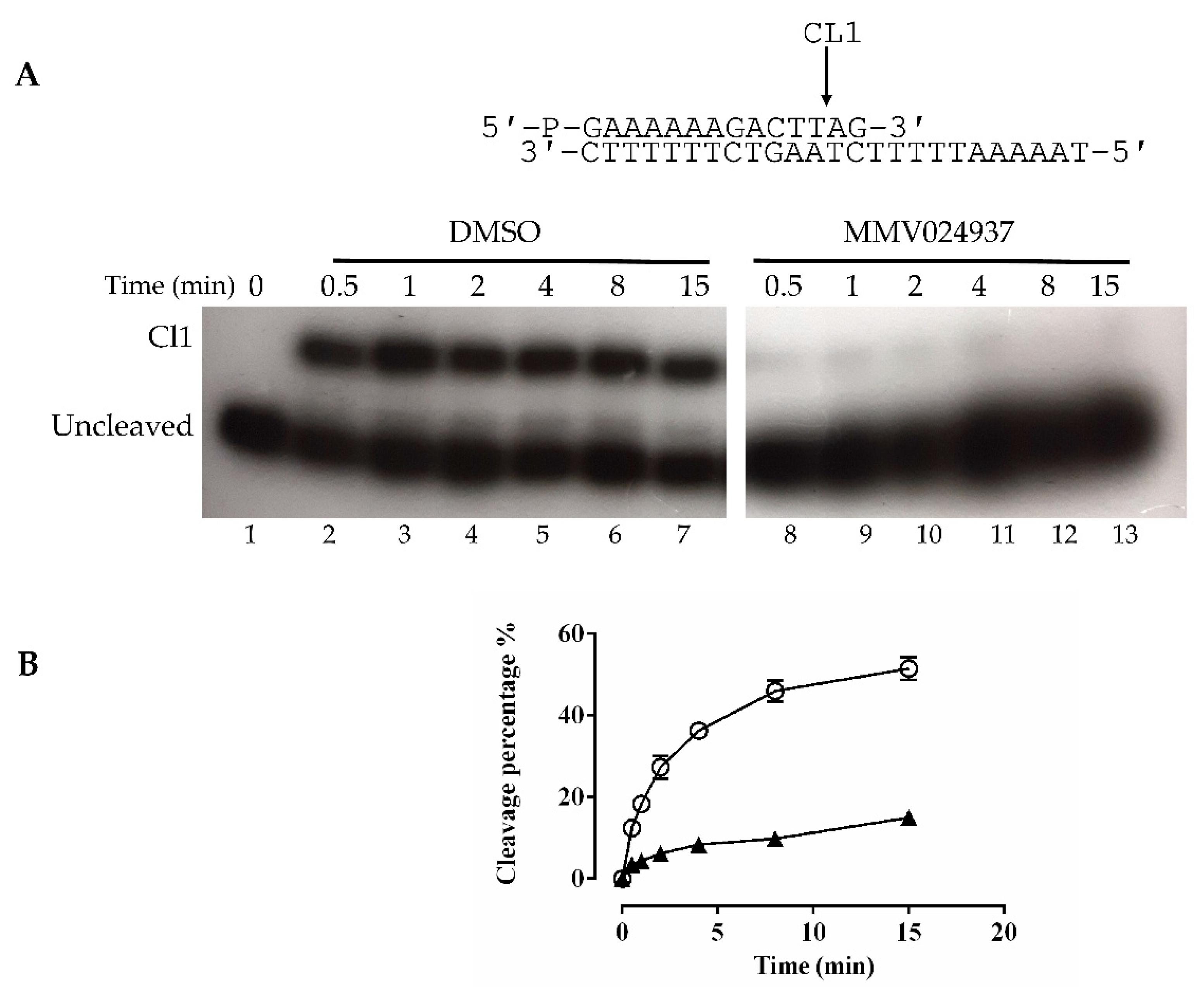
2.2. Cleavage with CL14/CP25 Suicide Substrate
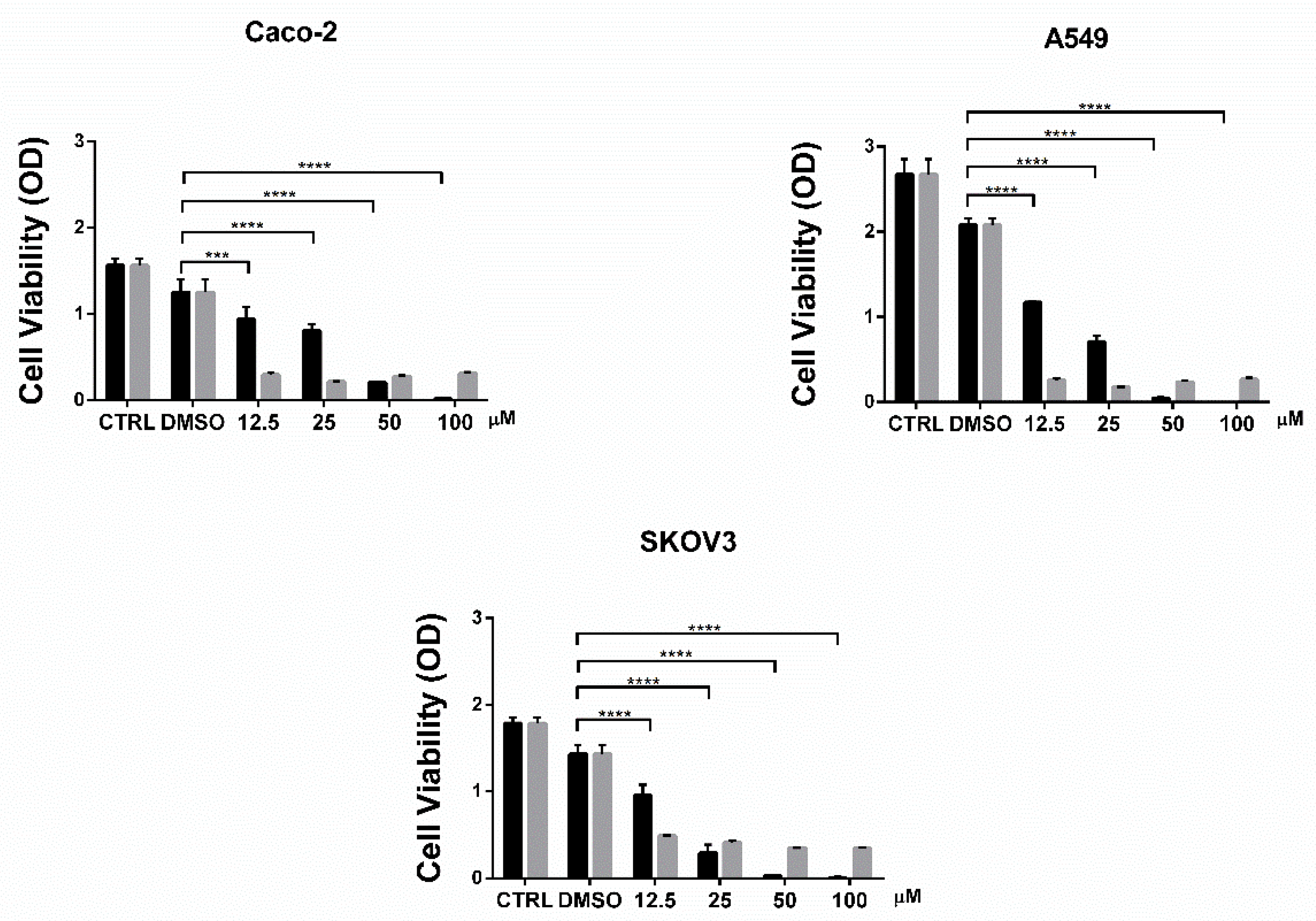
2.3. Cell Viability Assay
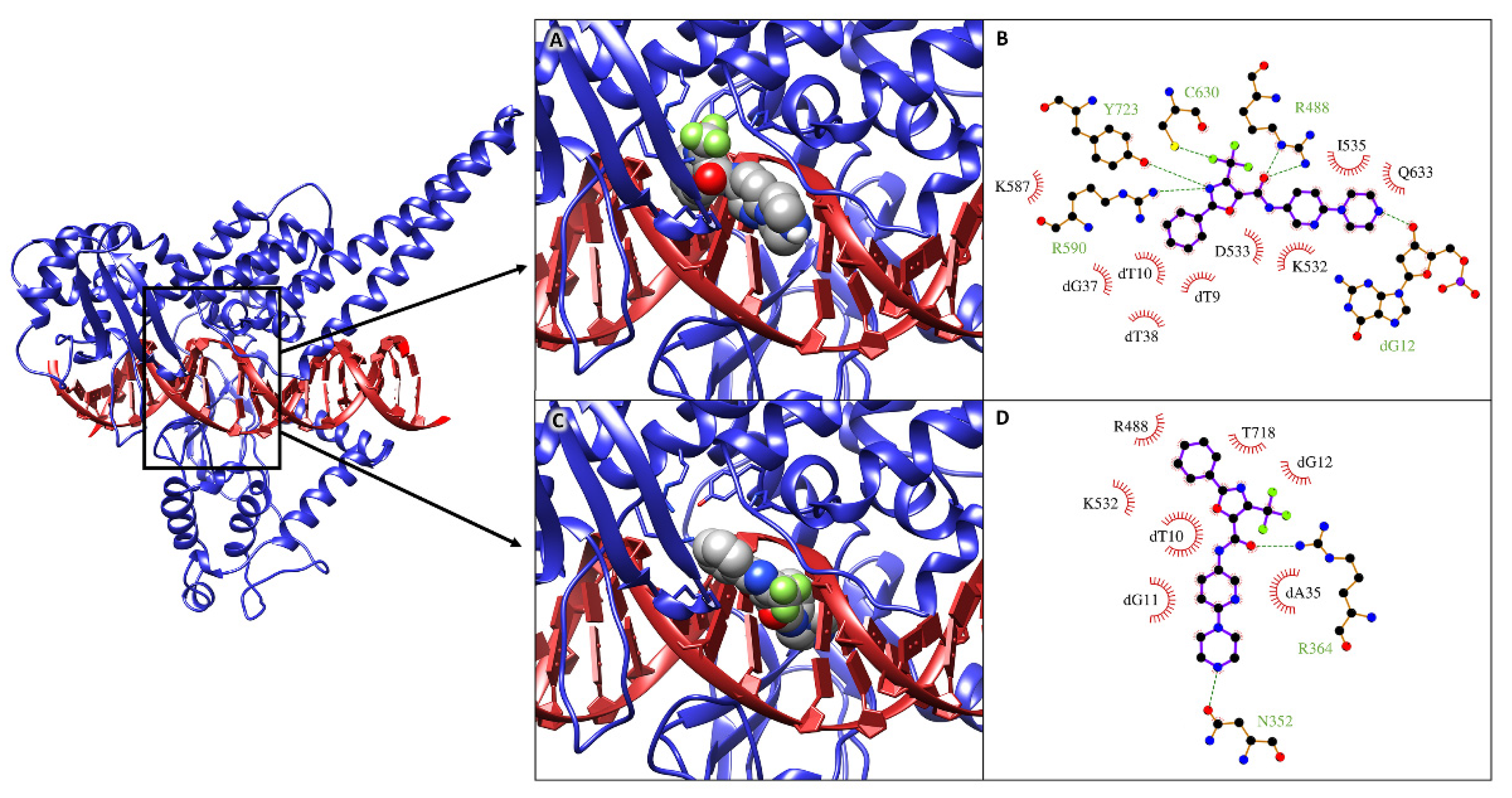
2.4. Molecular Docking Simulations
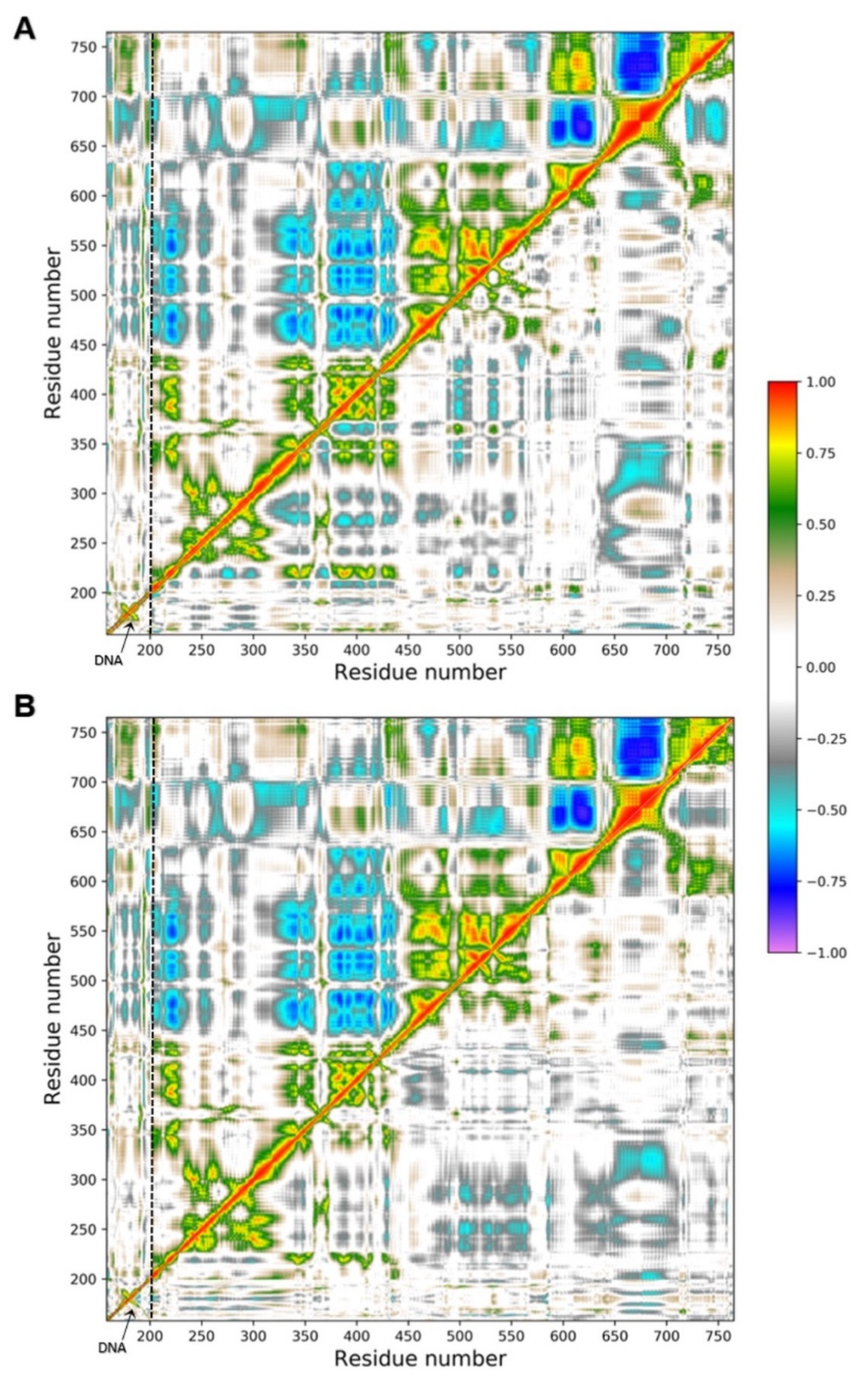
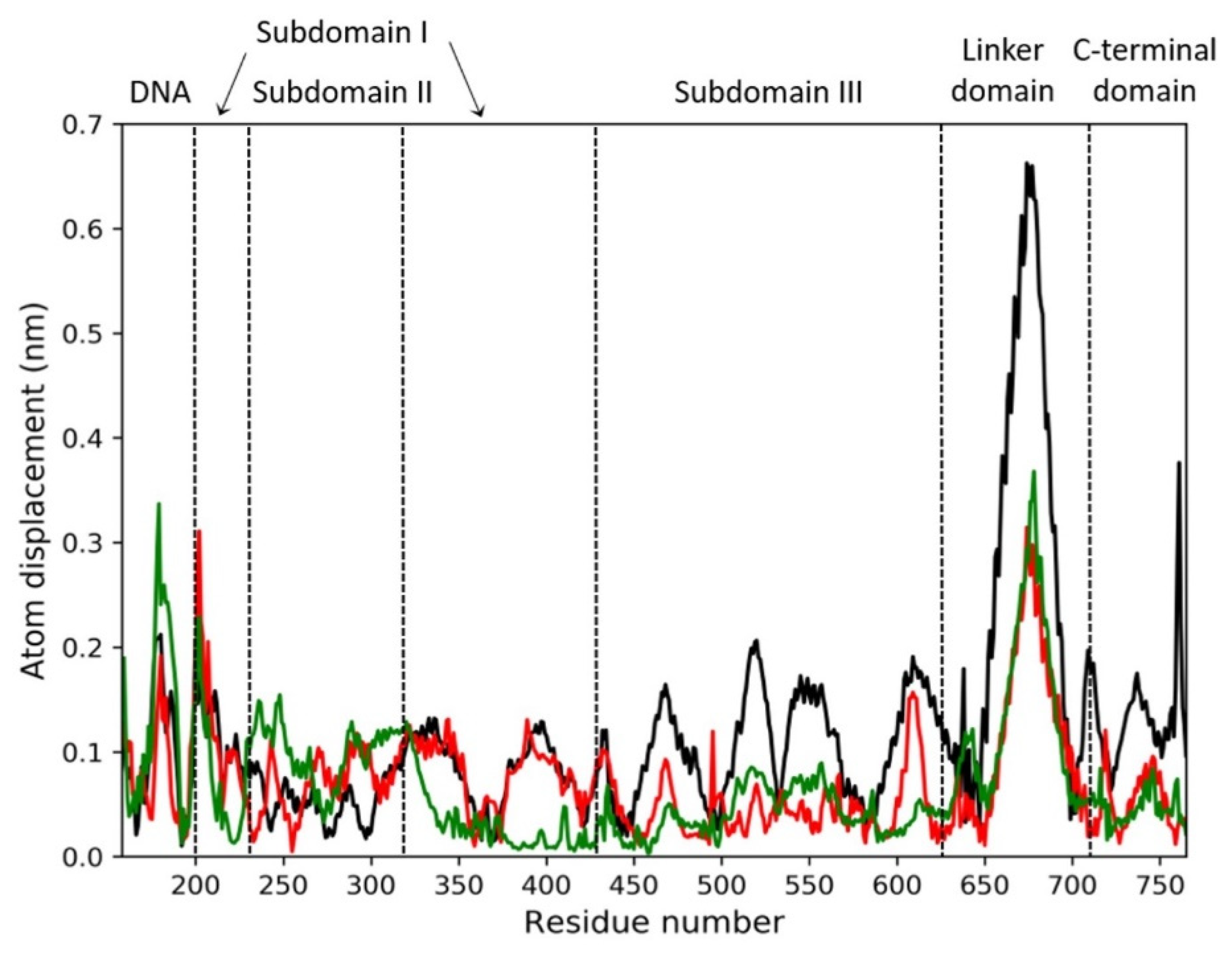
2.5. Molecular Dynamics Simulations and MM/GBSA Analysis
2.6. Principal Component Analysis
3. Discussion
4. Materials and Methods
4.1. Chemicals, Cells, Yeast Strains and Plasmids
4.2. Protein Purification
4.3. Relaxation Assay
4.4. Cleavage Kinetics Using CL14/CP25 Oligonucleotide Substrate
4.5. Cell Viability Assay
4.6. Molecular Docking Simulations
4.7. Classical Molecular Dynamics Simulations and Trajectory Analysis
4.8. Trajectory Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, J.C. DNA topoisomerases. Annu. Rev. Biochem. 1996, 65, 635–692. [Google Scholar] [CrossRef]
- Leppard, J.B.; Champoux, J.J. Human DNA topoisomerase I: Relaxation, roles, and damage control. Chromosoma 2005, 114, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soren, B.C.; Dasari, J.B.; Ottaviani, A.; Lacovelli, F.; Fiorani, P. Topoisomerase IB: A relaxing enzyme for stressed DNA. Cancer Drug Resist. 2019, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Fiorani, P.; Tesauro, C.; Mancini, G.; Chillemi, G.; D’Annessa, I.; Graziani, G.; Tentori, L.; Muzi, A.; Desideri, A. Evidence of the crucial role of the linker domain on the catalytic activity of human topoisomerase I by experimental and simulative characterization of the Lys681Ala mutant. Nucleic Acids Res. 2009, 37, 6849–6858. [Google Scholar] [CrossRef] [Green Version]
- Stewart, L.; Ireton, G.C.; Champoux, J.J. A Functional Linker in Human Topoisomerase I Is Required for Maximum Sensitivity to Camptothecin in a DNA Relaxation Assay. J. Biol. Chem. 1999, 274, 32950–32960. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; D’Annessa, I.; Tesauro, C.; Croce, S.; Ottaviani, A.; Fiorani, P.; Desideri, A. Mutation of Gly717Phe in human topoisomerase 1B has an effect on enzymatic function, reactivity to the camptothecin anticancer drug and on the linker domain orientation. Biochim. Biophys. Acta 2015, 1854, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Chillemi, G.; Fiorani, P.; Benedetti, P.; Desideri, A. Protein concerted motions in the DNA-human topoisomerase I complex. Nucleic Acids Res. 2003, 31, 1525–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wall, M.E.; Wani, M.C. Camptothecin and taxol: From discovery to clinic. Cancer Res. 1995, 51, 753–760. [Google Scholar] [CrossRef]
- Pizzolato, J.F.; Saltz, L.B. The camptothecins. Lancet 2003, 361, 2235–2242. [Google Scholar] [CrossRef]
- Ottaviani, A.; Iacovelli, F.; Fiorani, P.; Desideri, A. Natural compounds as therapeutic agents: The case of human topoisomerase ib. Int. J. Mol. Sci. 2021, 22, 4138. [Google Scholar] [CrossRef] [PubMed]
- Paton, F.; Paulden, M.; Saramago, P.; Manca, A.; Misso, K.; Palmer, S.; Eastwood, A. Topotecan for the treatment of recurrent and stage IVB carcinoma of the cervix. Health Technol. Assess. 2010, 14 (Suppl. S1), 55–62. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Giovannetti, E.; Cortesi, F.; Mey, V.; Nannizzi, S.; Gallegos Ruiz, M.I.; Ricciardi, S.; Del Tacca, M.; Peters, G.J.; Danesi, R. Epigenetic mechanisms of irinotecan sensitivity in colorectal cancer cell lines. Mol. Cancer Ther. 2009, 8, 1964–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, Y.; Figg, W.D. Irinotecan in Small-Cell Lung Cancer. N. Engl. J. Med. 2002, 346, 1414–1415. [Google Scholar] [CrossRef] [PubMed]
- Fiorani, P.; Amatruda, J.F.; Silvestri, A.; Butler, R.H.; Bjornsti, M.A.; Benedetti, P. Domain interactions affecting human DNA topoisomerase I catalysis and camptothecin sensitivity. Mol. Pharmacol. 1999, 56, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Tesauro, C.; Morozzo della Rocca, B.; Ottaviani, A.; Coletta, A.; Zuccaro, L.; Arnò, B.; D’Annessa, I.; Fiorani, P.; Desideri, A. Molecular mechanism of the camptothecin resistance of Glu710Gly topoisomerase IB mutant analyzed in vitro and in silico. Mol. Cancer 2013, 12, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Annessa, I.; Tesauro, C.; Fiorani, P.; Chillemi, G.; Castelli, S.; Vassallo, O.; Capranico, G.; Desideri, A. Role of Flexibility in Protein-DNA-Drug Recognition: The Case of Asp677Gly-Val703Ile Topoisomerase Mutant Hypersensitive to Camptothecin. J. Amino Acids 2012, 2012, 206083. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Tesauro, C.; Fiorani, P.; D’Annessa, I.; Chillemi, G.; Turchi, G.; Desideri, A. Erybraedin C, a natural compound from the plant Bituminaria bituminosa, inhibits both the cleavage and religation activities of human topoisomerase I. Biochem. J. 2010, 425, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.R.; Majumder, H.K. DNA Topoisomerases in Unicellular Pathogens: Structure, Function, and Druggability. Trends Biochem. Sci. 2019, 44, 415–432. [Google Scholar] [CrossRef]
- García-Estrada, C.; Prada, C.F.; Fernández-Rubio, C.; Rojo-Vázquez, F.; Balaña-Fouce, R. DNA topoisomerases in apicomplexan parasites: Promising targets for drug discovery. Proc. Biol. Sci. 2010, 277, 1777–1787. [Google Scholar] [CrossRef] [Green Version]
- Tesauro, C.; Juul, S.; Arnò, B.; Nielsen, C.J.F.; Fiorani, P.; Frøhlich, R.F.; Andersen, F.F.; Desideri, A.; Stougaard, M.; Petersen, E.; et al. Specific detection of topoisomerase I from the malaria causing P. falciparum parasite using isothermal rolling circle amplification. In Proceedings of the 2012 Annual International Conference of the IEEE Engineering in Medicine and Biology Society, San Diego, CA, USA, 28 August–1 September 2012; pp. 2416–2419. [Google Scholar] [CrossRef]
- Duffy, S.; Sykes, M.L.; Jones, A.J.; Shelper, T.B.; Simpson, M.; Lang, R.; Poulsen, S.A.; Sleebs, B.E.; Avery, V.M. Screening the medicines for malaria venture pathogen box across multiple pathogens reclassifies starting points for open-source drug discovery. Antimicrob. Agents Chemother 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Arnò, B.; D’Annessa, I.; Tesauro, C.; Zuccaro, L.; Ottaviani, A.; Knudsen, B.; Fiorani, P.; Desideri, A. Replacement of the human topoisomerase linker domain with the plasmodial counterpart renders the enzyme camptothecin resistant. PLoS ONE 2013, 8, e68404. [Google Scholar] [CrossRef] [PubMed]
- Staker, B.L.; Feese, M.D.; Cushman, M.; Pommier, Y.; Zembower, D.; Stewart, L.; Burgin, A.B. Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J. Med. Chem. 2005, 48, 2336–2345. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Stewart, L. A Model for the Mechanism of Human Topoisomerase I. Science 1998, 279, 1534–1541. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.M.; Berendsen, H.J.C. Essential dynamics of proteins. Proteins Struct. Funct. Genet. 1993, 17, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Veale, C.G.L. Unpacking the Pathogen Box—An Open Source Tool for Fighting Neglected Tropical Disease. ChemMedChem 2019, 14, 386–453. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Geng, F.; Guo, D. DGAT1 protects tumor from lipotoxicity, emerging as a promising metabolic target for cancer therapy. Mol. Cell. Oncol. 2020, 7, 4–6. [Google Scholar] [CrossRef]
- Chillemi, G.; Fiorani, P.; Castelli, S.; Bruselles, A.; Benedetti, P.; Desideri, A. Effect on DNA relaxation of the single Thr718Ala mutation in human topoisomerase I: A functional and molecular dynamics study. Nucleic Acids Res. 2005, 33, 3339–3350. [Google Scholar] [CrossRef] [Green Version]
- Fiorani, P.; Bruselles, A.; Falconi, M.; Chillemi, G.; Desideri, A.; Benedetti, P. Single mutation in the linker domain confers protein flexibility and camptothecin resistance to human topoisomerase I. J. Biol. Chem. 2003, 278, 43268–43275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ireton, G.C.; Stewart, L.; Parker, L.H.; Champoux, J.J. Expression of human topoisomerase I with a partial deletion of the linker region yields monomeric and dimeric enzymes that respond differently to camptothecin. J. Biol. Chem. 2000, 275, 25820–25830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An Overview of the Amber Biomolecular Simulation Package. WIREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2015, 13, 55–58. [Google Scholar] [CrossRef] [Green Version]
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 1 June 2021).
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Software news and updates AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. Ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Goga, N.; Rzepiela, A.J.; De Vries, A.H.; Marrink, S.J.; Berendsen, H.J.C. Efficient algorithms for langevin and DPD dynamics. J. Chem. Theory Comput. 2012, 8, 3637–3649. [Google Scholar] [CrossRef] [Green Version]
- Aoki, K.M.; Yoneya, M.; Yokoyama, H. Constant pressure MD simulation method. Mol. Cryst. Liq. Cryst. 2004, 413, 109–116. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Iannone, F.; Ambrosino, F.; Bracco, G.; De Rosa, M.; Funel, A.; Guarnieri, G.; Migliori, S.; Palombi, F.; Ponti, G.; Santomauro, G.; et al. CRESCO ENEA HPC clusters: A working example of a multifabric GPFS Spectrum Scale layout. In Proceedings of the 2019 International Conference on High Performance Computing and Simulation, HPCS 2019; Institute of Electrical and Electronics Engineers Inc.: Dublin, Ireland, 2019; pp. 1051–1052. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Binding Configuration | VdW (kcal/mol) | Electrostatic (kcal/mol) | Interaction Energy (kcal/mol) |
|---|---|---|---|
| non-intercalated | −63.7 ± 2.8 | −14.4 ± 9.5 | −49.0 ± 3.4 |
| intercalated | −67.1 ± 2.9 | −64.0 ± 12.3 | −55.1 ± 4.0 |
| Nucleotide/Residue | Non-Intercalated Drug Binding Energy (kcal/mol) | Intercalated Drug Binding Energy (kcal/mol) |
|---|---|---|
| dT10 | −2.0 ± 0.5 | −3.0 ± 1.8 |
| dG11 | −0.1 ± 0.1 | −5.8 ± 0.9 |
| dG12 | −3.0 ± 1.1 | −0.4 ± 0.2 |
| dA13 | −2.1 ± 1.7 | +0.1 ± 0.0 |
| dC34 | +0.1 ± 0.0 | −4.4 ± 1.0 |
| dA35 | 0.0 ± 0.0 | −6.1 ± 1.0 |
| dA36 | −0.1 ± 0.1 | −0.7 ± 0.2 |
| dG37 | −0.7 ± 0.4 | −0.2 ± 0.2 |
| Arg488 | −0.8 ± 0.6 | −0.9 ± 0.5 |
| Lys532 | −2.7 ± 0.6 | −0.8 ±1.0 |
| Arg590 | +0.1 ± 0.4 | −0.1 ± 0.1 |
| His632 | −1.8 ± 0.4 | −0.2 ± 0.2 |
| Tyr723 | −1.3 ± 0.5 | −0.3 ± 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soren, B.C.; Babu Dasari, J.; Ottaviani, A.; Messina, B.; Andreotti, G.; Romeo, A.; Iacovelli, F.; Falconi, M.; Desideri, A.; Fiorani, P. In Vitro and In Silico Characterization of an Antimalarial Compound with Antitumor Activity Targeting Human DNA Topoisomerase IB. Int. J. Mol. Sci. 2021, 22, 7455. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147455
Soren BC, Babu Dasari J, Ottaviani A, Messina B, Andreotti G, Romeo A, Iacovelli F, Falconi M, Desideri A, Fiorani P. In Vitro and In Silico Characterization of an Antimalarial Compound with Antitumor Activity Targeting Human DNA Topoisomerase IB. International Journal of Molecular Sciences. 2021; 22(14):7455. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147455
Chicago/Turabian StyleSoren, Bini Chhetri, Jagadish Babu Dasari, Alessio Ottaviani, Beatrice Messina, Giada Andreotti, Alice Romeo, Federico Iacovelli, Mattia Falconi, Alessandro Desideri, and Paola Fiorani. 2021. "In Vitro and In Silico Characterization of an Antimalarial Compound with Antitumor Activity Targeting Human DNA Topoisomerase IB" International Journal of Molecular Sciences 22, no. 14: 7455. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147455