
QTL Mapping and Diurnal Transcriptome Analysis Identify Candidate Genes Regulating Brassica napus Flowering Time

 , ,
, ,
Abstract
:1. Introduction
2. Results
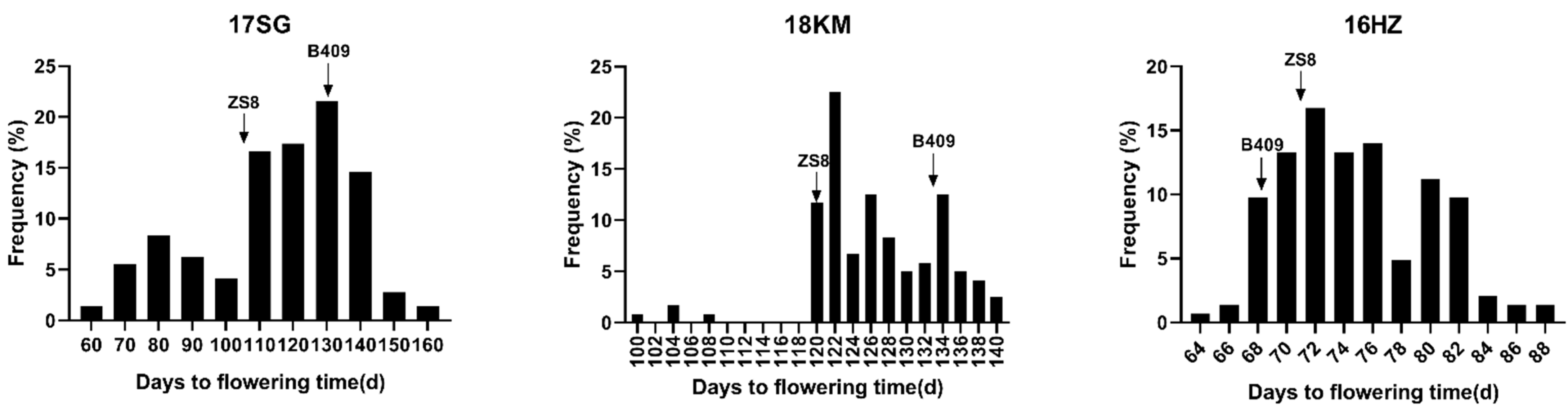
2.1. Analysis of Phenotypic Data in Double Haploid (DH) Population and Parents
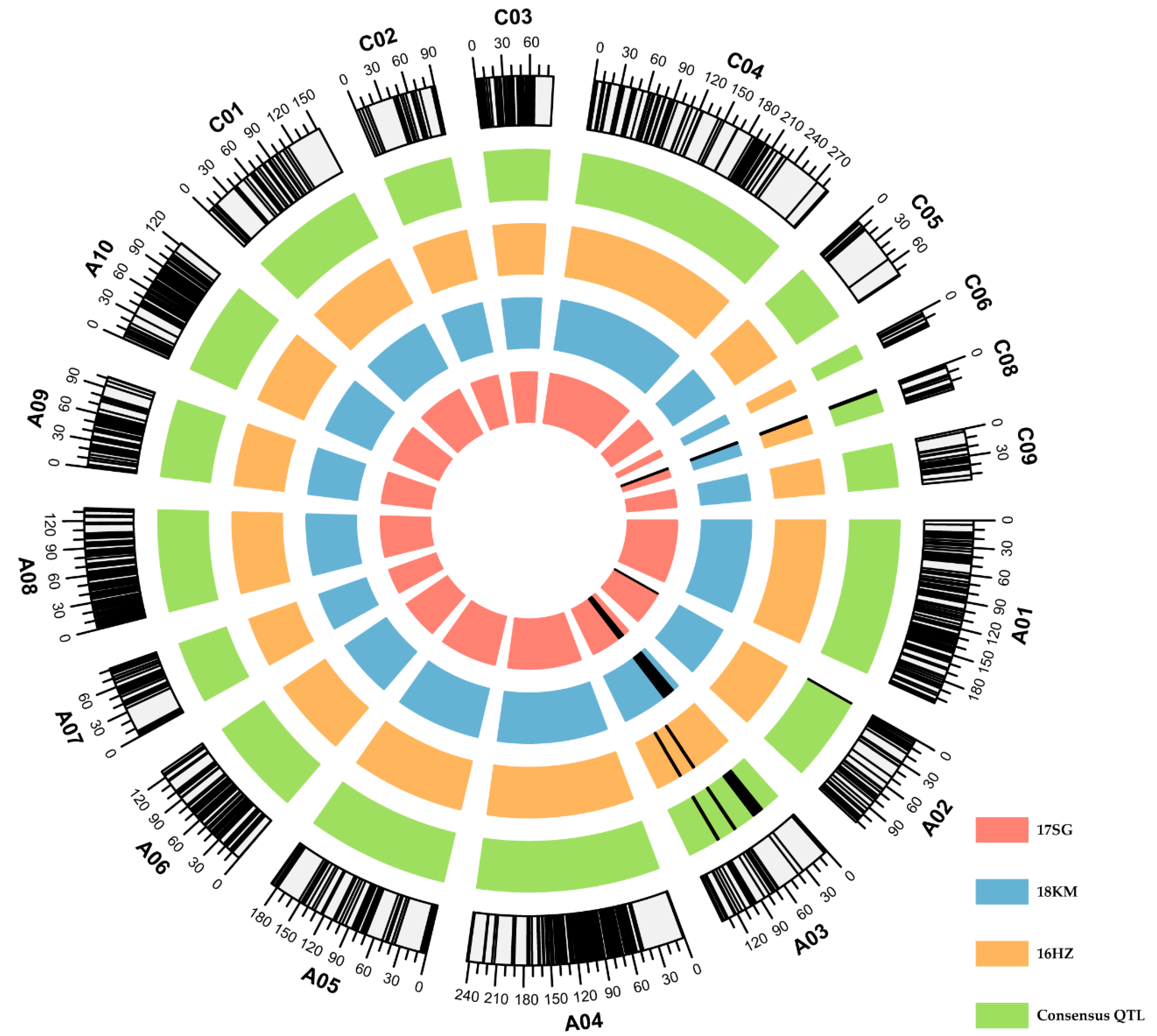
2.2. DH Population Genetic Linkage Map Construction
2.3. Flowering-Time Quantitative Trait Loci (QTL) Mapping
2.4. Gene Expression and Differential Expression Analysis in RNA Sequencing (RNA-Seq) Data
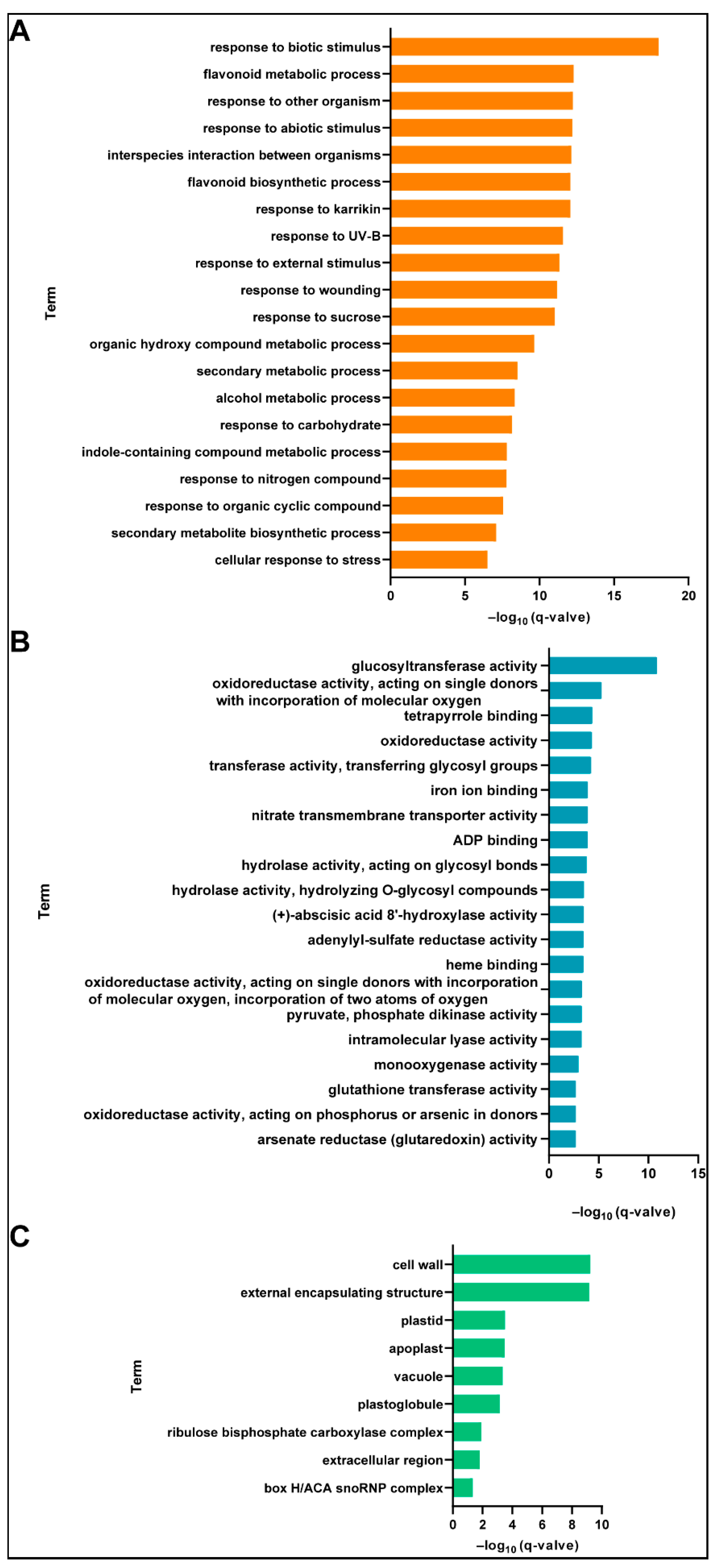
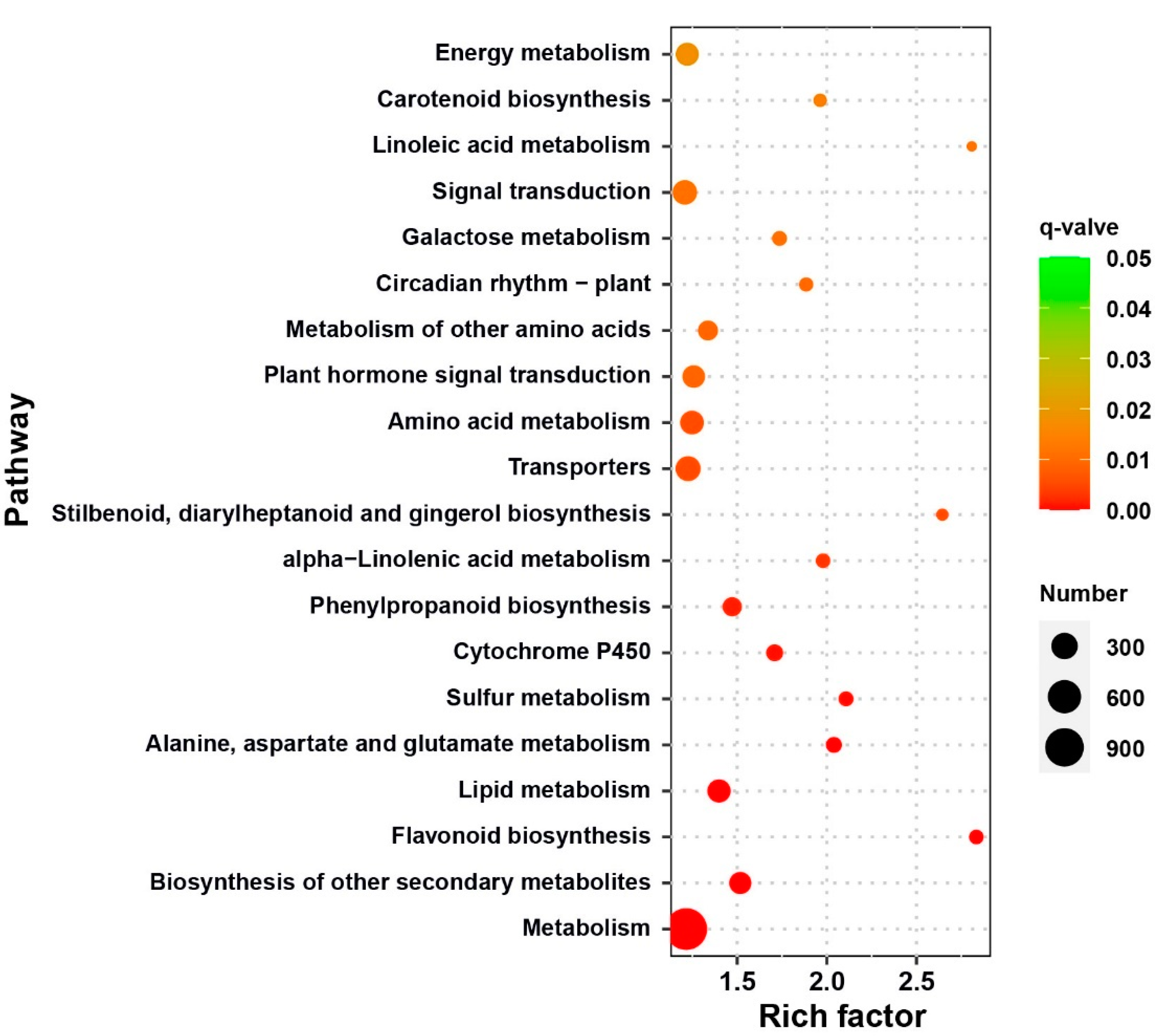
2.5. Functional Classification of Differentially Expressed Genes (DEGs)
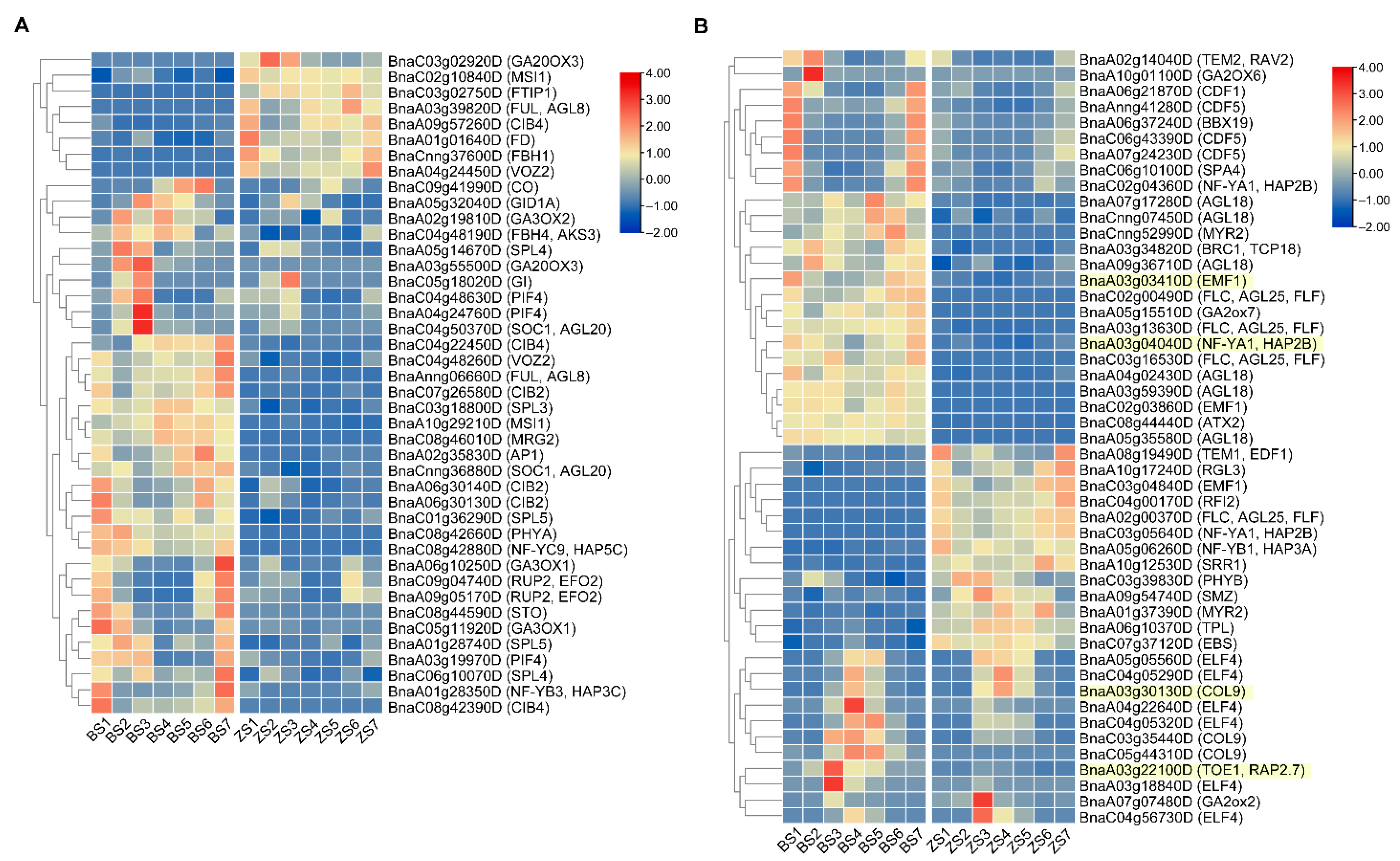
2.6. Candidate Flowering-Time Genes Analysis
2.7. Flowering-Time Related DEGs Analysis
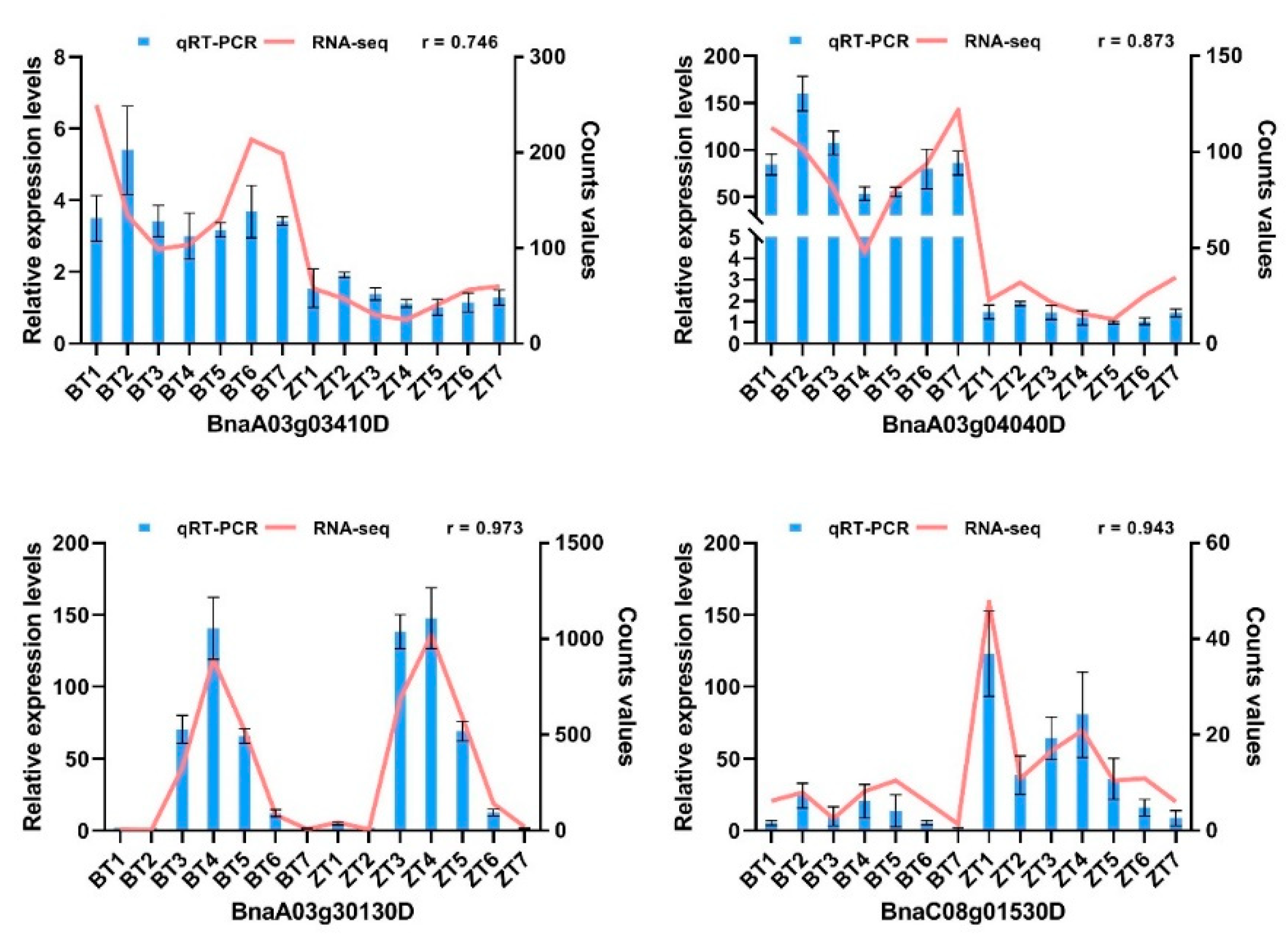
2.8. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Verification of RNA-Seq Data
3. Discussion
3.1. Flowering-Time QTLs Analysis in the DH Population
3.2. Candidate Flowering-Time Genes Analysis in QTL Intervals
3.3. Advantages of RNA-Seq Analysis at Multiple Time-Points in a Day
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Phenotype Data Measurement and Analysis
4.3. Genotyping and Markers Filtering
4.4. Genetic Linkage Map Construction and QTL Mapping
4.5. Leaf Sampling and RNA Isolation
4.6. RNA-Seq and Data Analysis
4.7. qRT-PCR Validation of RNA-seq Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| QTL | Quantitative trait loci |
| DH | Double haploid |
| ZS8 | Zhongshuang8hao |
| B409 | Bing409 |
| DNA | Deoxyribonucleic acid |
| SNP | Single nucleotide polymorphism |
| SSR | Simple sequence repeat |
| ICIM | Inclusive composite interval mapping |
| LOD | Logarithm of odds |
| FDR | False discovery rate |
| PVE | Phenotypic variation |
| RNA | Ribonucleic acid |
| RNA-seq | Ribonucleic acid sequencing |
| DEGs | Differentially expressed genes |
| A. thaliana | Arabidopsis thaliana |
| GA | Gibberellin |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
References
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.P.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blümel, M.; Dally, N.; Jung, C. Flowering time regulation in crops—What did we learn from Arabidopsis? Curr. Opin. Biotechnol. 2015, 32, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Müller, A.E. Flowering time control and applications in plant breeding. Trends Plant Sci. 2009, 14, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hu, K.; Zhang, Z.; Guan, C.; Chen, S.; Hua, W.; Li, J.; Wen, J.; Yi, B.; Shen, J.; et al. Genome-wide association study reveals the genetic architecture of flowering time in rapeseed (Brassica napus L.). DNA Res. 2015, 23, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murat, F.; Louis, A.; Maumus, F.; Armero, A.; Cooke, R.; Quesneville, H.; Crollius, H.R.; Salse, J. Understanding Brassicaceae evolution through ancestral genome reconstruction. Genome Biol. 2015, 16, 262. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Fan, Y.; Yu, J.; Chai, L.; Zhang, J.; Jiang, J.; Cui, C.; Zheng, B.; Jiang, L.; Lu, K. Genome-Wide Identification of Flowering-Time Genes in Brassica Species and Reveals a Correlation between Selective Pressure and Expression Patterns of Vernalization-Pathway Genes in Brassica napus. Int. J. Mol. Sci. 2018, 19, 3632. [Google Scholar] [CrossRef] [Green Version]
- Bouché, F.; Lobet, G.; Tocquin, P.; Périlleux, C. FLOR-ID: An interactive database of flowering-time gene networks in Arabidopsis thaliana. Nucleic Acids Res. 2016, 44, D1167–D1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.; Shu, C.; Chen, L.; Xu, J.; Wu, J.; Liu, K. Identification of a major QTL for silique length and seed weight in oilseed rape (Brassica napus L.). Theor. Appl. Genet. 2012, 125, 285–296. [Google Scholar] [CrossRef]
- Shen, Y.; Yang, Y.; Xu, E.; Ge, X.; Xiang, Y.; Li, Z. Novel and major QTL for branch angle detected by using DH population from an exotic introgression in rapeseed (Brassica napus L.). Theor. Appl. Genet. 2017, 131, 67–78. [Google Scholar] [CrossRef]
- He, Y.; Fu, Y.; Hu, D.; Wei, D.; Qian, W. QTL Mapping of Seed Glucosinolate Content Responsible for Environment in Brassica napus. Front. Plant Sci. 2018, 9, 891. [Google Scholar] [CrossRef]
- Raman, H.; Raman, R.; Eckermann, P.; Coombes, N.; Manoli, S.; Zou, X.; Edwards, D.; Meng, J.; Prangnell, R.; Stiller, J.; et al. Genetic and physical mapping of flowering time loci in canola (Brassica napus L.). Theor. Appl. Genet. 2012, 126, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Xiang, Y.; Xu, E.; Ge, X.; Li, Z. Major Co-localized QTL for Plant Height, Branch Initiation Height, Stem Diameter, and Flowering Time in an Alien Introgression Derived Brassica napus DH Population. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Zhang, B.; Ma, N.; Liu, X.; Qin, M.; Zhang, Y.; Wang, K.; Guo, N.; Zuo, K.; Liu, X.; et al. Quantitative Trait Locus Mapping and Identification of Candidate Genes Controlling Flowering Time in Brassica napus L. Front. Plant Sci. 2021, 11. [Google Scholar] [CrossRef]
- Zhang, Z.; Fan, Y.; Xiong, J.; Guo, X.; Hu, K.; Wang, Z.; Gao, J.; Wen, J.; Yi, B.; Shen, J.; et al. Two young genes reshape a novel interaction network in Brassica napus. New Phytol. 2019, 225, 530–545. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Hu, K.; Tian, T.; Li, X.; Chen, L.; Zhang, Y.; Yi, B.; Wen, J.; Ma, C.; Shen, J.; et al. Transcriptomic Analysis of Seed Coats in Yellow-Seeded Brassica napus Reveals Novel Genes That Influence Proanthocyanidin Biosynthesis. Front. Plant Sci. 2017, 8, 1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, H.; Zhang, A.; Ma, J.; Wang, T.; Yang, B.; Shuang, L.S.; Liu, M.; Li, J.; Xu, X.; Paterson, A.H.; et al. Joint QTL mapping and transcriptome sequencing analysis reveal candidate flowering time genes in Brassica napus L. BMC Genom. 2019, 20, 21. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Wang, X.; Chen, F.; Chen, S.; Peng, Q.; Li, H.; Zhang, W.; Hu, M.; Chu, P.; Zhang, J.; et al. Genome-wide transcriptomic analysis uncovers the molecular basis underlying early flowering and apetalous characteristic in Brassica napus L. Sci. Rep. 2016, 6, 30576. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.M.; Wells, R.; Pullen, N.; Trick, M.; Morris, R.J. Spatio-temporal expression dynamics differ between flowering time gene homologues in the allopolyploid Brassica napus. Plant J. 2018, 96, 103–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mure, L.S.; Le, H.D.; Benegiamo, G.; Chang, M.W.; Rios, L.; Jillani, N.; Ngotho, M.; Kariuki, T.; Dkhissi-Benyahya, O.; Cooper, H.M.; et al. Diurnal transcriptome atlas of a primate across major neural and peripheral tissues. Science 2018, 359, eaao0318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Qu, X.; Zhou, Y.; Song, G.; Abiri, N.; Xiao, Y.; Liang, F.; Jiang, D.; Hu, Z.; Yang, D. OsPRR37 confers an expanded regulation of the diurnal rhythms of the transcriptome and photoperiodic flowering pathways in rice. Plant Cell Environ. 2018, 41, 630–645. [Google Scholar] [CrossRef]
- Greenham, K.; Sartor, R.C.; Zorich, S.; Lou, P.; Mockler, T.C.; McClung, C.R. Expansion of the circadian transcriptome in Brassica rapa and genome-wide diversification of paralog expression patterns. Elife 2020, 9, e58993. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Shim, D.; Kumari, S.; Jung, H.; Jung, K.-H.; Jeong, H.; Kim, W.-Y.; Lee, S.I.; Jeong, M.-J. Transcriptome Analysis of Diurnal Gene Expression in Chinese Cabbage. Genes 2019, 10, 130. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zhao, W.; Li, D.; Chao, H.; Zhao, X.; Ta, N.; Li, Y.; Guan, Z.; Guo, L.; Zhang, L.; et al. Genetic dissection of the mechanism of flowering time based on an environmentally stable and specific QTL in Brassica napus. Plant Sci. 2018, 277, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Shi, J.; Qiu, D.; Li, R.; Zhang, C.; Wang, J.; Hou, J.; Zhao, J.; Shi, L.; Park, B.-S.; et al. Flowering Time Quantitative Trait Loci Analysis of Oilseed Brassica in Multiple Environments and Genomewide Alignment with Arabidopsis. Genetics 2007, 177, 2433–2444. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, R.; Kim, M.Y.; Calonje, M.; Moon, Y.-H.; Sung, Z.R. Temporal and Spatial Requirement of EMF1 Activity for Arabidopsis Vegetative and Reproductive Development. Mol. Plant 2009, 2, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Wenkel, S.; Turck, F.; Singer, K.; Gissot, L.; Le Gourrierec, J.; Samach, A.; Coupland, G. CONSTANS and the CCAAT Box Binding Complex Share a Functionally Important Domain and Interact to Regulate Flowering of Arabidopsis. Plant Cell 2006, 18, 2971–2984. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Wang, Z. Overexpression of COL9, a CONSTANS-LIKE gene, delays flowering by reducing expression of CO and FT in Arabidopsis thaliana. Plant J. 2005, 43, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Schiessl, S.; Samans, B.; Hüttel, B.; Reinhard, R.; Snowdon, R.J. Capturing sequence variation among flowering-time regulatory gene homologs in the allopolyploid crop species Brassica napus. Front. Plant Sci. 2014, 5, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raboanatahiry, N.; Chao, H.; Dalin, H.; Pu, S.; Yan, W.; Yu, L.; Wang, B.; Li, M. QTL Alignment for Seed Yield and Yield Related Traits in Brassica napus. Front. Plant Sci. 2018, 9, 1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Gu, X.; Yuan, W.; Schmitz Robert, J.; He, Y. Photoperiodic Control of the Floral Transition through a Distinct Polycomb Repressive Complex. Dev. Cell 2014, 28, 727–736. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Wu, D.; Kong, F.; Lin, K.; Zhang, H.; Li, G. The Arabidopsis thaliana Nuclear Factor Y Transcription Factors. Front. Plant Sci. 2017, 7, 2045. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Cao, M.; Ma, X.; Chen, W.; Zhao, J.; Sun, C.; Tan, L.; Liu, F. Integrated RNA Sequencing and QTL Mapping to Identify Candidate Genes from Oryza rufipogon Associated with Salt Tolerance at the Seedling Stage. Front. Plant Sci. 2017, 8, 1427. [Google Scholar] [CrossRef] [Green Version]
- Park, M.; Lee, J.-H.; Han, K.; Jang, S.; Han, J.; Lim, J.-H.; Jung, J.-W.; Kang, B.-C. A major QTL and candidate genes for capsaicinoid biosynthesis in the pericarp of Capsicum chinense revealed using QTL-seq and RNA-seq. Theor. Appl. Genet. 2018, 132, 515–529. [Google Scholar] [CrossRef]
- Montaigu, A.D.; Tóth, R.; Coupland, G. Plant development goes like clockwork. Trends Genet. 2010, 26, 296–306. [Google Scholar] [CrossRef]
- Song, Y.H.; Shim, J.S.; Kinmonth-Schultz, H.A.; Imaizumi, T. Photoperiodic Flowering: Time Measurement Mechanisms in Leaves. Annu. Rev. Plant Biol. 2015, 66, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Turck, F.; Fornara, F.; Coupland, G. Regulation and Identity of Florigen: FLOWERING LOCUS T Moves Center Stage. Annu. Rev. Plant Biol. 2008, 59, 573–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solanas, G.; Peixoto, F.O.; Perdiguero, E.; Jardí, M.; Ruiz-Bonilla, V.; Datta, D.; Symeonidi, A.; Castellanos, A.; Welz, P.-S.; Caballero, J.M.; et al. Aged Stem Cells Reprogram Their Daily Rhythmic Functions to Adapt to Stress. Cell 2017, 170, 678–692.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koda, S.; Onda, Y.; Matsui, H.; Takahagi, K.; Uehara-Yamaguchi, Y.; Shimizu, M.; Inoue, K.; Yoshida, T.; Sakurai, T.; Honda, H.; et al. Diurnal Transcriptome and Gene Network Represented through Sparse Modeling in Brachypodium distachyon. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef] [Green Version]
- George, D.; Mallery, P. IBM SPSS Statistics 26 Step by Step: A Simple Guide and Reference, 6th ed.; Routledge: New York, NY, USA, 2019. [Google Scholar]
- Doyle, J.J.T.; Doyle, J.L. Isolation of Plant DNA from fresh tissue. Focus 1990, 12, 13–15. [Google Scholar]
- Mason, A.S.; Higgins, E.E.; Snowdon, R.J.; Batley, J.; Stein, A.; Werner, C.; Parkin, I.A.P. A user guide to the Brassica 60K Illumina Infinium™ SNP genotyping array. Theor. Appl. Genet. 2017, 130, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Cai, G.; Yang, Q.; Yi, B.; Fan, C.; Zhang, C.; Edwards, D.; Batley, J.; Zhou, Y. A bi-filtering method for processing single nucleotide polymorphism array data improves the quality of genetic map and accuracy of quantitative trait locus mapping in doubled haploid populations of polyploid Brassica napus. BMC Genom. 2015, 16, 409. [Google Scholar] [CrossRef] [Green Version]
- Voorrips, R.; Van, D.; van den Heuvel, L.P.W.J.; Ooijen, J.; Van, J.W. JoinMap® 4.0: Software for the Calculation of Genetic Linkage Maps in Experimental Populations; Kyazma BV: Wageningen, The Netherlands, 2006. [Google Scholar]
- Sosnowski, O.; Charcosset, A.; Joets, J. BioMercator V3: An upgrade of genetic map compilation and quantitative trait loci meta-analysis algorithms. Bioinformatics 2012, 28, 2082–2083. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 7, 923–930. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Environment | Parents | DH Lines | ||||
|---|---|---|---|---|---|---|
| B409 a | ZS8 a | DH Lines Range | Mean ± SD | Skewness | Kurtosis | |
| 17 SG | 131 ± 2.94 | 106.6 ± 4.13 *** | 60–159 | 114.47 ± 22.95 | −0.59 | −0.52 |
| 18 KM | 133.43 ± 0.49 | 120.43 ± 0.73 **** | 100.5–139 | 126.03 ± 7.21 | −0.59 | 1.49 |
| 16 HZ | 68.44 ± 1.57 | 71 ± 2.24 *** | 64–87.3 | 74.73 ± 5.07 | 0.36 | −0.61 |
| Consensus QTLs | Identified QTLs | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| QTLs | Chr. a | CI (cM) b | PI (kb) c | QTLs | Peak (cM) | CI (cM) b | LOD d | PVE (%) e | Add f | Env. g |
| cqFT.A02-1 | A02 | 0–0.5 | 5672–5851 | qFT.A02-1 | 0 | 0–0.5 | 8.30 | 16.21 | 9.27 | 17 SG |
| cqFT.A03-1 | A03 | 26.33–40.39 | 574–4966 | qFT.A03-1 | 34 | 22.5–39.5 | 5.46 | 10.20 | −6.86 | 17 SG |
| qFT.A03-2 | 32 | 14.5–39.5 | 4.51 | 11.06 | −2.43 | 18 KM | ||||
| cqFT.A03-2 | A03 | 69.5–71.5 | 9834–10,261 | qFT.A03-3 | 71 | 69.5–71.5 | 11.82 | 17.56 | 2.82 | 16 HZ |
| cqFT.A03-3 | A03 | 95.5–97.5 | 12,981–15,100 | qFT.A03-4 | 97 | 95.5–97.5 | 5.16 | 6.99 | 1.80 | 16 HZ |
| cqFT.C08-1 | C08 | 0–2.5 | 802–1388 | qFT.C08-1 | 1 | 0–2.5 | 13.11 | 27.87 | −11.87 | 17 SG |
| qFT.C08-2 | 2 | 0–2.5 | 9.20 | 24.43 | −3.71 | 18 KM | ||||
| qFT.C08-3 | 0 | 0–0.5 | 4.13 | 5.32 | −1.64 | 16 HZ | ||||
| QTL Name | B. napus ID | TAIR ID | Gene | Regulator | Pathway |
|---|---|---|---|---|---|
| cqFT.A03-1 | BnaA03g03410D a | AT5G11530 | EMF1 | Negative | Autonomous pathway, photoperiod pathway |
| BnaA03g04040D | AT5G12840 | NF-YA1, HAP2B | Negative | Photoperiod pathway | |
| cqFT.A03-3 | BnaA03g30130D | AT3G07650 | COL9 | Negative | Photoperiod pathway |
| cqFT.C08-1 | BnaC08g01530D | AT1G05835 | PHD finger proteins | Unknown | Unknown |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, J.; Li, B.; Cui, Y.; Zhuo, C.; Gu, Y.; Hu, K.; Wen, J.; Yi, B.; Shen, J.; Ma, C.; et al. QTL Mapping and Diurnal Transcriptome Analysis Identify Candidate Genes Regulating Brassica napus Flowering Time. Int. J. Mol. Sci. 2021, 22, 7559. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147559
Song J, Li B, Cui Y, Zhuo C, Gu Y, Hu K, Wen J, Yi B, Shen J, Ma C, et al. QTL Mapping and Diurnal Transcriptome Analysis Identify Candidate Genes Regulating Brassica napus Flowering Time. International Journal of Molecular Sciences. 2021; 22(14):7559. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147559
Chicago/Turabian StyleSong, Jurong, Bao Li, Yanke Cui, Chenjian Zhuo, Yuanguo Gu, Kaining Hu, Jing Wen, Bin Yi, Jinxiong Shen, Chaozhi Ma, and et al. 2021. "QTL Mapping and Diurnal Transcriptome Analysis Identify Candidate Genes Regulating Brassica napus Flowering Time" International Journal of Molecular Sciences 22, no. 14: 7559. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147559